Coupling of HSP72 α-Helix Subdomains by the Unexpected Irreversible Targeting of Lysine-56 over Cysteine-17; Coevolution of Covalent Bonding


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methodology
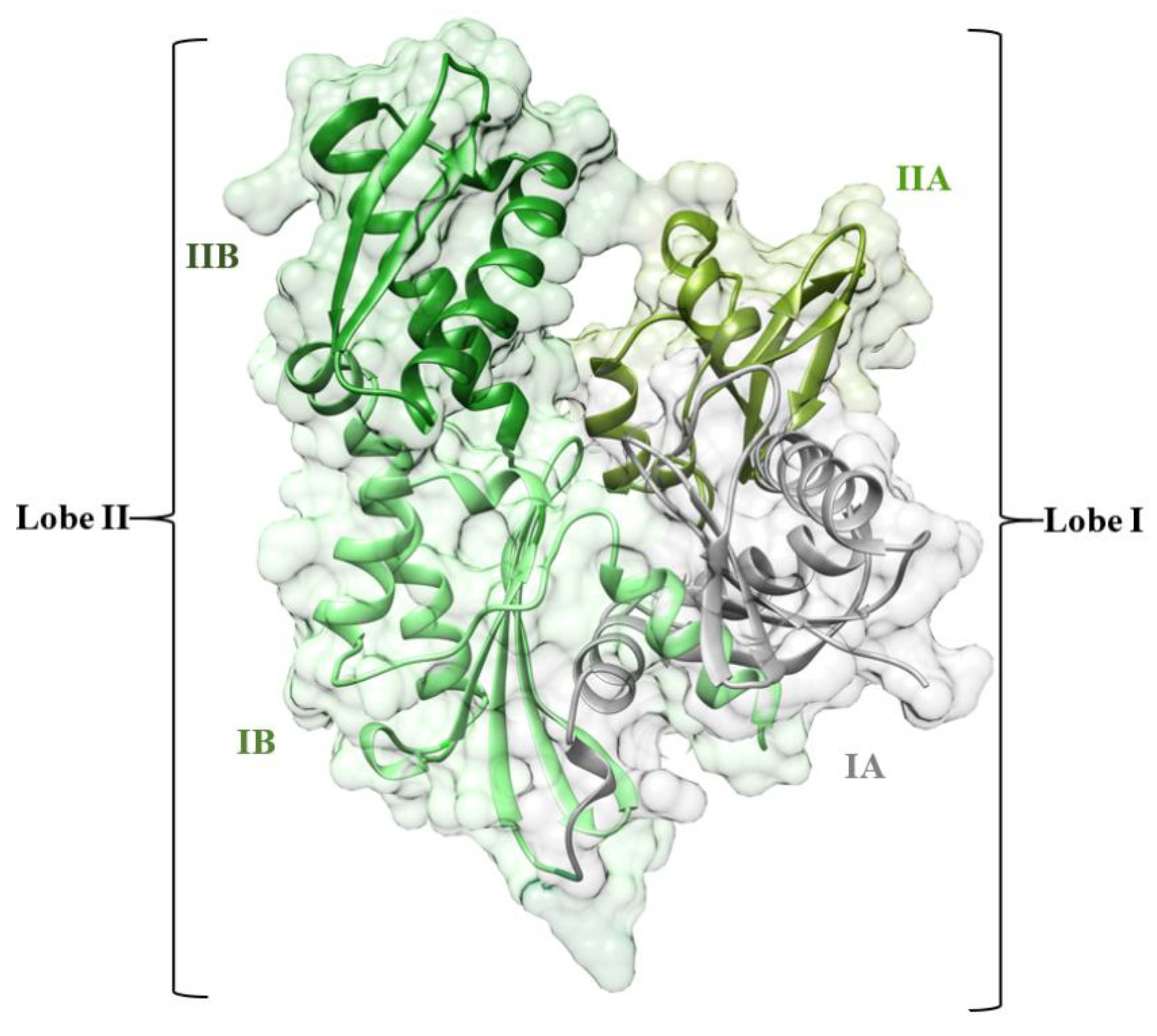
2.1. System Preparation
2.2. Covalent Docking
2.3. Covalent Molecular Dynamic Simulation
2.4. Clustering and Principal Component Analysis
3. Result and Discussion
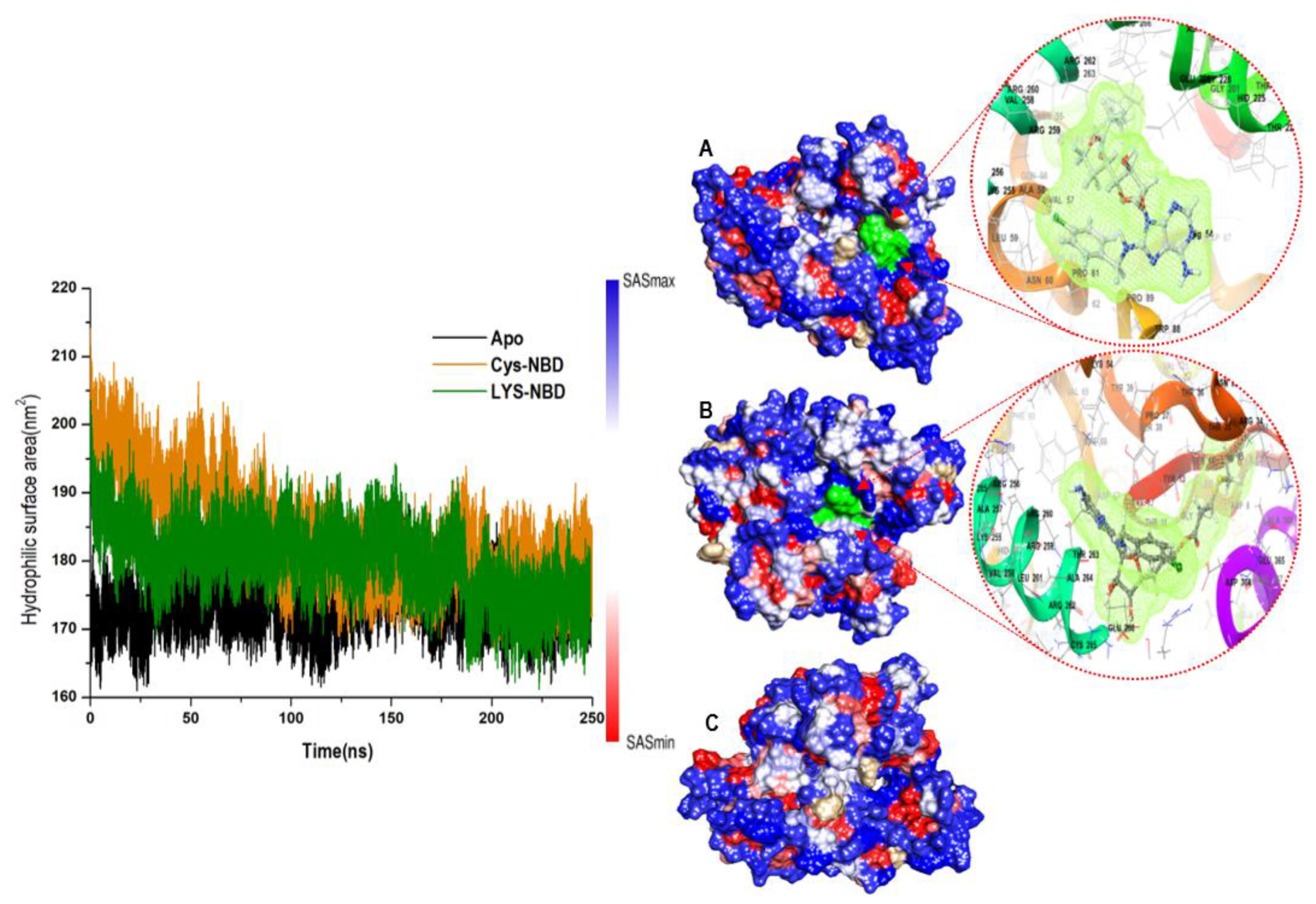
3.1. Overall Structural Stability and Dynamics of the Simulated Systems
3.2. Analysis of Secondary Structure Variation
3.3. Conformational Clustering and Principal Component Analysis
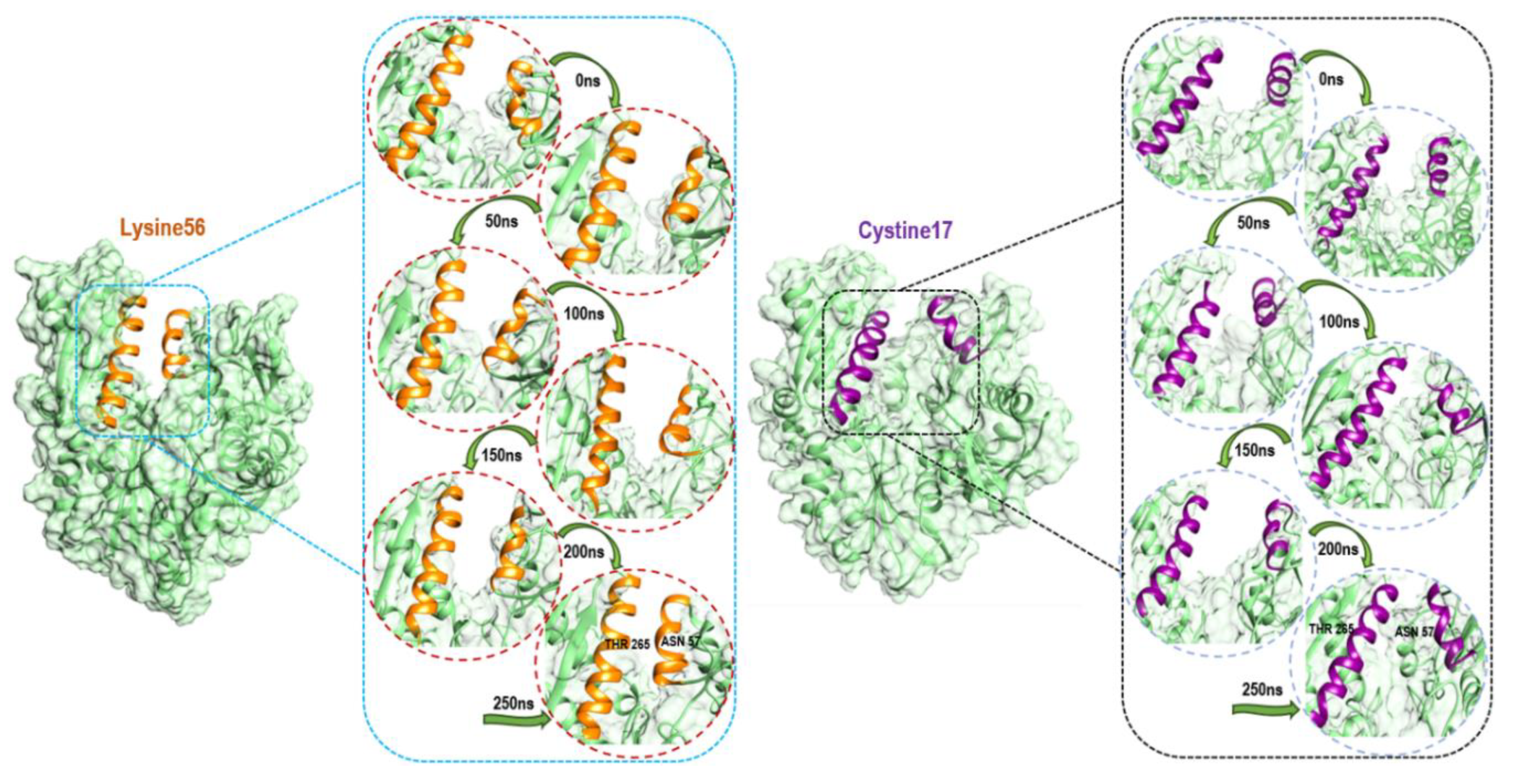
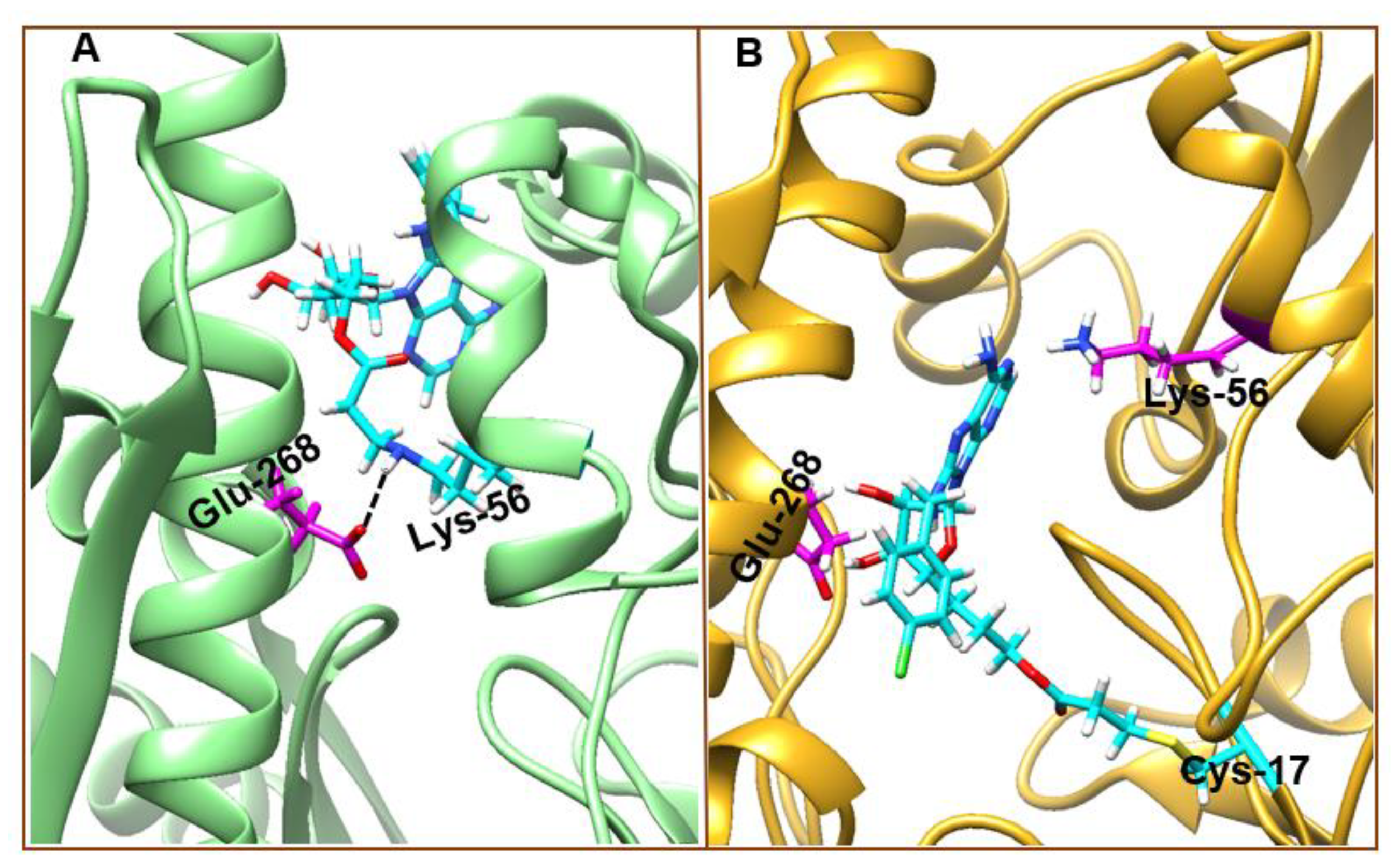
3.4. Understanding Structural Dynamics upon 8-N-Benzyladenosine Coupling to Lysine and Cysteine of HSP72-NBD α-Helices
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathew, A.; Morimoto, R.I. Role of the Heat-Shock Response in the Life and Death of Proteins. Ann. N. Y. Acad. Sci. 1998, 851, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Nollen, E.A.A.; Morimoto, R.I. Chaperoning signaling pathways: Molecular chaperones as stress-sensing “heat shock” proteins. J. Cell Sci. 2002, 115, 2809–2816. [Google Scholar] [PubMed]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.K.K.; Sakamoto, K.M.; Milani, M.; Aldana-Masankgay, G.; Fan, D.; Wu, K.; Lee, C.W.; Cho, C.H.; Yu, J.; Sung, J.J.Y. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist. Updat. 2010, 13, 87–92. [Google Scholar] [CrossRef]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Hunt, C.; Morimoto, R. Structure and expression of the human gene encoding major heat shock protein HSP70. Mol. Cell. Biol. 1985, 5, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [Green Version]
- Jäättelä, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar] [CrossRef]
- Nylandsted, J.; Rohde, M.; Brand, K.; Bastholm, L.; Elling, F.; Jaattela, M. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc. Natl. Acad. Sci. USA 2000, 97, 7871–7876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylandsted, J.; Wick, W.; Hirt, U.A.; Brand, K.; Rohde, M.; Leist, M.; Weller, M.; Jäättelä, M. Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res. 2002, 62, 7139–7142. [Google Scholar] [PubMed]
- Powers, M.V.; Jones, K.; Barillari, C.; Westwood, I.; van Montfort, R.L.M.; Workman, P. Targeting HSP70: The second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 2010, 9, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Penkler, D.; Sensoy, Ö.; Atilgan, C.; Tastan Bishop, Ö. Perturbation–Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017, 57, 1359–1374. [Google Scholar] [CrossRef]
- Sharma, D.; Masison, D.C. Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept. Lett. 2009, 16, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, F.; Merelli, I.; Colombo, G.; Milanesi, L.; Morra, G. Molecular mechanism of allosteric communication in Hsp70 revealed by molecular dynamics simulations. PLoS Comput. Biol. 2012, 8, e1002844. [Google Scholar] [CrossRef] [Green Version]
- Leu, J.I.-J.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L. Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin. Biochem. Pharmacol. 1999, 57, 877–880. [Google Scholar] [CrossRef]
- Ding, Y.; Ding, C.; Ye, N.; Liu, Z.; Wold, E.A.; Chen, H.; Wild, C.; Shen, Q.; Zhou, J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016, 122, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.Q.; Kirby, C.A.; Zhou, W.; Schuhmann, T.; Kityk, R.; Kipp, D.R.; Baird, J.; Chen, J.; Chen, Y.; Chung, F.; et