
Protein Adsorption on Solid Supported Membranes: Monitoring the Transport Activity of P-Type ATPases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
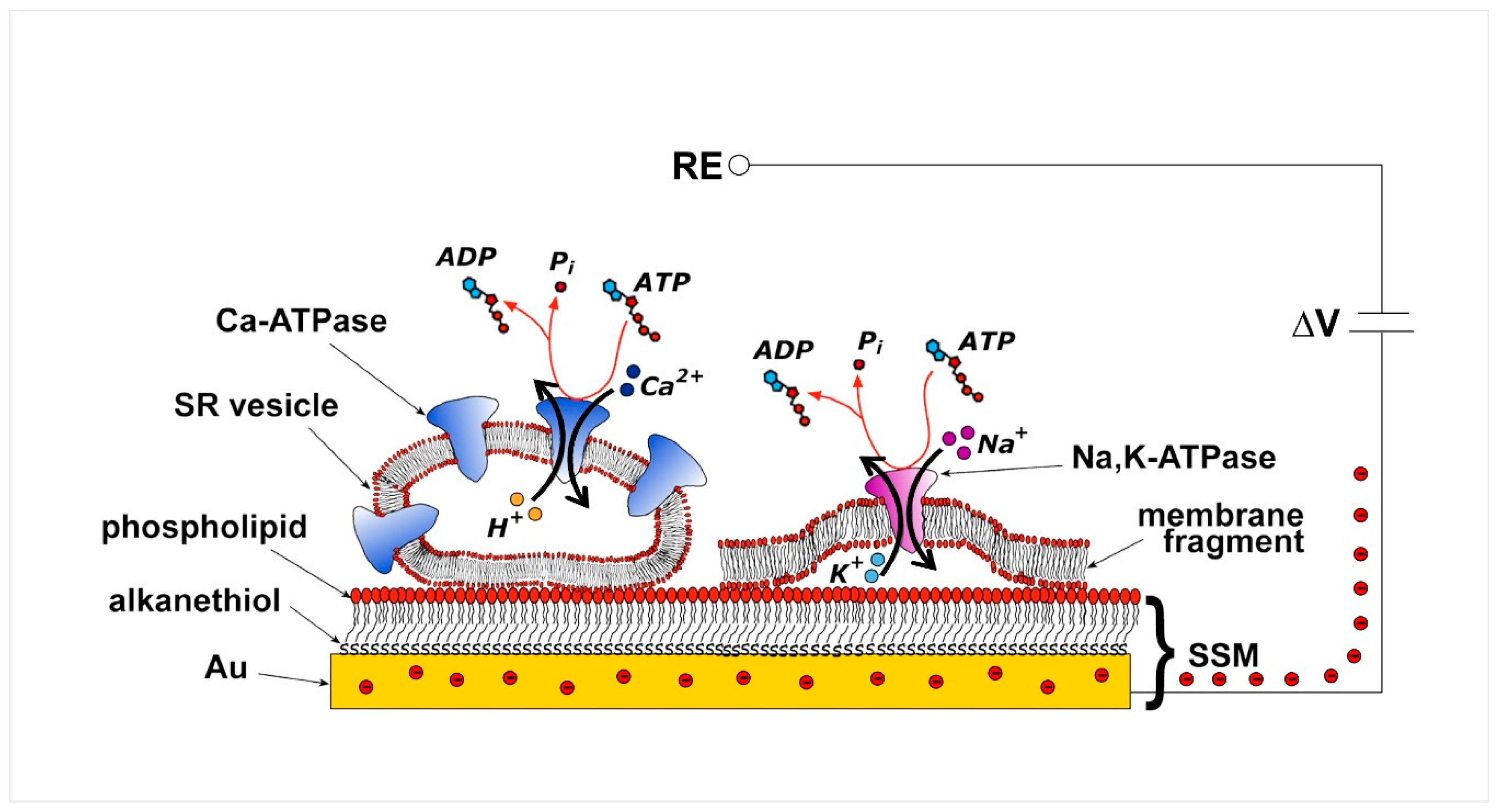
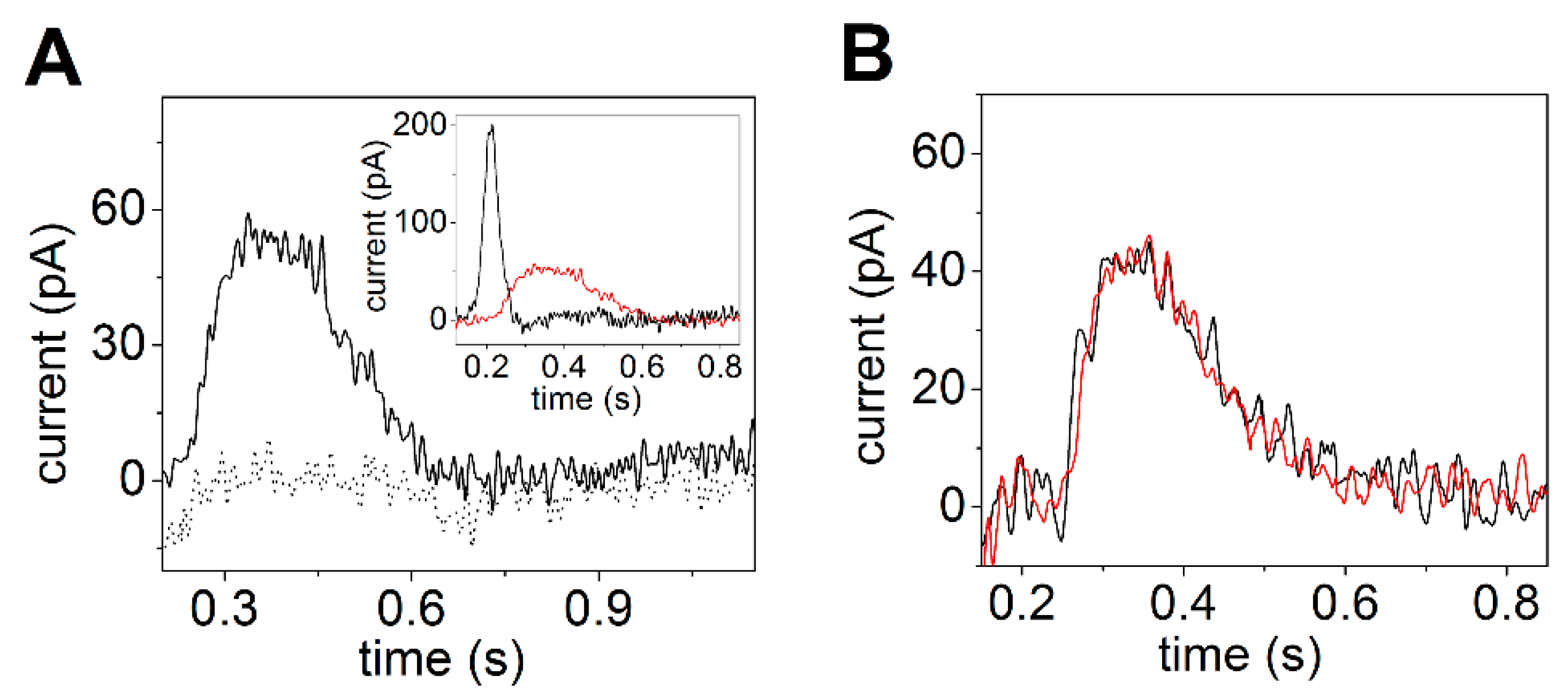
2. Current Measurements on Solid Supported Membranes
3. P-Type ATPases Investigated on Solid Supported Membranes
3.1. Sarcoplasmic Reticulum Ca2+-ATPase
3.2. Cu+-ATPases ATP7A and ATP7B
3.3. P4-ATPase ATP8A2
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kühlbrandt, W. Biology, structure and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Bublitz, M.; Morth, J.P.; Nissen, P. P-type ATPases at a glance. J. Cell Sci. 2011, 124, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Dyla, M.; Kjærgaard, M.; Poulsen, H.; Nissen, P. Structure and mechanism of P-type ATPase ion pumps. Annu. Rev. Biochem. 2020, 89, 583–603. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.; Clarke, R.J. Evolutionary analysis of the lysine-rich N-terminal cytoplasmic domains of the gastric H+,K+-ATPase and the Na+,K+-ATPase. J. Membr. Biol. 2018, 251, 653–666. [Google Scholar] [CrossRef]
- Albers, R.W. Biochemical aspects of active transport. Ann. Rev. Biochem. 1967, 36, 727–756. [Google Scholar] [CrossRef]
- Post, R.L.; Hegyvary, C.; Kume, S. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 1972, 247, 6530–6540. [Google Scholar]
- Apell, H.J. Structure-function relationship in P-type ATPases—A biophysical approach. Rev. Physiol. Biochem. Pharmacol. 2003, 150, 1–35. [Google Scholar] [CrossRef]
- Bublitz, M.; Poulsen, H.; Morth, J.P.; Nissen, P. In and out of the cation pumps: P-type ATPase structure revisited. Curr. Opin. Struct. Biol. 2010, 20, 431–439. [Google Scholar] [CrossRef]
- Toyoshima, C.; Cornelius, F. New crystal structures of PII-type ATPases: Excitement continues. Curr. Opin. Struct. Biol. 2013, 23, 507–514. [Google Scholar] [CrossRef]
- De Meis, L.; Vianna, A.L. Energy interconversion by the Ca2+-dependent ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 1979, 48, 275–292. [Google Scholar] [CrossRef]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Inesi, G. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 269–292. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C. Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 2008, 476, 3–11. [Google Scholar] [CrossRef]
- Toyoshima, C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 2009, 1793, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Møller, J.V.; Olesen, C.; Winther, A.-M.L.; Nissen, P. The sarcoplasmic Ca2+-ATPase: Design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 2010, 43, 501–566. [Google Scholar] [CrossRef]
- Bublitz, M.; Musgaard, M.; Poulsen, H.; Thøgersen, L.; Olesen, C.; Schiøtt, B.; Morth, J.P.; Møller, J.V.; Nissen, P. Ion pathways in the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 2013, 288, 10759–10765. [Google Scholar] [CrossRef] [PubMed]
