
Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications





Abstract
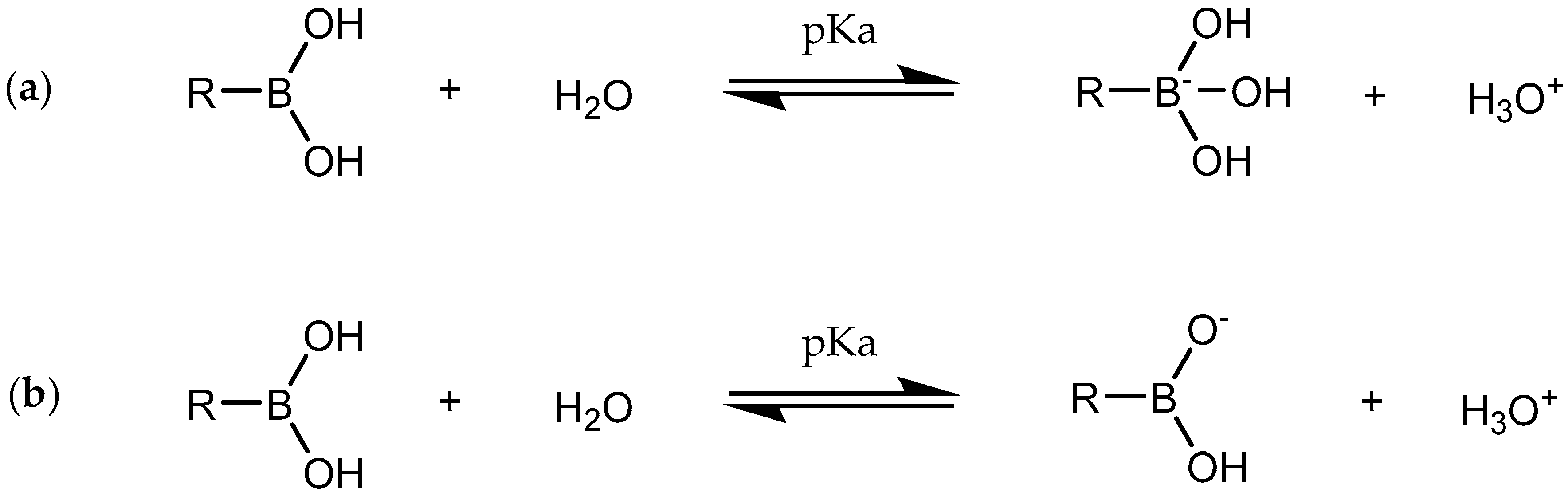
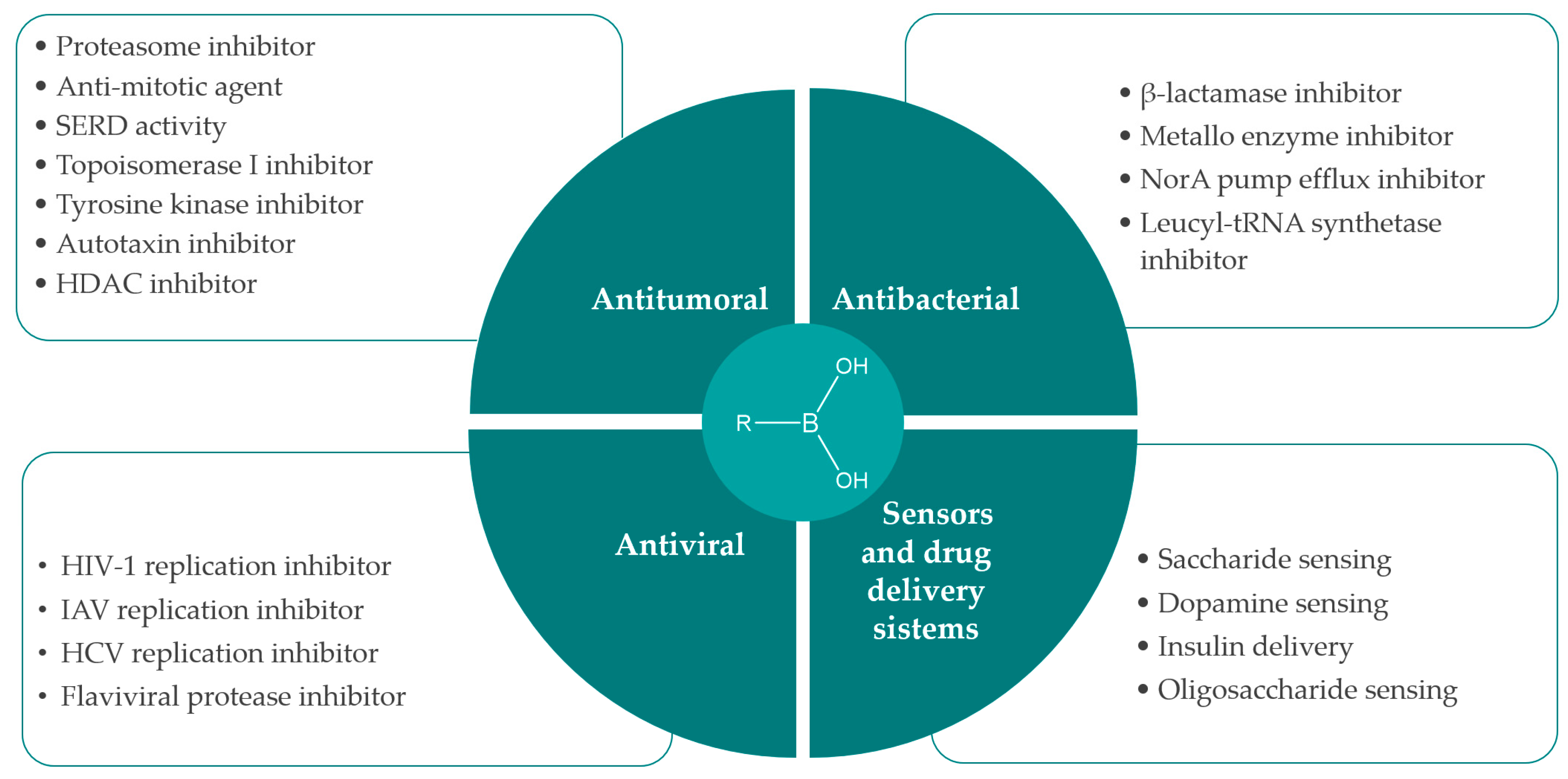
1. Introduction
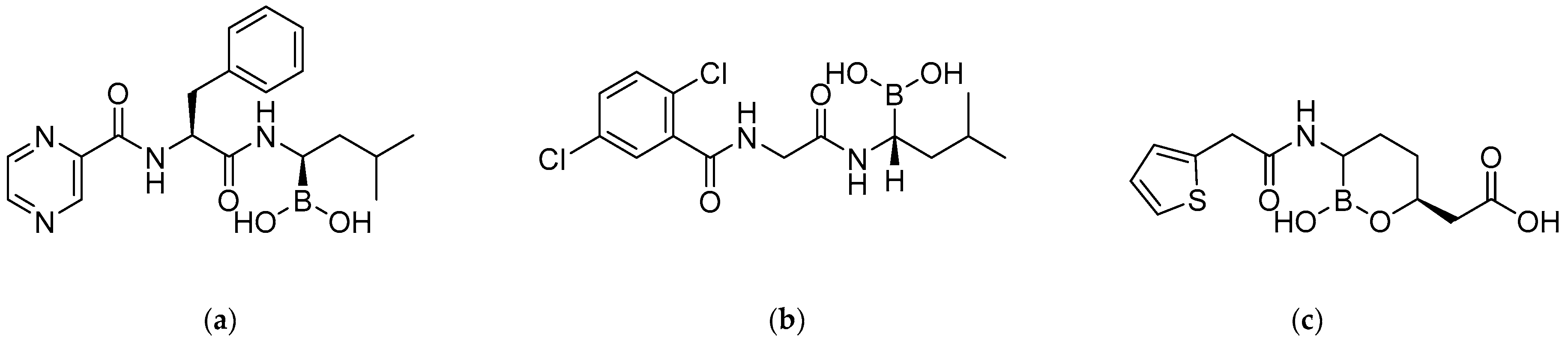
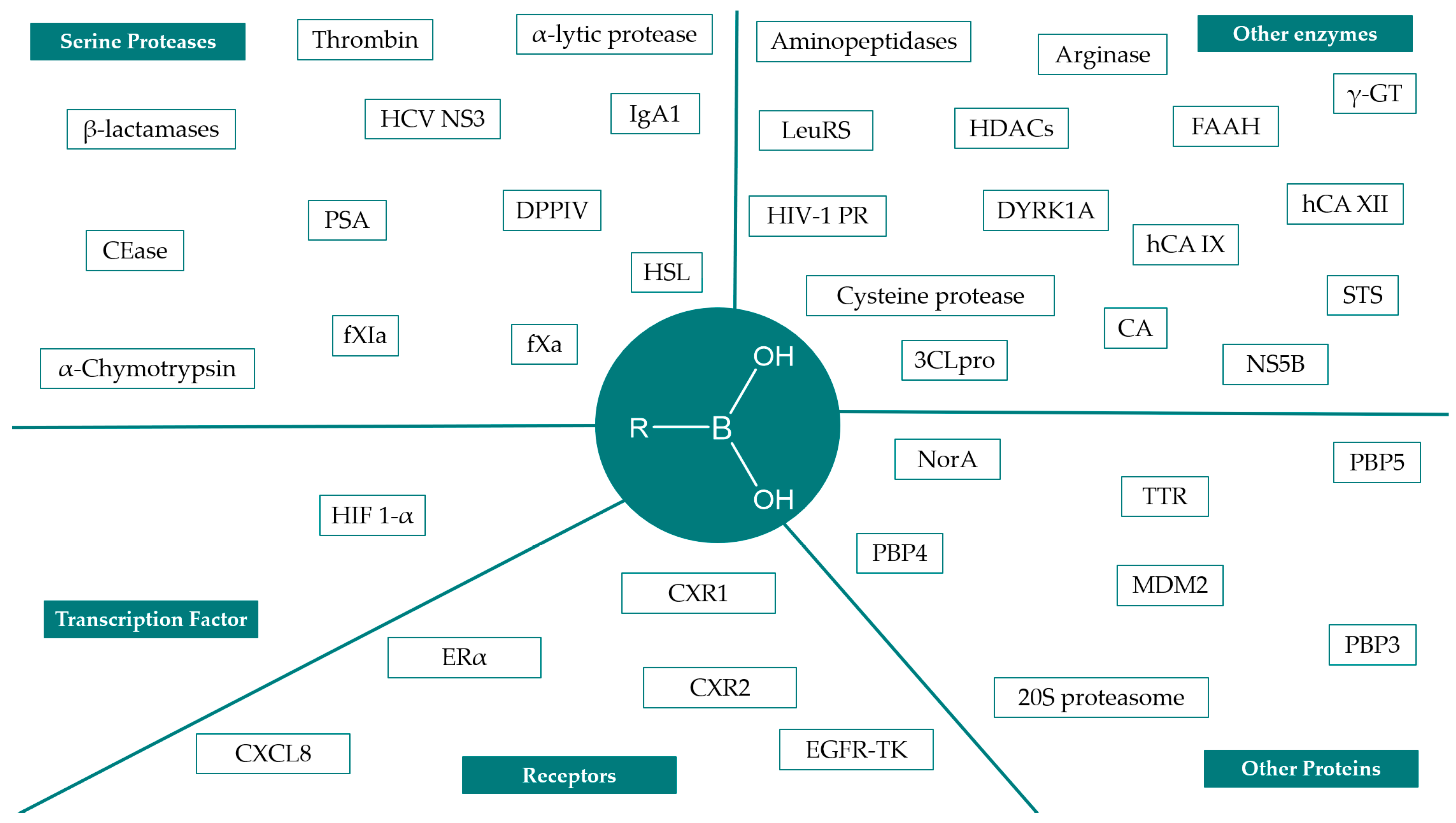
2. Synthesis and Biological Applications of Boronic Acids Derivatives
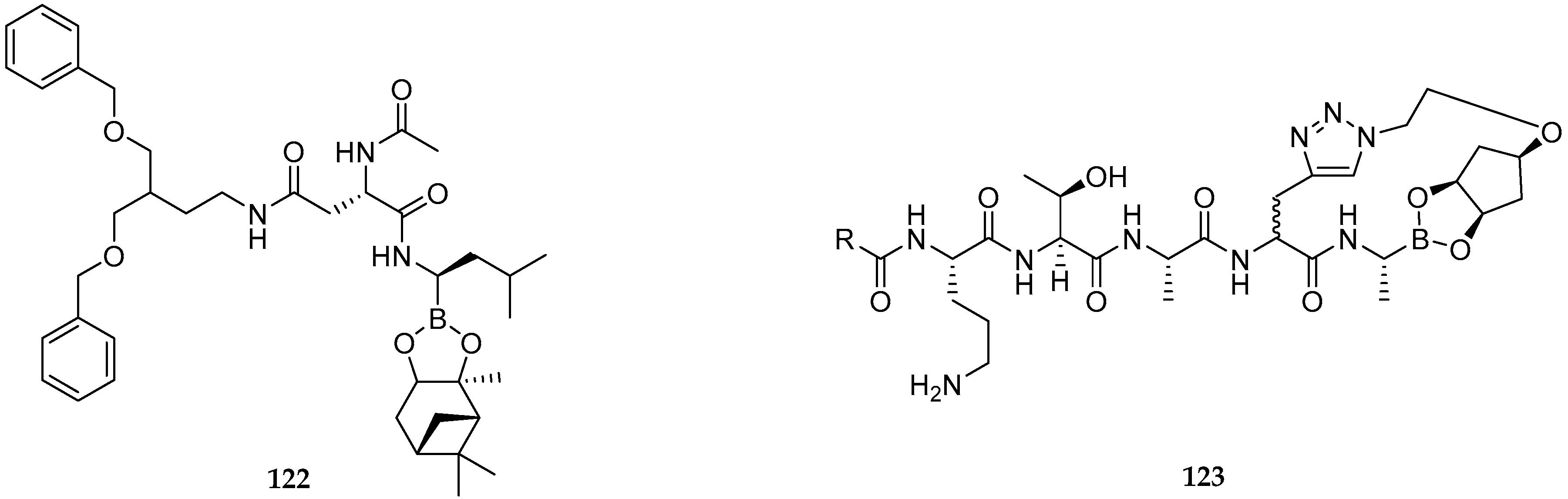
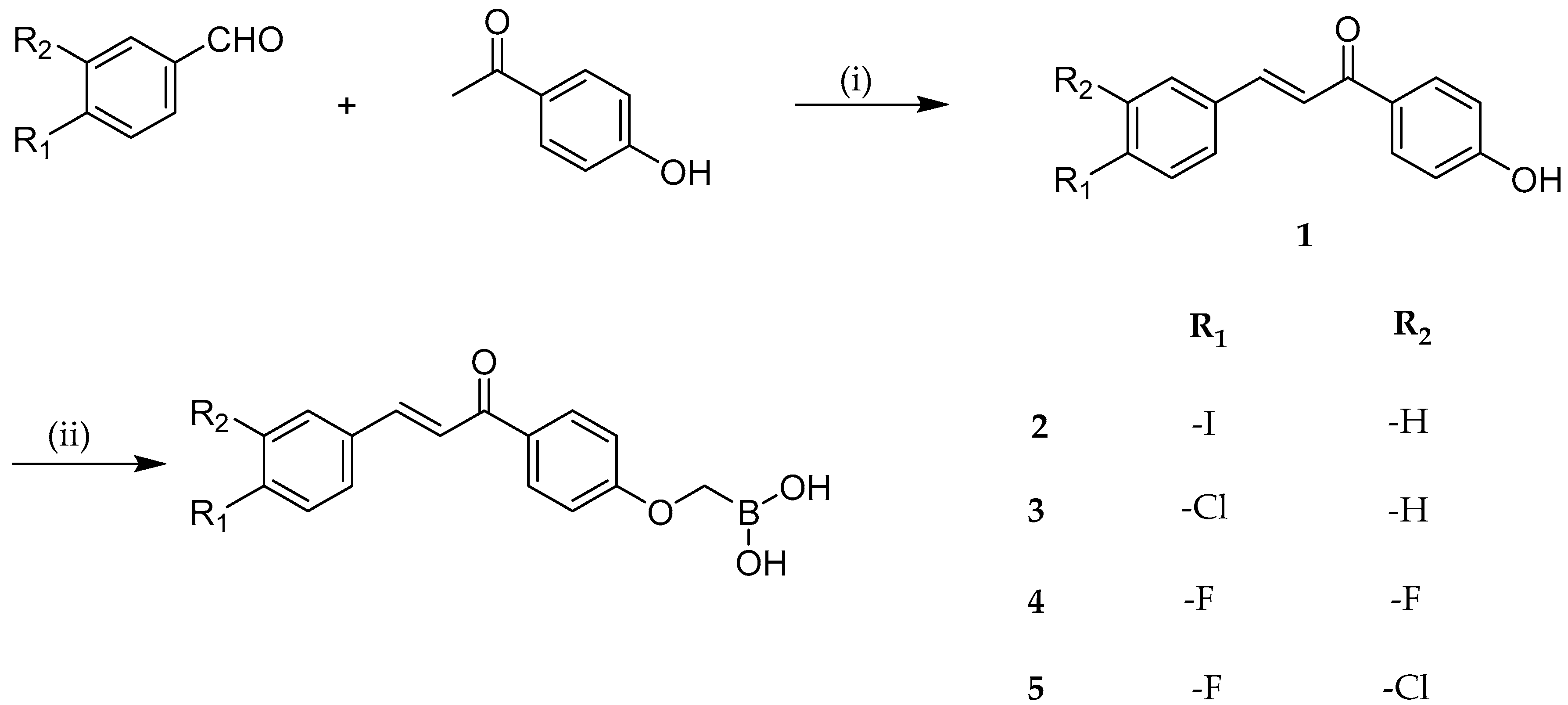
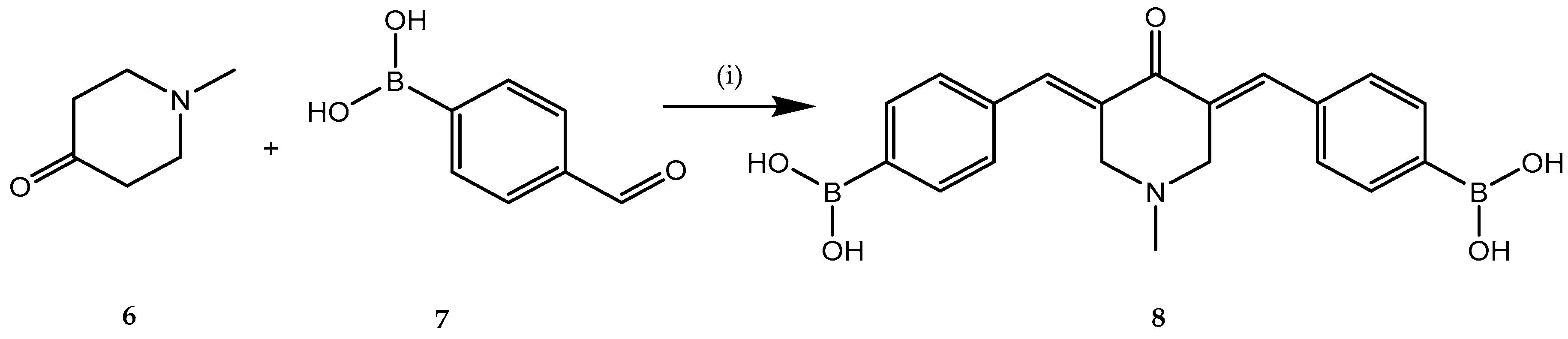
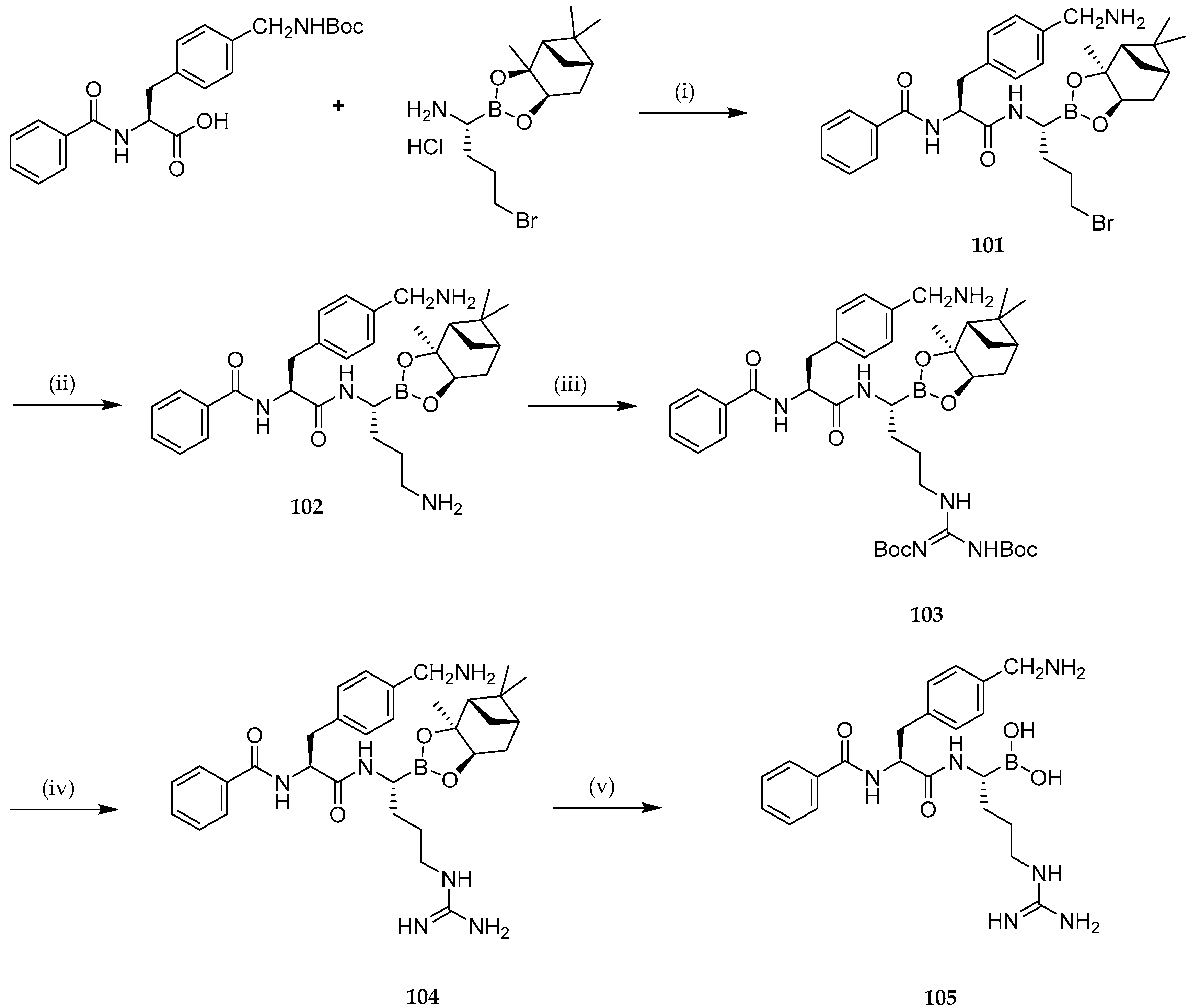
2.1. Anticancer Activity
2.2. Antibacterial Activity
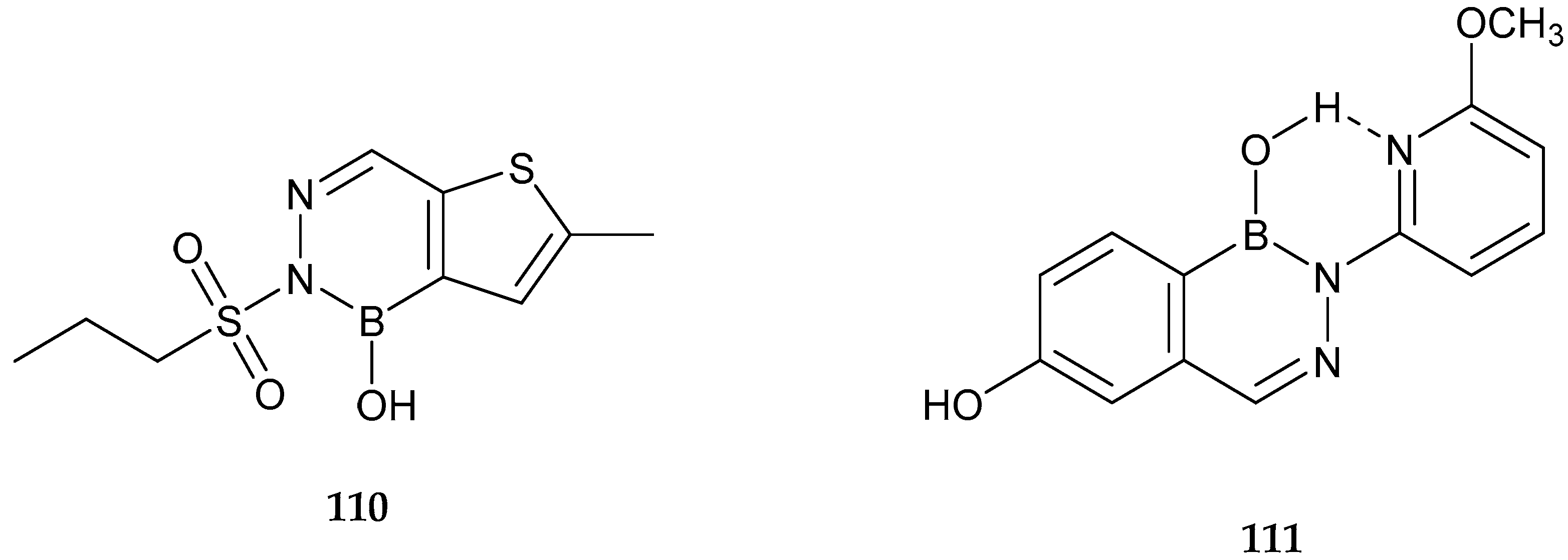
2.3. Antiviral Activity
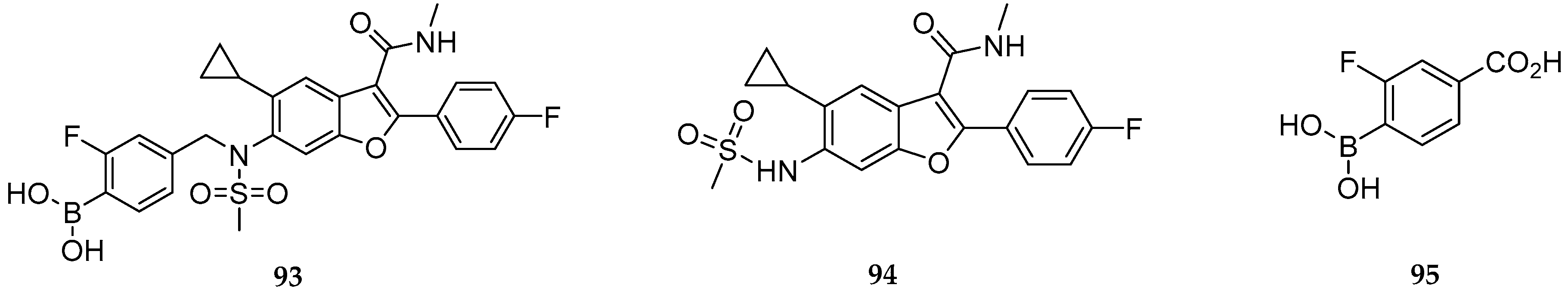
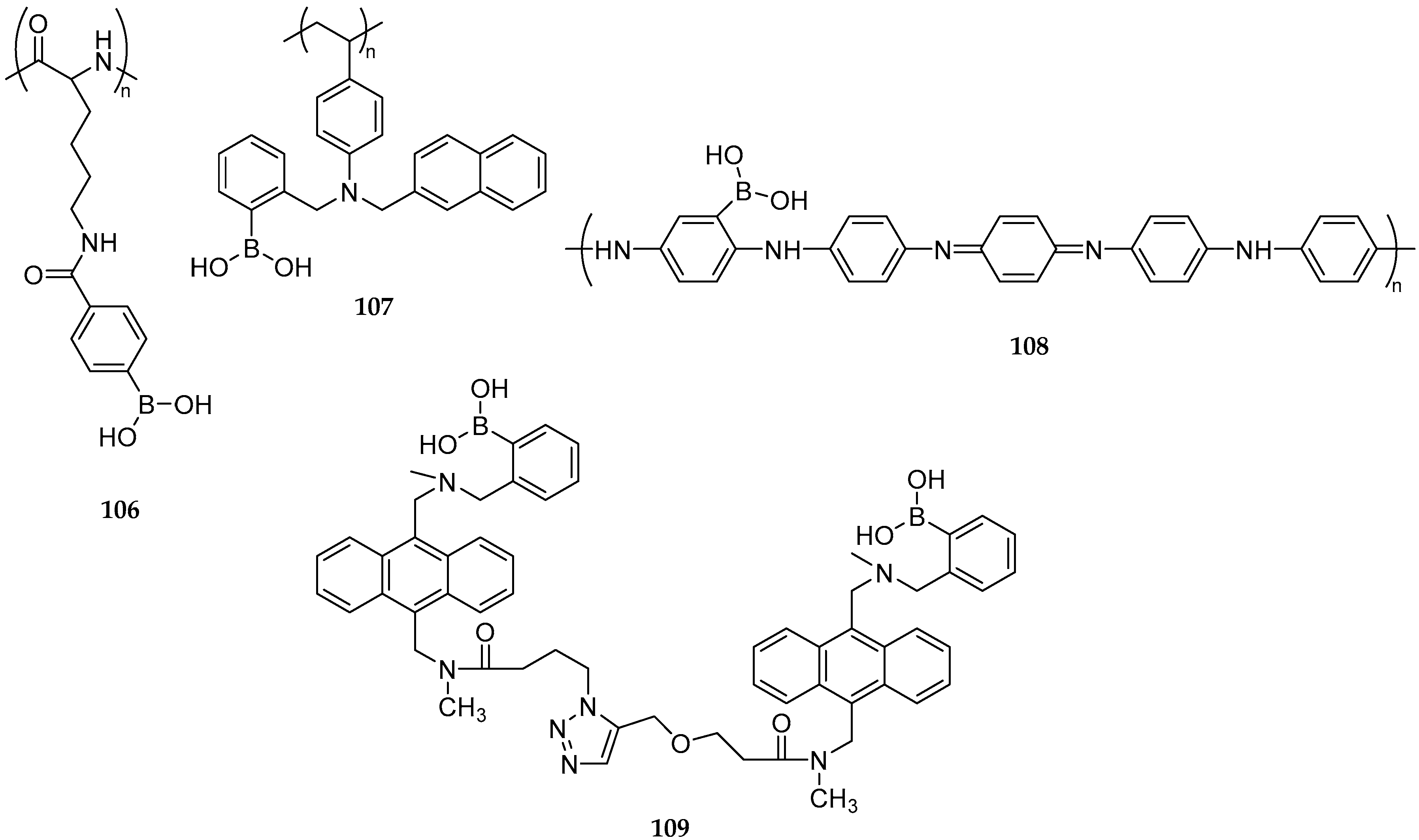
2.4. Sensors and Delivery Systems
3. Biological Applications of Other Boron Containing Compounds
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in drug design: Recent advances in the development of new therapeutic agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Ding, C.Z.; Zhang, Y.K.; Hernandez, V.; Xia, Y. Therapeutic