al. The Novolactone Natural Product Disrupts the Allosteric Regulation of Hsp70. Chem. Biol. 2015, 22, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Regan, L.; Sampson, J.; Richards, M.W.; Knebel, A.; Roth, D.; Hood, F.E.; Straube, A.; Royle, S.J.; Bayliss, R.; Fry, A.M. Hsp72 is targeted to the mitotic spindle by Nek6 to promote K-fiber assembly and mitotic progression. J. Cell Biol. 2015, 209, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- 27. Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Pettinger, J.; Le Bihan, Y.V.; Widya, M.; van Montfort, R.L.M.; Jones, K.; Cheeseman, M.D. An Irreversible Inhibitor of HSP72 that Unexpectedly Targets Lysine-56. Angew. Chemie—Int. Ed. 2017, 56, 3536–3540. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Philip, E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Schlecht, R.; Scholz, S.R.; Dahmen, H.; Wegener, A.; Sirrenberg, C.; Musil, D.; Bomke, J.; Eggenweiler, H.M.; Mayer, M.P.; Bukau, B. Functional analysis of Hsp70 inhibitors. PLoS ONE 2013, 8, e78443. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar]
- MMV Molegro Molecular Viewer. Available online: http://molexus.io/molegro-molecular-viewer/ (accessed on 7 April 2020).
- Kusumaningrum, S.; Budianto, E.; Kosela, S.; Sumaryono, W.; Juniarti, F. The molecular docking of 1,4-naphthoquinone derivatives as inhibitors of Polo-like kinase 1 using Molegro Virtual Docker. J. Appl. Pharm. Sci. 2014, 4, 47–53. [Google Scholar]
- Trott, O.; Olson, A. Autodock vina: Improving the speed and accuracy of docking. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mhlongo, N.N.; Ebrahim, M.; Skelton, A.A.; Kruger, H.G.; Williams, I.H.; Soliman, M.E.S. Dynamics of the thumb-finger regions in a GH11 xylanase Bacillus circulans: Comparison between the Michaelis and covalent intermediate. RSC Adv. 2015, 5, 82381–82394. [Google Scholar] [CrossRef]
- Ramharack, P.; Oguntade, S.; Soliman, M.E.S. Delving into Zika virus structural dynamics-a closer look at NS3 helicase loop flexibility and its role in drug discovery. RSC Adv. 2017, 7, 22133–22144. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Betz, R. Dabble (Version v2.6.3). Zenodo. Available online: http://dabble.robinbetz.com/index.html (accessed on 7 April 2020).