- Dyla, M.; Basse Hansen, S.; Nissen, P.; Kjaergaard, M. Structural dynamics of P-type ATPase ion pumps. Biochem. Soc. Trans. 2019, 47, 1247–1257. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Espinoza-Fonseca, L.M. Linking biochemical and structural states of SERCA: Achievements, challenges and new opportunities. Int. J. Mol. Sci. 2020, 21, 4146. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Fendler, K. Charge transfer in P-type ATPases investigated on planar membranes. Arch. Biochem. Biophys. 2008, 476, 75–86. [Google Scholar] [CrossRef]
- Seifert, K.; Fendler, K.; Bamberg, E. Charge transport by ion translocating membrane proteins on solid supported membranes. Biophys. J. 1993, 64, 384–391. [Google Scholar] [CrossRef]
- Pintschovius, J.; Fendler, K. Charge Translocation by the Na+/K+-ATPase investigated on solid supported membranes: Rapid solution exchange with a new technique. Biophys. J. 1999, 76, 814–826. [Google Scholar] [CrossRef]
- Bartolommei, G.; Moncelli, M.R.; Rispoli, G.; Kelety, B.; Tadini-Buoninsegni, F. Electrogenic ion pumps investigated on a solid supported membrane: Comparison of current and voltage measurements. Langmuir 2009, 25, 10925–10931. [Google Scholar] [CrossRef]
- Schulz, P.; Garcia-Celma, J.J.; Fendler, K. SSM-based electrophysiology. Methods 2008, 46, 97–103. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Fendler, K. Recording of pump and transporter activity using solid-supported membranes (SSM-based electrophysiology). In Pumps, Channels and Transporters: Methods of Functional Analysis; Clarke, R.J., Khalid, M.A.A., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015; pp. 147–177. [Google Scholar] [CrossRef]
- Pintschovius, J.; Fendler, K.; Bamberg, E. Charge translocation by the Na+/K+-ATPase investigated on solid supported membranes: Cytoplasmic cation binding and release. Biophys. J. 1999, 76, 827–836. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Guidelli, R.; Inesi, G. Pre-steady state electrogenic events of Ca2+/H+ exchange and transport by the Ca2+-ATPase. J. Biol. Chem. 2006, 281, 37720–37727. [Google Scholar] [CrossRef]
- Liu, Y.; Pilankatta, R.; Lewis, D.; Inesi, G.; Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R. High-yield heterologous expression of wild type and mutant Ca2+ ATPase: Characterization of Ca2+ binding sites by charge transfer. J. Mol. Biol. 2009, 391, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Kelety, B.; Diekert, K.; Tobien, J.; Watzke, N.; Dörner, W.; Obrdlik, P.; Fendler, K. Transporter assays using solid supported membranes: A novel screening platform for drug discovery. Assay Drug Dev. Technol. 2006, 4, 575–582. [Google Scholar] [CrossRef]