potential of boron-containing compounds. Future Med. Chem. 2009, 1, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Frankland, E.; Duppa, B.F. Vorläufige Notiz über Boräthyl. Justus Liebigs Ann. Chem. 1860, 115, 319–322. [Google Scholar] [CrossRef]
- Yang, W.; Gao, X.; Wang, B. Boronic Acid Compounds as Potential Pharmaceutical Agents. Med. Res. Rev. 2003, 23, 346–368. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G. (Ed.) Structure, Properties, and Preparation of Boronic Acid Derivatives. Overview of Their Reactions and Applications. In Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley-VCH: Weinheim, Germany, 2005; pp. 1–99. [Google Scholar]
- Cambre, J.N.; Sumerlin, B.S. Biomedical Applications of Boronic Acid Polymers. Polymer (Guildf) 2011, 52, 4631–4643. [Google Scholar] [CrossRef]
- Trippier, P.C.; McGuigan, C. Boronic Acids in Medicinal Chemistry: Anticancer, Antibacterial and Antiviral Applications. Medchemcomm 2010, 1, 183–198. [Google Scholar] [CrossRef]
- Petasis, N.A. Expanding Roles for Organoboron Compounds Versatile and Valuable Molecules for Synthetic, Biological and Medicinal Chemistry. Aust. J. Chem. 2007, 60, 795–798. [Google Scholar] [CrossRef]
- Ebdrup, S.; Jacobsen, P.; Farrington, A.D.; Vedsø, P. Structure–activity Relationship for Aryl and Heteroaryl Boronic Acid Inhibitors of Hormone-Sensitive Lipase. Bioorganic Med. Chem. 2005, 13, 2305–2312. [Google Scholar] [CrossRef]
- Brooks, W.L.A.; Sumerlin, B.S. Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine. Chem. Rev. 2016, 116, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Velcade. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/velcade (accessed on 16 March 2020).
- Novel Drug Approvals for 2015. Available online: https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2015 (accessed on 16 March 2020).
- Ninlaro. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ninlaro (accessed on 16 March 2020).