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Gonnet, P. P-SHAKE: A quadratically convergent SHAKE in O (n2). J. Comput. Phys. 2007, 220, 740–750. [Google Scholar] [CrossRef]
- Khan, S.; Bjij, I.; Betz, R.M.; Soliman, M.E.S. Reversible versus irreversible inhibition modes of ERK2: A comparative analysis for ERK2 protein kinase in cancer therapy. Future Med. Chem. 2018, 10, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Seifert, E. OriginPro 9.1: Scientific data analysis and graphing software—Software review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J. Solvent accessible surface area and excluded volume in proteins. Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Mol. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Wisniewska, M.; Karlberg, T.; Lehtiö, L.; Johansson, I.; Kotenyova, T.; Moche, M.; Schüler, H. Crystal Structures of the ATPase Domains of Four Human Hsp70 Isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70-2, HSPA6/Hsp70B’, and HSPA5/BiP/GRP78. PLoS ONE 2010, 5, e8625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, M.; Osipiuk, J.; Freeman, B.; Morimoto, R.; Joachimiak, A. Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure 1997, 5, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Shida, M.; Arakawa, A.; Ishii, R.; Kishishita, S.; Takagi, T.; Kukimoto-Niino, M.; Sugano, S.; Tanaka, A.; Shirouzu, M.; Yokoyama, S. Direct inter-subdomain interactions switch between the closed and open forms of the Hsp70 nucleotide-binding domain in the nucleotide-free state. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 223–232. [Google Scholar] [CrossRef]
- Cheeseman, M.D.; Westwood, I.M.; Barbeau, O.; Rowlands, M.; Dobson, S.; Jones, A.M.; Jeganathan, F.; uBurke, R.; Kadi, N.; Workman, P.; et al. Exploiting Protein Conformational Change to Optimize Adenosine-Derived Inhibitors of HSP70. J. Med. Chem. 2016, 59, 4625–4636. [Google Scholar] [CrossRef]
- Martínez-Archundia, M.; Correa-Basurto, J.; Montaño, S.; Rosas-Trigueros, J.L. Studying the collective motions of the adenosine A2A receptor as a result of ligand binding using principal component analysis. J. Biomol. Struct. Dyn. 2019, 37, 4685–4700. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aljoundi, A.; El Rashedy, A.; Appiah-Kubi, P.; Soliman, M.E.S. Coupling of HSP72 α-Helix Subdomains by the Unexpected Irreversible Targeting of Lysine-56 over Cysteine-17; Coevolution of Covalent Bonding. Molecules 2020, 25, 4239. https://doi.org/10.3390/molecules25184239
Aljoundi A, El Rashedy A, Appiah-Kubi P, Soliman MES. Coupling of HSP72 α-Helix Subdomains by the Unexpected Irreversible Targeting of Lysine-56 over Cysteine-17; Coevolution of Covalent Bonding. Molecules. 2020; 25(18):4239. https://doi.org/10.3390/molecules25184239
Chicago/Turabian StyleAljoundi, Aimen, Ahmed El Rashedy, Patrick Appiah-Kubi, and Mahmoud E. S. Soliman. 2020. "Coupling of HSP72 α-Helix Subdomains by the Unexpected Irreversible Targeting of Lysine-56 over Cysteine-17; Coevolution of Covalent Bonding" Molecules 25, no. 18: 4239. https://doi.org/10.3390/molecules25184239
APA StyleAljoundi, A., El Rashedy, A., Appiah-Kubi, P., & Soliman, M. E. S. (2020). Coupling of HSP72 α-Helix Subdomains by the Unexpected Irreversible Targeting of Lysine-56 over Cysteine-17; Coevolution of Covalent Bonding. Molecules, 25(18), 4239. https://doi.org/10.3390/molecules25184239