- Mattle, D.; Zhang, L.; Sitsel, O.; Pedersen, L.T.; Moncelli, M.R.; Tadini-Buoninsegni, F.; Gourdon, P.; Rees, D.C.; Nissen, P.; Meloni, G. A sulfur-based transport pathway in Cu+-ATPases. EMBO Rep. 2015, 16, 728–740. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Pilankatta, R.; Lewis, D.; Inesi, G. ATP dependent charge movement in ATP7B Cu+-ATPase is demonstrated by pre-steady state electrical measurements. FEBS Lett. 2010, 584, 4619–4622. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Mikkelsen, S.A.; Mogensen, L.S.; Molday, R.S.; Andersen, J.P. Phosphatidylserine flipping by the P4-ATPase ATP8A2 is electrogenic. Proc. Natl. Acad. Sci. USA 2019, 116, 16332–16337. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Palchetti, I. Label-Free Bioelectrochemical methods for evaluation of anticancer drug effects at a molecular level. Sensors 2020, 20, 1812. [Google Scholar] [CrossRef] [PubMed]
- Geibel, S.; Flores-Herr, N.; Licher, T.; Vollert, H. Establishment of cell-free electrophysiology for ion transporters: Application for pharmacological profiling. J. Biomol. Screen. 2006, 11, 262–268. [Google Scholar] [CrossRef]
- Barthmes, M.; Liao, J.; Jiang, Y.; Brüggemann, A.; Wahl-Schott, C. Electrophysiological characterization of the archaeal transporter NCX_Mj using solid supported membrane technology. J. Gen. Physiol. 2016, 147, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Bazzone, A.; Barthmes, M.; Fendler, K. SSM-based electrophysiology for transporter research. Methods Enzymol. 2017, 594, 31–83. [Google Scholar] [CrossRef] [PubMed]
- Inesi, G.; Tadini-Buoninsegni, F. Ca2+/H+ exchange, lumenal Ca2+ release and Ca2+/ATP coupling ratios in the sarcoplasmic reticulum ATPase. J. Cell Commun. Signal. 2014, 8, 5–11. [Google Scholar] [CrossRef]
- Primeau, J.O.; Armanious, G.P.; Fisher, M.E.; Young, H.S. The SarcoEndoplasmic Reticulum Calcium ATPase. Subcell. Biochem. 2018, 87, 229–258. [Google Scholar] [CrossRef]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.R.; Vangheluwe, P. Primary active Ca2+ transport systems in health and disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035113. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef]
- Grover, A.K.; Khan, I. Calcium pump isoforms: Diversity, selectivity and plasticity. Cell Calcium 1992, 13, 9–17. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C.; Ferguson, D.G. Density and disposition of Ca2+-ATPase in sarcoplasmic reticulum membrane as determined by shadowing techniques. Biophys. J. 1985, 48, 607–615. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Tal, D.M.; Lewis, D.; Inesi, G. Effects of high-affinity inhibitors on partial reactions, charge movements, and conformational states of the Ca2+ transport ATPase (sarco-endoplasmic reticulum Ca2+ ATPase). Mol. Pharmacol. 2008, 73, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y.; Inesi, G. Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J. Biol. Chem. 1991, 266, 13503–13506. [Google Scholar]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar]