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef] [PubMed]
- 2017 New Drug Therapy Approvals. Available online: https://www.fda.gov/media/110526/download (accessed on 16 March 2020).
- Vaborem. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vaborem (accessed on 16 March 2020).
- Tsivkovski, R.; Lomovskayaa, O. Biochemical Activity of Vaborbactam. Antimicrob. Agents Chemother. 2020, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Milo, L.J.; Lai, J.H. Recent Progress in the Synthesis of Pyridinylboronic Acids and Esters. Arkivoc 2013, 2013, 135–153. [Google Scholar]
- Yang, F.; Zhu, M.; Zhang, J.; Zhou, H. Synthesis of Biologically Active Boron-Containing Compounds. Medchemcomm 2018, 9, 201–211. [Google Scholar] [CrossRef]
- Matteson, D.S. α-Amido Boronic Acids: A Synthetic Challenge and Their Properties as Serine Protease Inhibitors. Med. Res. Rev. 2008, 28, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Primas, N.; Bouillon, A.; Rault, S. Recent Progress in the Synthesis of Five-Membered Heterocycle Boronic Acids and Esters. Tetrahedron 2010, 66, 8121–8136. [Google Scholar] [CrossRef]
- Smoum, R.; Rubinstein, A.; Dembitsky, V.M.; Srebnik, M. Boron Containing Compounds as Protease Inhibitors. Chem. Rev. 2012, 112, 4156–4220. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Miyaura, N. (Ed.) Organoboron Compounds. In Cross-Coupling Reactions. Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2002; pp. 11–59. [Google Scholar]
- Lam, P.Y.S.; Clark, C.G.; Saubern, S.; Adams, J.; Winters, M.P.; Chan, D.M.T.; Combs, A. New Aryl/Heteroaryl C-N Bond Cross-Coupling Reactions via Arylboronic Acid/Cupric Acetate Arylation. Tetrahedron Lett. 1998, 39, 2941–2944. [Google Scholar] [CrossRef]
- Fagnou, K.; Lautens, M. Rhodium-Catalyzed Carbon-Carbon Bond Forming Reactions of Organometallic Compounds. Chem. Rev. 2003, 103, 169–196. [Google Scholar] [CrossRef]
- Yang, H.; Li, H.; Wittenberg, R.; Egi, M.; Huang, W.; Liebeskind, L.S. Ambient Temperature Synthesis of High Enantiopurity N-Protected Peptidyl Ketones by Peptidyl Thiol Ester-Boronic Acid Cross-Coupling. J. Am. Chem. Soc. 2007, 129, 1132–1140. [Google Scholar] [CrossRef]
- Molander, G.A.; Trice, S.L.J.; Kennedy, S.M. Scope of the Two-Step, One-Pot Palladium-Catalyzed Borylation/Suzuki Cross-Coupling Reaction Utilizing Bis-Boronic Acid. J. Org. Chem. 2012, 77, 8678–8688. [Google Scholar] [CrossRef] [PubMed]
- Khotinsky, E.; Melamed, M. Die Wirkung der magnesiumorganischen Verbindungen auf die Borsäureester. Chem. Ber. 1909, 42, 3090–3096. [Google Scholar] [CrossRef]
- Lawesson, S.-O.; Krabisch, L.; Zimmer, E.C. Plant Growth Regulators. II. Methyl- and Methoxysubstituted Naphthylboronic Acids. Acta Chem. Scand. 1957, 11, 1075–1076. [Google Scholar] [CrossRef]
- Li, W.; Nelson, D.P.; Jensen, M.S.; Hoerrner, R.S.; Cai, D.; Larsen, R.D.; Reider, P.J. An Improved Protocol for the Preparation of 3-Pyridyl- and Some Arylboronic Acids. J. Org. Chem. 2002, 67, 5394–5397. [Google Scholar] [CrossRef]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N.R.; Hartwig, J.F. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef]
- Shimada, S.; Batsanov, A.S.; Howard, J.A.K.; Marder, T.B. Formation of Aryl- and Benzylboronate Esters by Rhodium-Catalyzed C-H Bond Functionalization with Pinacolborane. Angew. Chem. Int. Ed. 2001, 40, 2168–2171. [Google Scholar] [CrossRef]
- Usutani, H.; Cork, D.G. Effective Utilization of Flow Chemistry: Use of Unstable Intermediates, Inhibition of Side Reactions, and Scale-Up for Boronic Acid Synthesis. Org. Process Res. Dev. 2018, 22, 741–746. [Google Scholar] [CrossRef]
- Sharp, M.J.; Cheng, W.; Snieckus, V. Synthetic Connections to the Aromatic Directed Metalation Reaction. Functionalized Aryl Boronic Acids by Ipso Borodesilylation. General Syntheses of Unsymmetrical Biphenyls and m-Terphenyls. Tetrahedron Lett. 1987, 28, 5093–5096. [Google Scholar] [CrossRef]
- Achanta, G.; Modzelewska, A.; Feng, L.; Khan, S.R.; Huang, P. A Boronic-Chalcone Derivative Exhibits Potent Anticancer Activity through Inhibition of the Proteasome. Mol. Pharmacol. 2006, 70, 426–433. [Google Scholar] [CrossRef]
- Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.-T.; Plamondon, L.; Stein, R.L. ChemInform Abstract: Potent and Selective Inhibitors of the Proteasome: Dipeptidyl Boronic Acids. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hideshima, T.; Anderson, K.C. Bortezomib (PS-341): A Novel, First-in-Class Proteasome Inhibitor for the Treatment of Multiple Myeloma and Other Cancers. Cancer Control 2003, 10, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Feng, H.; Wang, C.; Li, H.; Shi, J.; Wang, J.; Liu, Z.; Chen, S.; Hu, S.; Zhu, Y. 3D-QSAR-Aided Design, Synthesis, in Vitro and in Vivo Evaluation of Dipeptidyl Boronic Acid Proteasome Inhibitors and Mechanism Studies. Bioorganic Med. Chem. 2016, 24, 2576–2588. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.H.G.; Van Meeteren, L.A.; Egan, D.A.; Van Tilburg, E.W.; Moolenaar, W.H.; Ovaa, H. Discovery and Optimization of Boronic Acid Based Inhibitors of Autotaxin. J. Med. Chem. 2010, 53, 4958–4967. [Google Scholar] [CrossRef]
- Kumar, S.K.; Hager, E.; Pettit, C.; Gurulingappa, H.; Davidson, N.E.; Khan, S.R. Design, Synthesis, and Evaluation of Novel Boronic-Chalcone Derivatives as Antitumor Agents. J. Med. Chem. 2003, 46, 2813–2815. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 Conceals the Activation Domain of Tumor Suppressor P53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Lei, M.; Feng, H.; Bai, E.; Zhou, H.; Wang, J.; Shi, J.; Wang, X.; Hu, S.; Liu, Z.; Zhu, Y. Design, synthesis, in vitro and in vivo evaluation, and structure–activity relationship (SAR) discussion of novel dipeptidyl boronic acid proteasome inhibitors as orally available anti-cancer agents for the treatment of multiple myeloma and mechanism studies. Bioorganic Med. Chem. 2018, 26, 3975–3981. [Google Scholar]
- Han, L.Q.; Yuan, X.; Wu, X.Y.; Li, R.D.; Xu, B.; Cheng, Q.; Liu, Z.M.; Zhou, T.Y.; An, H.Y.; Wang, X.; et al. Urea-Containing Peptide Boronic Acids as Potent Proteasome Inhibitors. Eur. J. Med. Chem. 2017, 125, 925–939. [Google Scholar] [CrossRef]
- Kong, Y.; Grembecka, J.; Edler, M.C.; Hamel, E.; Mooberry, S.L.; Sabat, M.; Rieger, J.; Brown, M.L. Structure-Based Discovery of a Boronic Acid Bioisostere of Combretastatin A-4. Chem. Biol. 2005, 12, 1007–1014. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, K.; Edler, M.C.; Hamel, E.; Mooberry, S.L.; Paige, M.A.; Brown, M.L. A Boronic Acid Chalcone Analog of Combretastatin A-4 as a Potent Anti-Proliferation Agent. Bioorganic Med. Chem. 2010, 18, 971–977. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, S.; Akerstrom, V.L.; Yuan, C.; Ma, Y.; Zhong, Q.; Zhang, C.; Zhang, Q.; Guo, S.; Ma, P.; et al. Fulvestrant-3 Boronic Acid (ZB716): An Orally Bioavailable Selective Estrogen Receptor Downregulator (SERD). J. Med. Chem. 2016, 59, 8134–8140. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, S.; Ma, L.; Chen, Y.; Lu, W. 10-Boronic Acid Substituted Camptothecin as Prodrug of SN-38. Eur. J. Med. Chem. 2016, 116, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.A.; Palte, M.J.; Raines, R.T. Boronate-Mediated Biologic Delivery. J. Am. Chem. Soc. 2012, 134, 3631–3634. [Google Scholar] [CrossRef]
- Peiró Cadahía, J.; Bondebjerg, J.; Hansen, C.A.; Previtali, V.; Hansen, A.E.; Andresen, T.L.; Clausen, M.H. Synthesis and Evaluation of Hydrogen Peroxide Sensitive Prodrugs of Methotrexate and Aminopterin for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2018, 61, 3503–3515. [Google Scholar] [CrossRef]
- Bielec, B.; Poetsch, I.; Ahmed, E.; Heffeter, P.; Keppler, B.K.; Kowol, C.R. Reactive Oxygen Species (ROS)-Sensitive Prodrugs of the Tyrosine Kinase Inhibitor Crizotinib. Molecules 2020, 25, 1149. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef]
- Albers, H.M.H.G.; Hendrickx, L.J.D.; Van Tol, R.J.P.; Hausmann, J.; Perrakis, A.; Ovaa, H. Structure-Based Design of Novel Boronic Acid-Based Inhibitors of Autotaxin. J. Med. Chem. 2011, 54, 4619–4626. [Google Scholar] [CrossRef]