- Inesi, G.; Lewis, D.; Toyoshima, C.; Hirata, A.; de Meis, L. Conformational fluctuations of the Ca2+-ATPase in the native membrane environment. Effects of pH, temperature, catalytic substrates, and thapsigargin. J. Biol. Chem. 2008, 283, 1189–1196. [Google Scholar] [CrossRef]
- Lewis, D.; Pilankatta, R.; Inesi, G.; Bartolommei, G.; Moncelli, M.R.; Tadini-Buoninsegni, F. Distinctive features of catalytic and transport mechanisms in mammalian sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) and Cu+ (ATP7A/B) ATPases. J. Biol. Chem. 2012, 287, 32717–32727. [Google Scholar] [CrossRef]
- Yu, X.; Hao, L.; Inesi, G. A pK change of acidic residues contributes to cation countertransport in the Ca-ATPase of sarcoplasmic reticulum. Role of H+ in Ca2+-ATPase countertransport. J. Biol. Chem. 1994, 269, 16656–16661. [Google Scholar]
- Cornelius, F.; Møller, J.V. Electrogenic pump current of sarcoplasmic reticulum Ca2+-ATPase reconstituted at high lipid/protein ratio. FEBS Lett. 1991, 284, 46–50. [Google Scholar] [CrossRef]
- Levy, D.; Seigneuret, M.; Bluzat, A.; Rigaud, J.L. Evidence for proton countertransport by the sarcoplasmic reticulum Ca2+-ATPase during calcium transport in reconstituted proteoliposomes with low ionic permeability. J. Biol. Chem. 1990, 265, 19524–19534. [Google Scholar]
- Yu, X.; Carroll, S.; Rigaud, J.L.; Inesi, G. H+ countertransport and electrogenicity of the sarcoplasmic reticulum Ca2+ pump in reconstituted proteoliposomes. Biophys. J. 1993, 64, 1232–1242. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.A.; Sahoo, S.K.; Periasamy, M. Phospholamban and sarcolipin: Are they functionally redundant or distinct regulators of the Sarco (Endo)Plasmic Reticulum Calcium ATPase? J. Mol. Cell. Cardiol. 2016, 91, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Glaves, J.P.; Trieber, C.A.; Ceholski, D.K.; Stokes, D.L.; Young, H.S. Phosphorylation and mutation of phospholamban alter physical interactions with the sarcoplasmic reticulum calcium pump. J. Mol. Biol. 2011, 405, 707–723. [Google Scholar] [CrossRef]
- Glaves, J.P.; Primeau, J.O.; Espinoza-Fonseca, L.M.; Lemieux, M.J.; Young, H.S. The phospholamban pentamer alters function of the sarcoplasmic reticulum calcium pump SERCA. Biophys. J. 2019, 116, 633–647. [Google Scholar] [CrossRef]
- Smeazzetto, S.; Armanious, G.P.; Moncelli, M.R.; Bak, J.J.; Lemieux, M.J.; Young, H.S.; Tadini-Buoninsegni, F. Conformational memory in the association of the transmembrane protein phospholamban with the sarcoplasmic reticulum calcium pump SERCA. J. Biol. Chem. 2017, 292, 21330–21339. [Google Scholar] [CrossRef]
- Schörner, M.; Beyer, S.R.; Southall, J.; Cogdell, R.J.; Köhler, J. Conformational memory of a protein revealed by single-molecule spectroscopy. J. Phys. Chem. B 2015, 119, 13964–13970. [Google Scholar] [CrossRef]
- Csermely, P.; Kunsic, N.; Mendik, P.; Kerestély, M.; Faragó, T.; Veres, D.V.; Tompa, P. Learning of signaling networks: Molecular mechanisms. Trends Biochem. Sci. 2020, 45, 284–294. [Google Scholar] [CrossRef]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef]
- Anderson, D.M.; Makarewich, C.A.; Anderson, K.M.; Shelton, J.M.; Bezprozvannaya, S.; Bassel-Duby, R.; Olson, E.N. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci. Signal. 2016, 9, ra119. [Google Scholar] [CrossRef]
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016, 351, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R.; Dalton, M.P.; Cho, E.E.; Pribadi, M.P.; Zak, T.J.; Šeflová, J.; Makarewich, C.A.; Olson, E.N.; Robia, S.L. Newly discovered micropeptide regulators of SERCA form oligomers but bind to the pump as monomers. J. Mol. Biol. 2019, 431, 4429–4443. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H.; Lutsenko, S. Copper transport in mammalian cells: Special care for a metal with special needs. J. Biol. Chem. 2009, 284, 25461–25465. [Google Scholar] [CrossRef]
- La Fontaine, S.; Ackland, M.L.; Mercer, J.F. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: Emerging roles. Int. J. Biochem. Cell Biol. 2010, 42, 206–209. [Google Scholar] [CrossRef]
- Gourdon, P.; Sitsel, O.; Lykkegaard Karlsen, J.; Birk Møller, L.; Nissen, P. Structural models of the human copper P-type ATPases ATP7A and ATP7B. Biol. Chem. 2012, 393, 205–216. [Google Scholar] [CrossRef]
- Petruzzelli, R.; Polishchuk, R.S. Activity and trafficking of copper-transporting ATPases in tumor development and defense against platinum-based drugs. Cells 2019, 8, 1080. [Google Scholar] [CrossRef]
- Yu, C.H.; Dolgova, N.V.; Dmitriev, O.Y. Dynamics of the metal binding domains and regulation of the human copper transporters ATP7B and ATP7A. IUBMB Life 2017, 69, 226–235. [Google Scholar] [CrossRef]
- Argüello, J.M.; González-Guerrero, M.; Raimunda, D. Bacterial transition metal P1B-ATPases: Transport mechanism and roles in virulence. Biochemistry 2011, 50, 9940–9949. [Google Scholar] [CrossRef]
- Argüello, J.M.; Raimunda, D.; González-Guerrero, M. Metal transport across biomembranes: Emerging models for a distinct chemistry. J. Biol. Chem. 2012, 287, 13510–13517. [Google Scholar] [CrossRef]
- Mattle, D.; Sitsel, O.; Autzen, H.E.; Meloni, G.; Gourdon, P.; Nissen, P. On allosteric modulation of P-type Cu+-ATPases. J. Mol. Biol. 2013, 425, 2299–2308. [Google Scholar] [CrossRef]
- Sitsel, O.; Grønberg, C.; Autzen, H.E.; Wang, K.; Meloni, G.; Nissen, P.; Gourdon, P. Structure and function of Cu(I)- and Zn(II)-ATPases. Biochemistry 2015, 54, 5673–5683. [Google Scholar] [CrossRef] [PubMed]