- Suzuki, N.; Suzuki, T.; Ota, Y.; Nakano, T.; Kurihara, M.; Okuda, H.; Yamori, T.; Tsumoto, H.; Nakagawa, H.; Naoki, M. Design, Synthesis, and Biological Activity of Boronic Acid-Based Histone Deacetylase Inhibitors. J. Med. Chem. 2009, 52, 2909–2922. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, X.; Xue, J.; Liu, L.; Liang, T.; Li, W.; Yang, X.; Hou, X.; Fang, H. Discovery of Peptide Boronate Derivatives as Histone Deacetylase and Proteasome Dual Inhibitors for Overcoming Bortezomib Resistance of Multiple Myeloma. J. Med. Chem. 2020, 63, 4701–4715. [Google Scholar] [CrossRef]
- Caselli, E.; Romagnoli, C.; Vahabi, R.; Taracila, M.A.; Bonomo, R.A.; Prati, F. Click Chemistry in Lead Optimization of Boronic Acids as β-Lactamase Inhibitors. J. Med. Chem. 2015, 58, 5445–5458. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs. Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Peppoloni, S.; Pericolini, E.; Colombari, B.; Pinetti, D.; Cermelli, C.; Fini, F.; Prati, F.; Caselli, E.; Blasi, E. The β-Lactamase Inhibitor Boronic Acid Derivative SM23 as a New Anti-Pseudomonas Aeruginosa Biofilm. Front. Microbiol. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Pan, J.; Root, Y.; Scapin, G.; Xiao, L.; Su, J. Serendipitous Discovery of Aryl Boronic Acids as β-Lactamase Inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 126795. [Google Scholar] [CrossRef] [PubMed]
- Kiener, P.A.; Waley, S.G. Reversible inhibitors of penicillinases. Biochem. J. 1978, 169, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Caselli, E.; Morandi, S.; Focia, P.J.; Blázquez, J.; Shoichet, B.K.; Prati, F. Nanomolar Inhibitors of AmpC β-Lactamase. J. Am. Chem. Soc. 2003, 125, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Romagnoli, C.; Powers, R.A.; Taracila, M.A.; Bouza, A.A.; Swanson, H.C.; Smolen, K.A.; Fini, F.; Wallar, B.J.; Bonomo, R.A.; et al. Inhibition of Acinetobacter -Derived Cephalosporinase: Exploring the Carboxylate Recognition Site Using Novel β-Lactamase Inhibitors. ACS Infect. Dis. 2018, 4, 337–348. [Google Scholar] [CrossRef]
- Eidam, O.; Romagnoli, C.; Dalmasso, G.; Barelier, S.; Caselli, E.; Bonnet, R.; Shoichet, B.K.; Prati, F. Fragment-Guided Design of Subnanomolar β-Lactamase Inhibitors Active in Vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 17448–17453. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Lomovskaya, O.; Griffith, D.C.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Sun, D.; Sabet, M.; Tarazi, Z.; et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultra-Broad-Spectrum Inhibitor of Serine and Metallo Beta-Lactamases. J. Med. Chem. 2020, 63, 7491–7507. [Google Scholar] [CrossRef]
- Fontaine, F.; Héquet, A.; Voisin-Chiret, A.S.; Bouillon, A.; Lesnard, A.; Cresteil, T.; Jolivalt, C.; Rault, S. Boronic Species as Promising Inhibitors of the Staphylococcus Aureus NorA Efflux Pump: Study of 6-Substituted Pyridine-3-Boronic Acid Derivatives. Eur. J. Med. Chem. 2015, 95, 185–198. [Google Scholar] [CrossRef]
- Durães, F.; Pinto, M.; Sousa, E. Medicinal Chemistry Updates on Bacterial Efflux Pump Modulators. Curr. Med. Chem. 2018, 25, 6030–6069. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.M.; Pegu, R.; Porto, W.F.; Franco, O.L.; Pratihar, S. Novel Boronic Acid Derivatives of Bis(Indolyl) Methane as Anti-MRSA Agents. Bioorganic Med. Chem. Lett. 2017, 27, 2135–2138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bryson, D.I.; Crumpton, J.B.; Wynn, J.; Santos, W.L. Branched Peptide Boronic Acids (BPBAs): A Novel Mode of Binding towards RNA. Chem. Commun. 2013, 49, 2436–2438. [Google Scholar] [CrossRef]
- Wynn, J.E.; Zhang, W.; Tebit, D.M.; Gray, L.R.; Hammarskjold, M.L.; Rekosh, D.; Santos, W.L. Characterization and in Vitro Activity of a Branched Peptide Boronic Acid That Interacts with HIV-1 RRE RNA. Bioorganic Med. Chem. 2016, 24, 3947–3952. [Google Scholar] [CrossRef] [PubMed]
- Windsor, I.W.; Palte, M.J.; Lukesh, J.C.; Gold, B.; Forest, K.T.; Raines, R.T. Sub-Picomolar Inhibition of HIV-1 Protease with a Boronic Acid. J. Am. Chem. Soc. 2018, 140, 14015–14018. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Xia, Z.; Kovela, S.; Robinson, W.L.; Johnson, M.E.; Kneller, D.W.; Wang, Y.F.; Aoki, M.; Takamatsu, Y.; Weber, I.T.; et al. Potent HIV-1 Protease Inhibitors Containing Carboxylic and Boronic Acids: Effect on Enzyme Inhibition and Antiviral Activity and Protein-Ligand X-ray Structural Studies. ChemMedChem 2019, 14, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yin, R.; Zhang, M.; Yu, R.; Hao, C.; Zhang, L.; Jiang, T. Boronic Acid Modifications Enhance the Anti-Influenza A Virus Activities of Novel Quindoline Derivatives. J. Med. Chem. 2017, 60, 2840–2852. [Google Scholar] [CrossRef]