- Wittung-Stafshede, P. Unresolved questions in human copper pump mechanisms. Q. Rev. Biophys. 2015, 48, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, P.; Liu, X.Y.; Skjørringe, T.; Morth, J.P.; Møller, L.B.; Pedersen, B.P.; Nissen, P. Crystal structure of a copper-transporting PIB-type ATPase. Nature 2011, 475, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Mattle, D.; Sitsel, O.; Klymchuk, T.; Nielsen, A.M.; Møller, L.B.; White, S.H.; Nissen, P.; Gourdon, P. Copper-transporting P-type ATPases use a unique ion-release pathway. Nat. Struct. Mol. Biol. 2014, 21, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Bartolommei, G.; Moncelli, M.R.; Inesi, G.; Galliani, A.; Sinisi, M.; Losacco, M.; Natile, G.; Arnesano, F. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew. Chem. Int. Ed. Engl. 2014, 53, 1297–1301. [Google Scholar] [CrossRef]
- Verjovski-Almeida, S.; Kurzmack, M.; Inesi, G. Partial reactions in the catalytic and transport cycle of sarcoplasmic reticulum ATPase. Biochemistry 1978, 17, 5006–5013. [Google Scholar] [CrossRef]
- González-Guerrero, M.; Eren, E.; Rawat, S.; Stemmler, T.L.; Argüello, J.M. Structure of the two transmembrane Cu+ transport sites of the Cu+-ATPases. J. Biol. Chem. 2008, 283, 29753–29759. [Google Scholar] [CrossRef]
- Obara, K.; Miyashita, N.; Xu, C.; Toyoshima, I.; Sugita, Y.; Inesi, G.; Toyoshima, C. Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+. Proc. Natl. Acad. Sci. USA 2005, 102, 14489–14496. [Google Scholar] [CrossRef]
- Wang, K.; Sitsel, O.; Meloni, G.; Autzen, H.E.; Andersson, M.; Klymchuk, T.; Nielsen, A.M.; Rees, D.C.; Nissen, P.; Gourdon, P. Structure and mechanism of Zn2+-transporting P-type ATPases. Nature 2014, 514, 518–522. [Google Scholar] [CrossRef]
- Abeyrathna, N.; Abeyrathna, S.; Morgan, M.T.; Fahrni, C.J.; Meloni, G. Transmembrane Cu(I) P-type ATPase pumps are electrogenic uniporters. Dalton Trans. 2020. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.; Levine, T.P. Lipid traffic: Floppy drives and a superhighway. Nat. Rev. Mol. Cell Biol. 2005, 6, 209–220. [Google Scholar] [CrossRef]
- Lenoir, G.; Williamson, P.; Holthuis, J.C. On the origin of lipid asymmetry: The flip side of ion transport. Curr. Opin. Chem. Biol. 2007, 11, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Sakuragi, T.; Segawa, K. Flippase and scramblase for phosphatidylserine exposure. Curr. Opin. Immunol. 2020, 62, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.J.; Hossain, K.R.; Cao, K. Physiological roles of transverse lipid asymmetry of animal membranes. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183382. [Google Scholar] [CrossRef] [PubMed]
- Puts, C.F.; Holthuis, J.C. Mechanism and significance of P4 ATPase-catalyzed lipid transport: Lessons from a Na+/K+-pump. Biochim. Biophys. Acta 2009, 1791, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Hankins, H.M.; Baldridge, R.D.; Xu, P.; Graham, T.R. Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic 2015, 16, 35–47. [Google Scholar] [CrossRef]