- Richter, S.; Parolin, C.; Palumbo, M.; Palù, G. Antiviral Properties of Quinolone-Based Drugs. Curr. Drug Targets Infect. Disord. 2004, 4, 111–116. [Google Scholar] [CrossRef]
- Maynard, A.; Crosby, R.M.; Ellis, B.; Hamatake, R.; Hong, Z.; Johns, B.A.; Kahler, K.M.; Koble, C.; Leivers, A.; Leivers, M.R.; et al. Discovery of a Potent Boronic Acid Derived Inhibitor of the HCV RNA-Dependent RNA Polymerase. J. Med. Chem. 2014, 57, 1902–1913. [Google Scholar] [CrossRef]
- Wilfret, D.A.; Walker, J.; Voitenleitner, C.; Baptiste-Brown, S.; Lovern, M.; Kim, J.; Adkison, K.; Shotwell, B.; Mathis, A.; Moss, L.; et al. A Randomized, Double Blind, Dose Escalation, First Time in Human Study to Assess the Safety, Tolerability, Pharmacokinetics, and Antiviral Activity of Single Doses of GSK2485852 in Chronically Infected Hepatitis C Subjects. Clin. Pharmacol. Drug Dev. 2014, 3, 439–448. [Google Scholar] [CrossRef]
- Chong, P.Y.; Shotwell, J.B.; Miller, J.; Price, D.J.; Maynard, A.; Voitenleitner, C.; Mathis, A.; Williams, S.; Pouliot, J.J.; Creech, K.; et al. Design of N-Benzoxaborole Benzofuran GSK8175—Optimization of Human Pharmacokinetics Inspired by Metabolites of a Failed Clinical HCV Inhibitor. J. Med. Chem. 2019, 62, 3254–3267. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Zhang, L.; Weigel, L.F.; Schilz, J.; Graf, D.; Bartenschlager, R.; Hilgenfeld, R.; Klein, C.D. Peptide-Boronic Acid Inhibitors of Flaviviral Proteases: Medicinal Chemistry and Structural Biology. J. Med. Chem. 2017, 60, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Liu, A.; Li, Y.; Fang, G.; Yao, Q.; Zhang, G.; Wu, Z. Boronic Acid Sensors with Double Recognition Sites: A Review. Analyst 2020, 145, 719–744. [Google Scholar] [CrossRef] [PubMed]
- Resendez, A.; Malhotra, S.V. Boronic Acid Appended Naphthyl-Pyridinium Receptors as Chemosensors for Sugars. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gronowitz, S.; Dhlgren, T.; Namtvedt, J.; Roos, C.; Sjöberg, B.; Forsgren, U. Antibacterial borazaro derivatives. I. 5-Arylsulphonyl-4-hydroxy-4,5-borazarothieno(2,3-c)pyridines and 6-arylsulphonyl-7-hydroxy-7,6-borazarothieno(3,2-c)pyridines. Acta Pharm. Suec. 1971, 8, 377–390. [Google Scholar]
- Gronowitz, S.; Dhlgren, T.; Namtvedt, J.; Roos, C.; Sjöberg, B.; Forsgren, U. Antibacterial borazaro derivatives. II. Effect of substituents on the antibacterial activity of 5-arylsulfonyl-4hydroxy-4,5-borazarothieno(2,3-c)pyridines and 6-arylsulfonyl-7-hydroxy-7,6- borazarothieno(3,2-c)pyridines. Acta Pharm. Suec. 1971, 8, 623–638. [Google Scholar]
- Grassberger, M.A.; Turnowsky, F.; Hildebrandt, J. Preparation and Antibacterial Activities of New 1, 2, 3-Diazaborine Derivatives and Analogues. J. Med. Chem. 1984, 27, 947–953. [Google Scholar] [CrossRef]
- Groziak, M.P. Boron Therapeutics on the Horizon. Am. J. Ther. 2001, 8, 321–328. [Google Scholar] [CrossRef]
- Torssell, K. Arylboronic acids. III. Bromination of tolylboronic acids according to WohlZiegler. Ark. Kemi. 1957, 10, 507–511. [Google Scholar]
- Nocentini, A.; Supuran, C.T.; Winum, J.-Y. Benzoxaborole compounds for therapeutic uses: A patent review (2010–2018). Expert Opin. Ther. Pat. 2018, 28, 493–504. [Google Scholar] [CrossRef]
- Sharma, N.; Sharma, D. An upcoming drug for onychomycosis: Tavaborole. J. Pharmacol. Pharmacother. 2015, 6, 236–239. [Google Scholar] [CrossRef]
- Adamczyk-Wozniak, A.; Cyranski, M.K.; Zubrowska, A.; Sporzynski, A. Benzoxaboroles—Old compounds with new applications. J. Organomet. Chem. 2009, 694, 3533–3541. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crépin, T.; Zhou, H.; Zhang, Y.-K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–6171. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hernandez, V.; Rock, F.L.; Choi, W.; Mak, Y.S.L.; Mohan, M.; Mao, W.; Zhou, Y.; Easom, E.E.; Plattner, J.J.; et al. Discovery of a Potent and Specific, M. Tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-Chloro-7-(2-Hydroxyethoxy)Benzo[c][1,2]Oxaborol-1(3H)-ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. [Google Scholar] [CrossRef]