- Andersen, J.P.; Vestergaard, A.L.; Mikkelsen, S.A.; Mogensen, L.S.; Chalat, M.; Molday, R.S. P4-ATPases as phospholipid flippases-structure, function, and enigmas. Front. Physiol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Devaux, P.F.; López-Montero, I.; Bryde, S. Proteins involved in lipid translocation in eukaryotic cells. Chem. Phys. Lipids 2006, 141, 119–132. [Google Scholar] [CrossRef]
- Van der Mark, V.A.; Elferink, R.P.; Paulusma, C.C. P4 ATPases: Flippases in health and disease. Int. J. Mol. Sci. 2013, 14, 7897–7922. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Marques, R.L.; Theorin, L.; Palmgren, M.G.; Pomorski, T.G. P4-ATPases: Lipid flippases in cell membranes. Pflugers Arch. 2014, 466, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Panatala, R.; Hennrich, H.; Holthuis, J.C. Inner workings and biological impact of phospholipid flippases. J. Cell Sci. 2015, 128, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Roland, B.P.; Graham, T.R. Decoding P4-ATPase substrate interactions. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 513–527. [Google Scholar] [CrossRef]
- Bryde, S.; Hennrich, H.; Verhulst, P.M.; Devaux, P.F.; Lenoir, G.; Holthuis, J.C. CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 2010, 285, 40562–40572. [Google Scholar] [CrossRef]
- Coleman, J.A.; Molday, R.S. Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem. 2011, 286, 17205–17216. [Google Scholar] [CrossRef]
- Timcenko, M.; Lyons, J.A.; Januliene, D.; Ulstrup, J.J.; Dieudonné, T.; Montigny, C.; Ash, M.R.; Karlsen, J.L.; Boesen, T.; Kühlbrandt, W.; et al. Structure and autoregulation of a P4-ATPase lipid flippase. Nature 2019, 571, 366–370. [Google Scholar] [CrossRef]
- Bai, L.; Kovach, A.; You, Q.; Hsu, H.C.; Zhao, G.; Li, H. Autoinhibition and activation mechanisms of the eukaryotic lipid flippase Drs2p-Cdc50p. Nat. Commun. 2019, 10, 4142. [Google Scholar] [CrossRef]
- Hiraizumi, M.; Yamashita, K.; Nishizawa, T.; Nureki, O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 2019, 365, 1149–1155. [Google Scholar] [CrossRef]
- Lyons, J.A.; Timcenko, M.; Dieudonné, T.; Lenoir, G.; Nissen, P. P4-ATPases: How an old dog learnt new tricks-structure and mechanism of lipid flippases. Curr. Opin. Struct. Biol. 2020, 63, 65–73. [Google Scholar] [CrossRef]
- Nakanishi, H.; Irie, K.; Segawa, K.; Hasegawa, K.; Fujiyoshi, Y.; Nagata, S.; Abe, K. Crystal structure of a human plasma membrane phospholipid flippase. J. Biol. Chem. 2020, 295, 10180–10194. [Google Scholar] [CrossRef]
- Baldridge, R.D.; Graham, T.R. Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc. Natl. Acad. Sci. USA 2013, 110, E358–E367. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, A.L.; Coleman, J.A.; Lemmin, T.; Mikkelsen, S.A.; Molday, L.L.; Vilsen, B.; Molday, R.S.; Dal Peraro, M.; Andersen, J.P. Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. USA 2014, 111, E1334–E1343. [Google Scholar] [CrossRef] [PubMed]
- Montigny, C.; Lyons, J.; Champeil, P.; Nissen, P.; Lenoir, G. On the molecular mechanism of flippase- and scramblase-mediated phospholipid transport. Biochim. Biophys. Acta 2016, 1861, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.S.; Costa, S.R.; Duelli, A.S.; Andersen, P.A.; Poulsen, L.R.; Stanchev, L.D.; Gourdon, P.; Palmgren, M.; Günther Pomorski, T.; López-Marqués, R.L. Phospholipid flipping involves a central cavity in P4 ATPases. Sci. Rep. 2017, 7, 17621. [Google Scholar] [CrossRef] [PubMed]
- Gantzel, R.H.; Mogensen, L.S.; Mikkelsen, S.A.; Vilsen, B.; Molday, R.S.; Vestergaard, A.L.; Andersen, J.P. Disease mutations reveal residues critical to the interaction of P4-ATPases with lipid substrates. Sci. Rep. 2017, 7, 10418. [Google Scholar] [CrossRef]
- Coleman, J.A.; Vestergaard, A.L.; Molday, R.S.; Vilsen, B.; Andersen, J.P. Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. USA 2012, 109, 1449–1454. [Google Scholar] [CrossRef]
- Coleman, J.A.; Kwok, M.C.; Molday, R.S. Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem. 2009, 284, 32670–32679. [Google Scholar] [CrossRef]
- Hossain, K.R.; Clarke, R.J. General and specific interactions of the phospholipid bilayer with P-type ATPases. Biophys. Rev. 2019, 11, 353–364. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug interactions with the Ca2+-ATPase from sarco(endo)plasmic reticulum (SERCA). Front. Mol. Biosci. 2018, 5, 36. [Google Scholar] [CrossRef]
- Yatime, L.; Buch-Pedersen, M.J.; Musgaard, M.; Morth, J.P.; Winther, A.-M.L.; Pedersen, B.P.; Olesen, C.; Andersen, J.P.; Vilsen, B.; Schiøtt, B.; et al. P-type ATPases as drug targets: Tools for medicine and science. Biochim. Biophys. Acta-Bioenerg. 2009, 1787, 207–220. [Google Scholar] [CrossRef]
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011, 39, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/endoplasmic reticulum calcium ATPase inhibitors: Beyond anticancer perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tadini-Buoninsegni, F. Protein Adsorption on Solid Supported Membranes: Monitoring the Transport Activity of P-Type ATPases. Molecules 2020, 25, 4167. https://doi.org/10.3390/molecules25184167
Tadini-Buoninsegni F. Protein Adsorption on Solid Supported Membranes: Monitoring the Transport Activity of P-Type ATPases. Molecules. 2020; 25(18):4167. https://doi.org/10.3390/molecules25184167
Chicago/Turabian StyleTadini-Buoninsegni, Francesco. 2020. "Protein Adsorption on Solid Supported Membranes: Monitoring the Transport Activity of P-Type ATPases" Molecules 25, no. 18: 4167. https://doi.org/10.3390/molecules25184167
APA StyleTadini-Buoninsegni, F. (2020). Protein Adsorption on Solid Supported Membranes: Monitoring the Transport Activity of P-Type ATPases. Molecules, 25(18), 4167. https://doi.org/10.3390/molecules25184167