- Alterio, V.; Cadoni, R.; Esposito, D.; Vullo, D.; Di Fiore, A.; Monti, S.M.; Caporale, A.; Ruvo, M.; Sechi, M.; Dumy, P.; et al. Benzoxaborole as a new chemotype for carbonic anhydrase inhibition. Chem. Commun. 2016, 52, 1198311986. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Cadoni, R.; del Prete, S.; Capasso, C.; Dumy, P.; Gratteri, P.; Supuran, C.T.; Winum, J.-Y. Benzoxaboroles as efficient inhibitors of the β-carbonic anhydrases from pathogenic fungi: Activity and modeling study. ACS Med. Chem. Lett. 2017, 8, 11941198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Jacobs, R.T.; Guo, D.; Freund, Y.R.; Berry, P.; Ciaravino, V.; Erve, J.C.L.; Rosenthal, P.J.; et al. Benzoxaborole antimalarial agents. Part 5. Lead optimization of novel amide pyrazinyloxy benzoxaboroles and identification of a preclinical candidate. J. Med. Chem. 2017, 60, 5889–5908. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Akama, T.; Zhou, Y.; White, W.H.; Defauw, J.M.; Winkle, J.R.; Balko, T.W.; Cao, J.; et al. Optimization of isoxazoline amide benzoxaboroles for identification of a development candidate as an oral long acting animal ectoparasiticide. Bioorg. Med. Chem. Lett. 2016, 26, 3182–3186. [Google Scholar] [CrossRef]
- Van Bocxlaer, K.; Gaukel, E.; Hauser, D.; Park, S.H.; Schock, S.; Yardley, V.; Randolph, R.; Plattner, J.J.; Merchant, T.; Croft, S.L.; et al. Topical treatment for cutaneous Leishmaniasis: Dermato-pharmacokinetic lead optimization of benzoxaboroles. Antimicrob. Agents Chemother. 2018, 62, e02419-17. [Google Scholar] [CrossRef]
- McKinney, D.C.; Zhou, F.; Eyermann, C.J.; Ferguson, A.D.; Prince, D.B.; Breen, J.; Giacobbe, R.A.; Lahiri, S.; Verheijen, J.C. 4,5-Disubstituted 6-Aryloxy-1,3dihydrobenzo[c][1,2]oxaboroles are broad-spectrum serine β-lactamase inhibitors. ACS Infect. Dis. 2015, 1, 310–316. [Google Scholar] [CrossRef]
- Psurski, M.; Łupicka-Słowik, A.; Adamczyk-Wozniak, A.; Wietrzyk, J.; Sporzynski, A. Discovering simple phenylboronic acid and benzoxaborole derivatives for experimental oncology e phase cycle-specific inducers of apoptosis in A2780 ovarian cancer cells. Investig. New Drugs. 2019, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Hashim, P.W.; Chima, M.; Kim, H.J.; Bares, J.; Yao, C.J.; Singer, G.; Chen, T.; Genece, J.; Baum, D.; Kimmel, G.W.; et al. Crisaborole 2% Ointment for the Treatment of Intertriginous, Anogenital, and Facial Psoriasis: A DoubleBlind, Randomized, Vehicle-Controlled Trial. J. Am. Acad. Dermatol. 2020, 82, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, S.; Unno, Y.; Asai, A.; Arisawa, M.; Shuto, S. Structurally novel highly potent proteasome inhibitors created by the structure-based hybridization of nonpeptidic belactosin derivatives and peptide boronates. J. Med. Chem. 2014, 57, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Szałaj, N.; Lu, L.; Benediktsdottir, A.; Zamaratski, E.; Cao, S.; Olanders, G.; Hedgecock, C.; Karlén, A.; Erdélyi, M.; Hughes, D.; et al. Boronic Ester-Linked Macrocyclic Lipopeptides as Serine Protease Inhibitors Targeting Escherichia Coli Type I Signal Peptidase. Eur. J. Med. Chem. 2018, 157, 1346–1360. [Google Scholar] [CrossRef]




































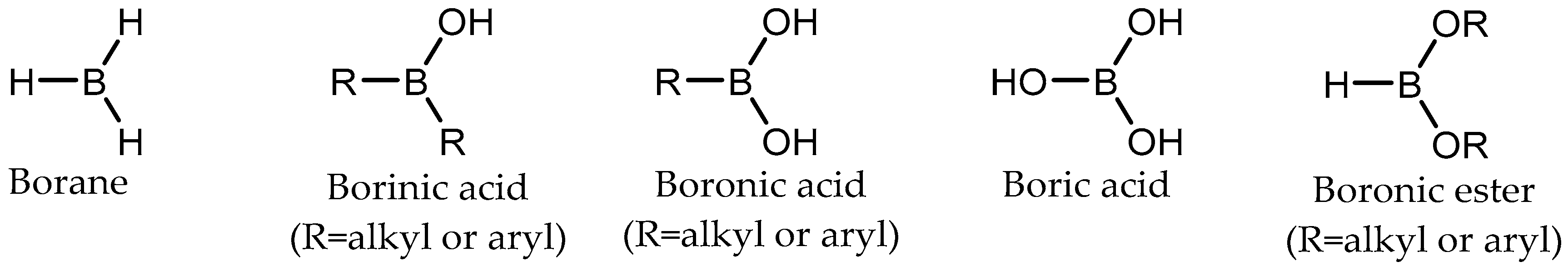{kind=link}
{kind=link}
{kind=link}
{kind=link}
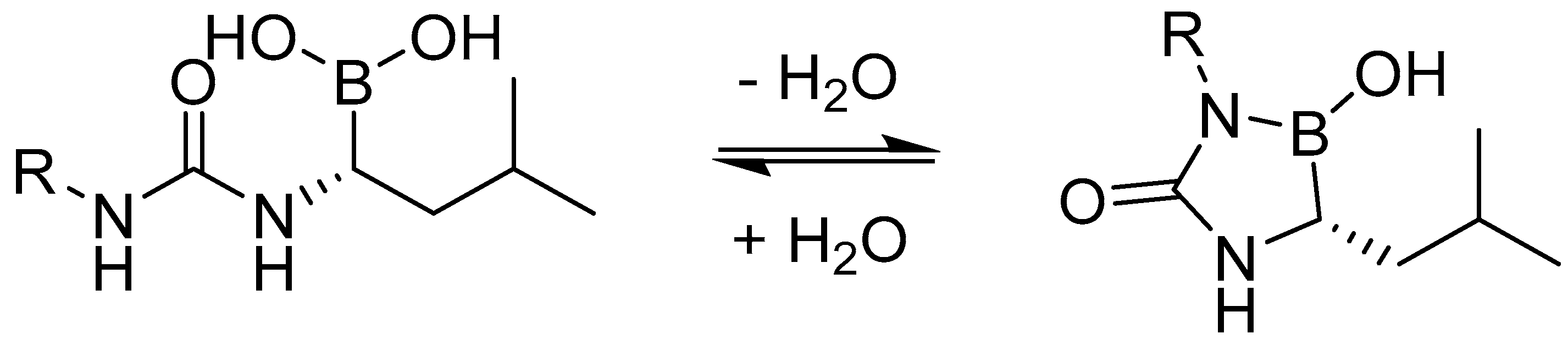{kind=link}
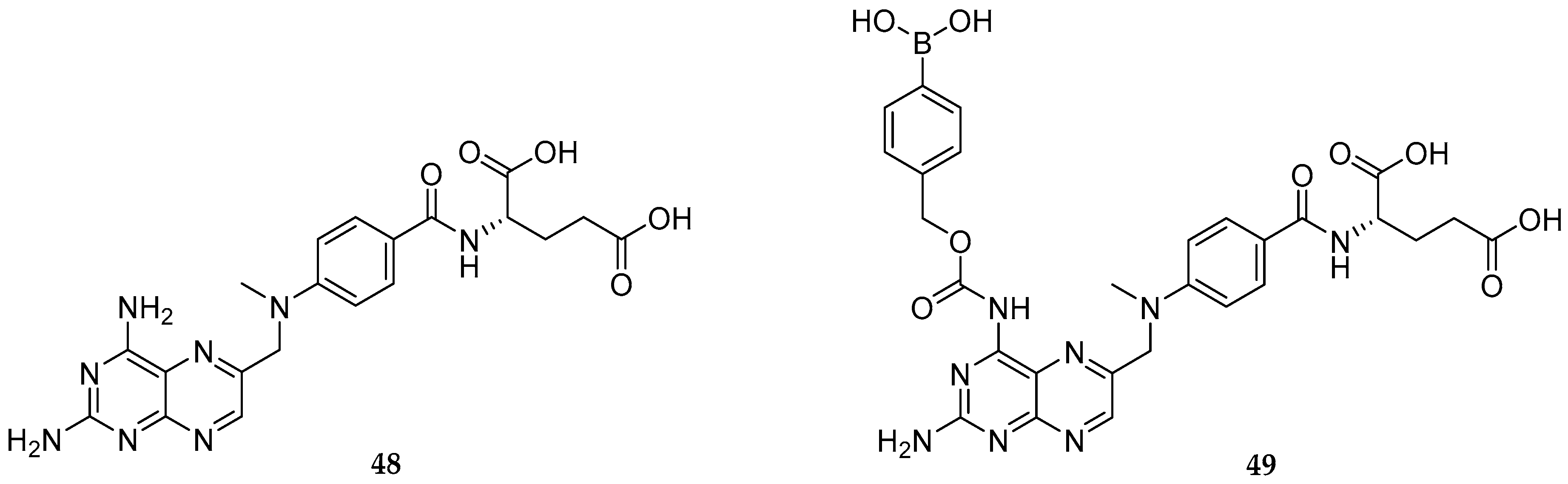{kind=link}
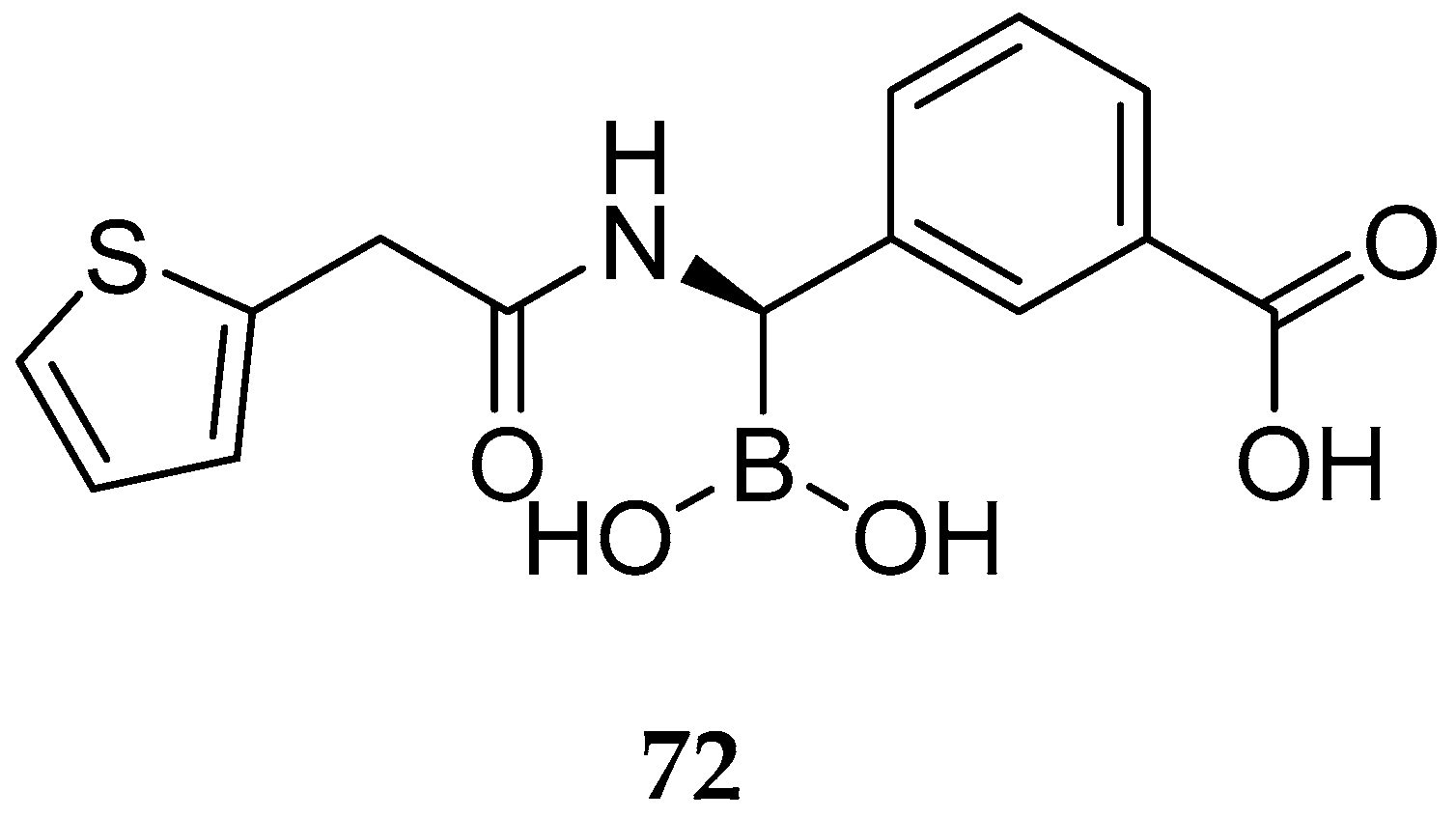{kind=link}
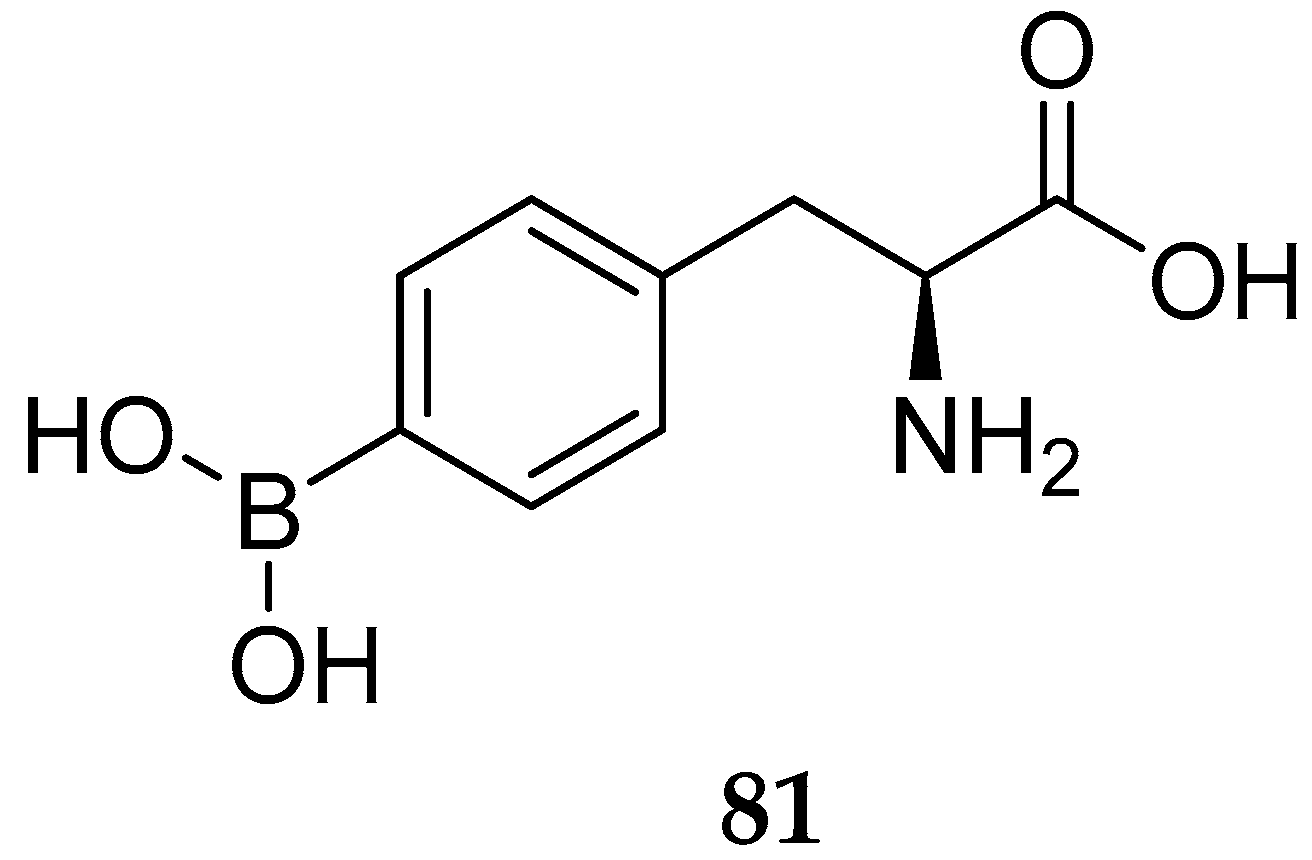{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
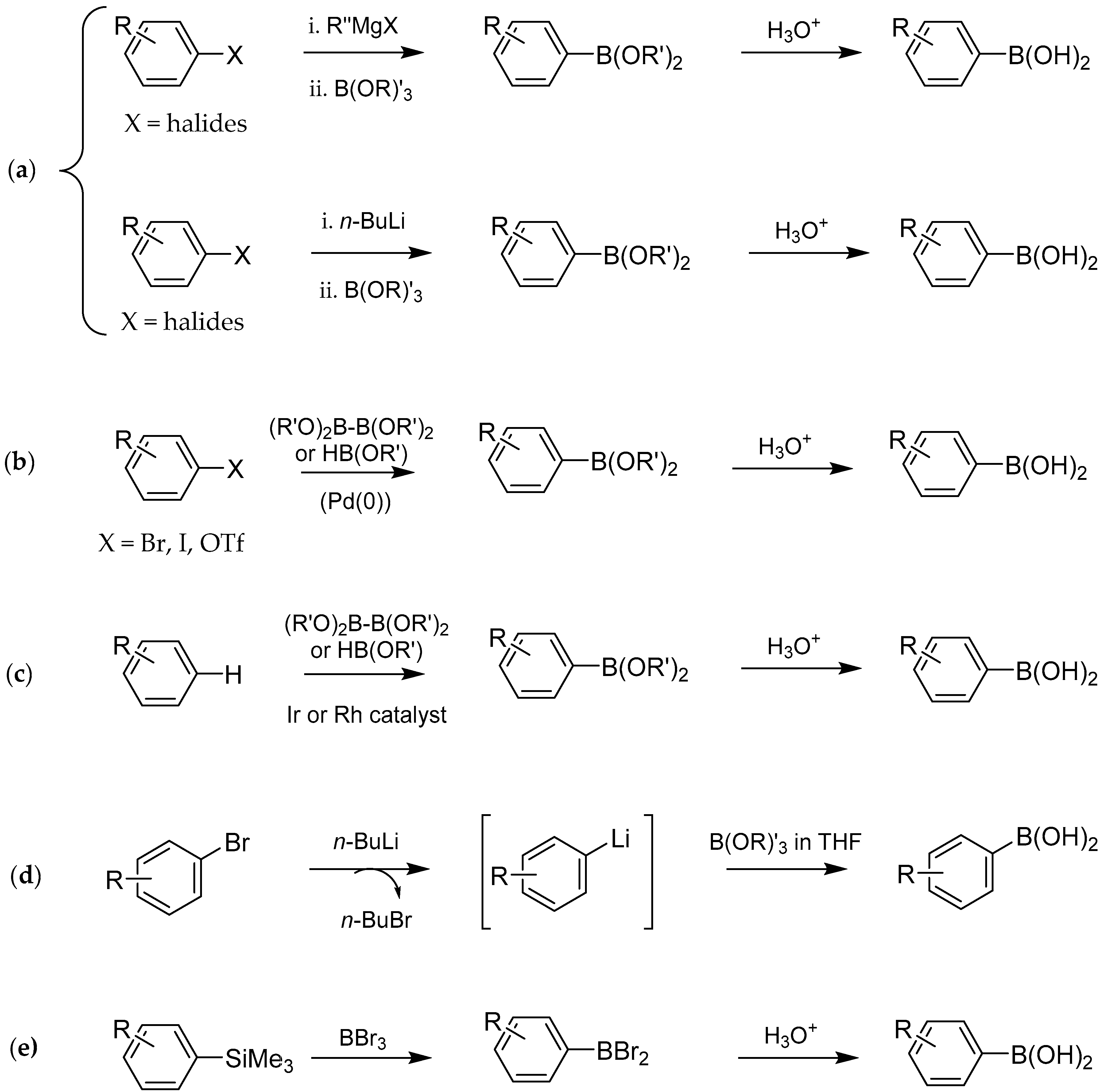{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
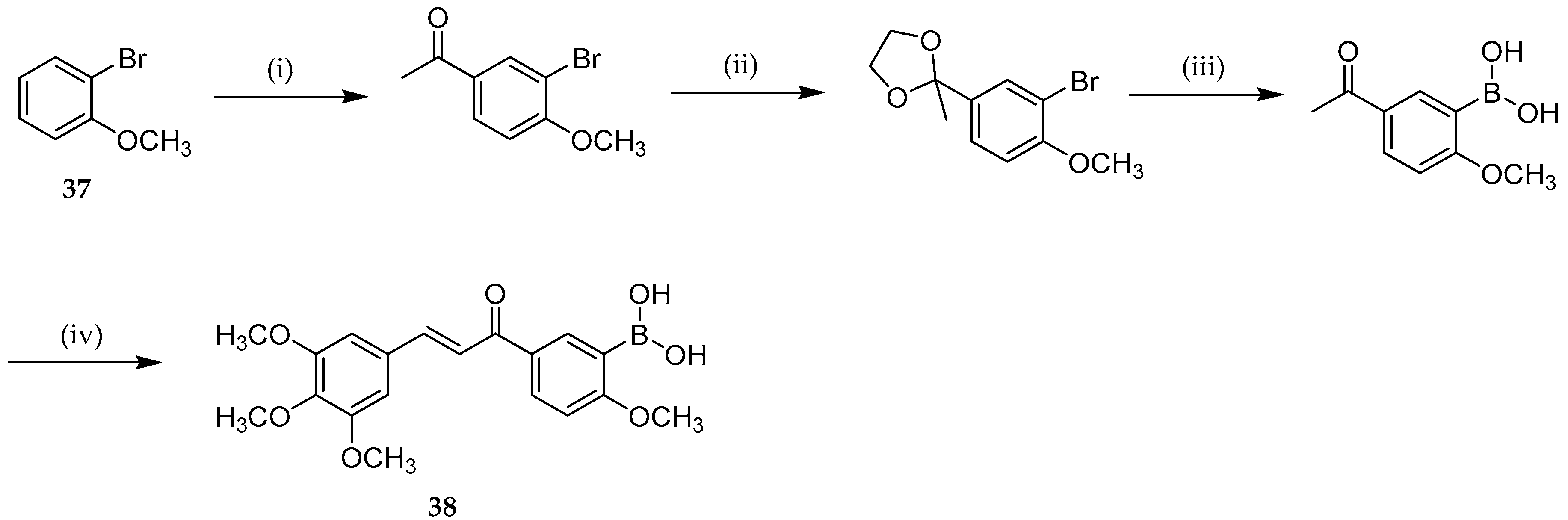{kind=link}
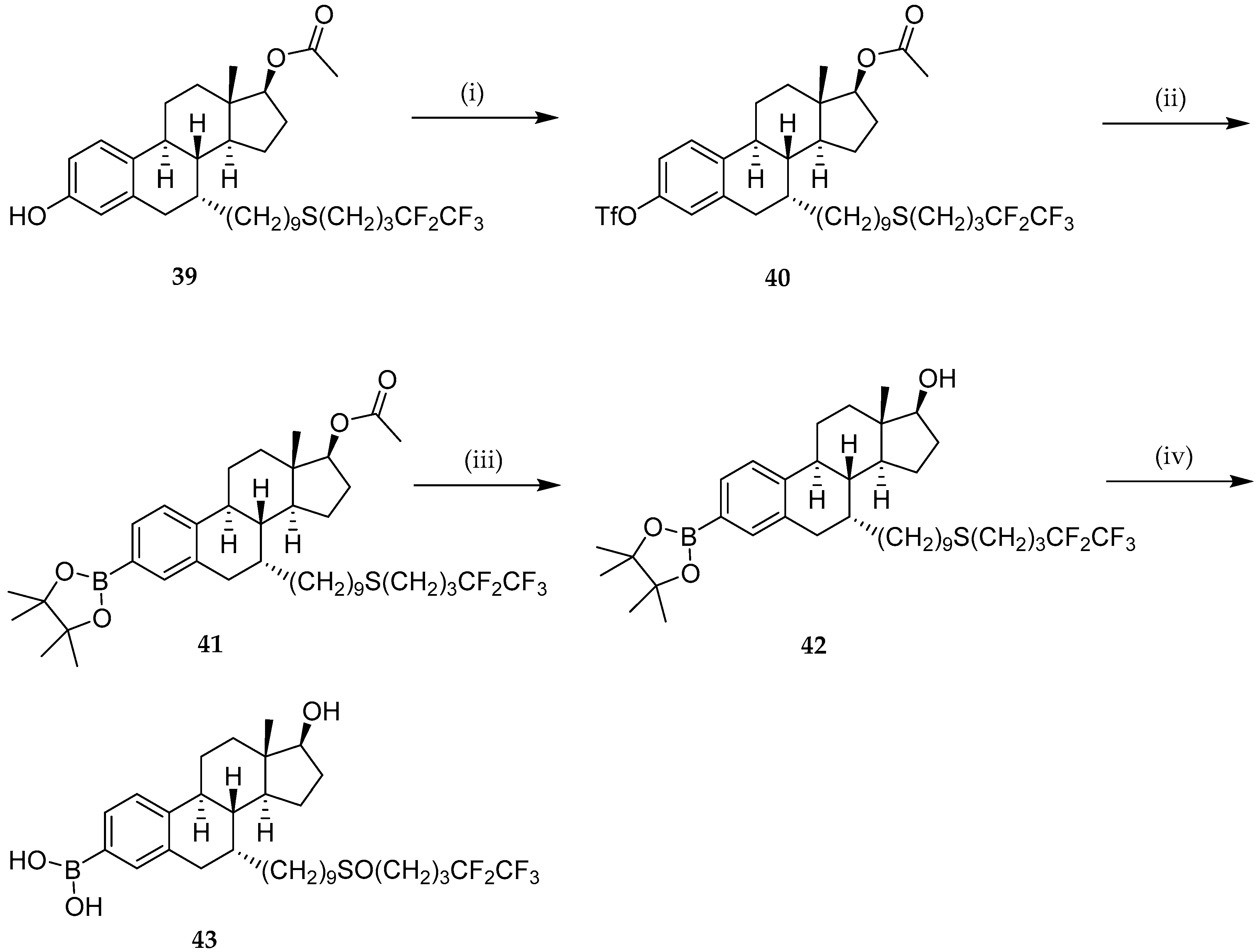{kind=link}
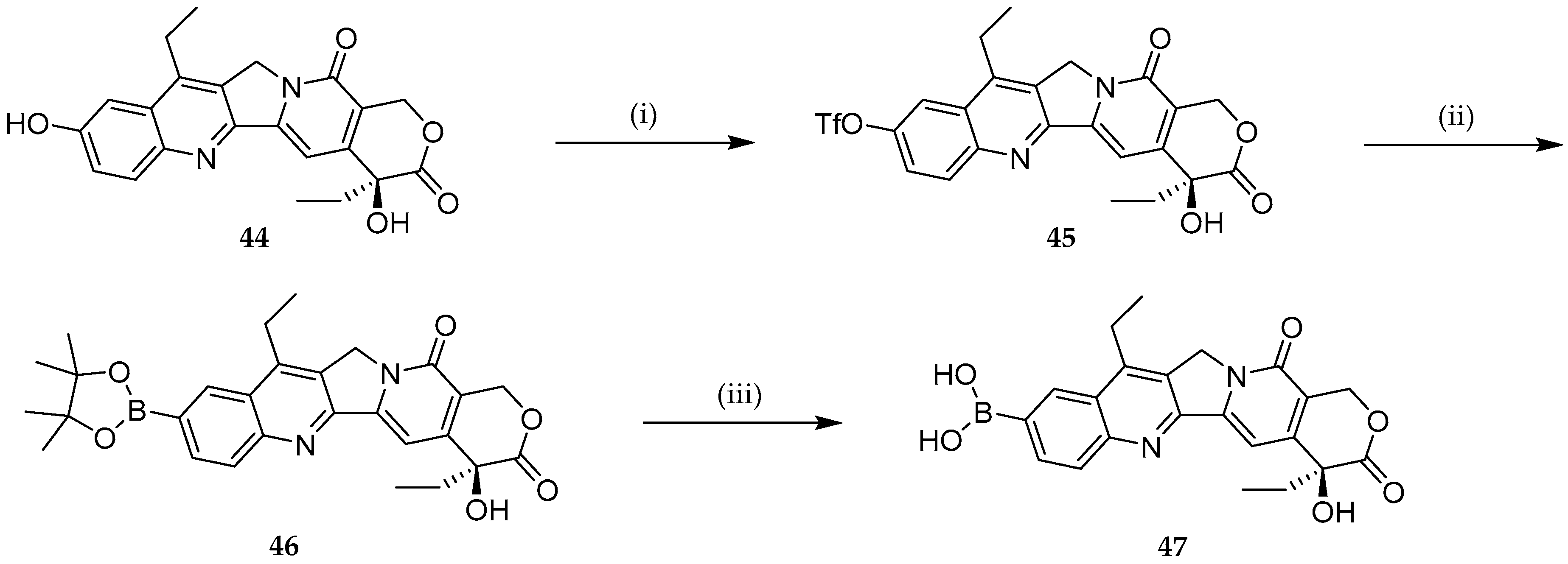{kind=link}
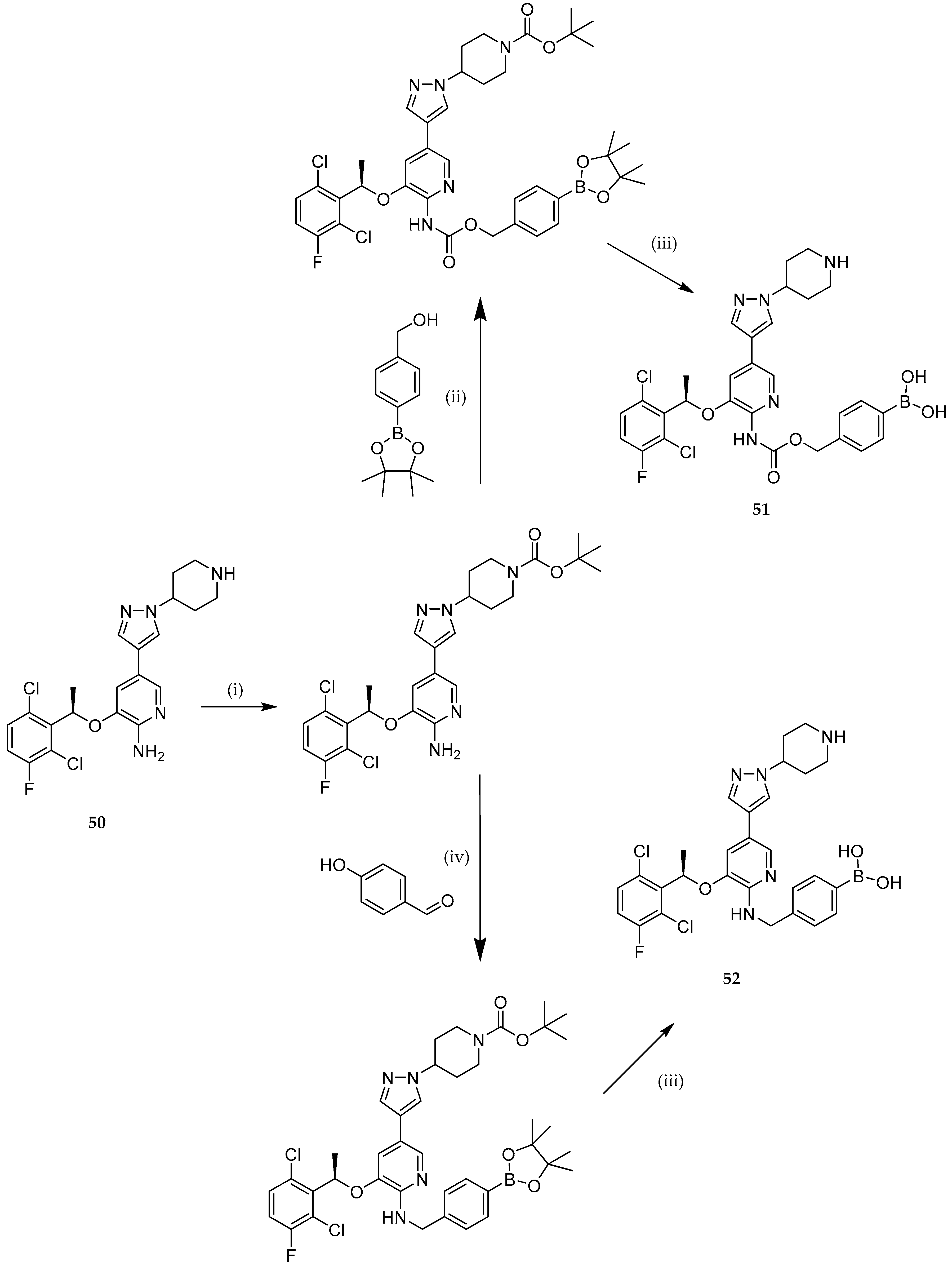{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
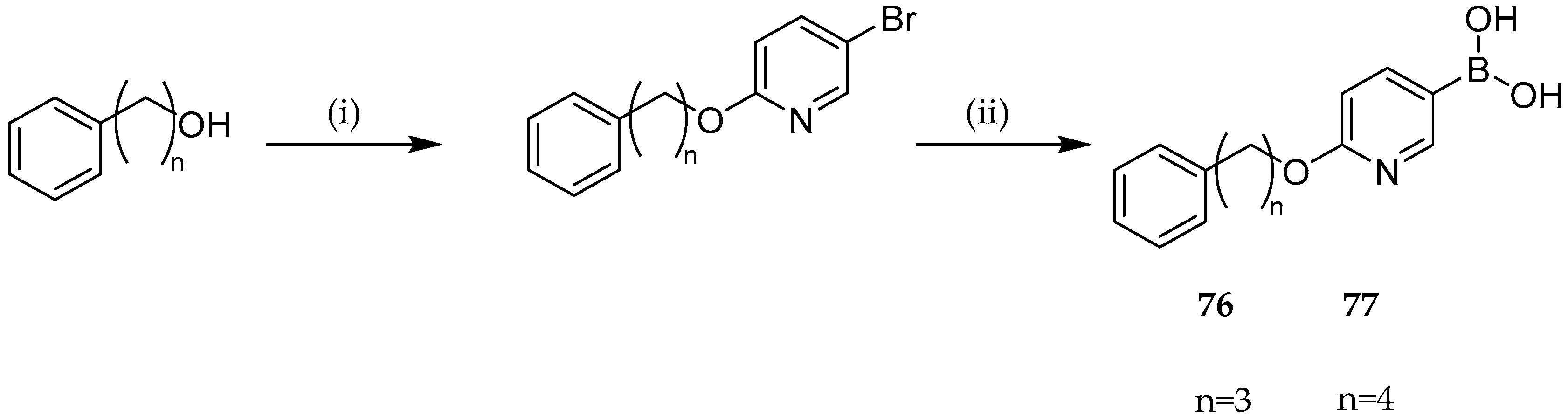{kind=link}
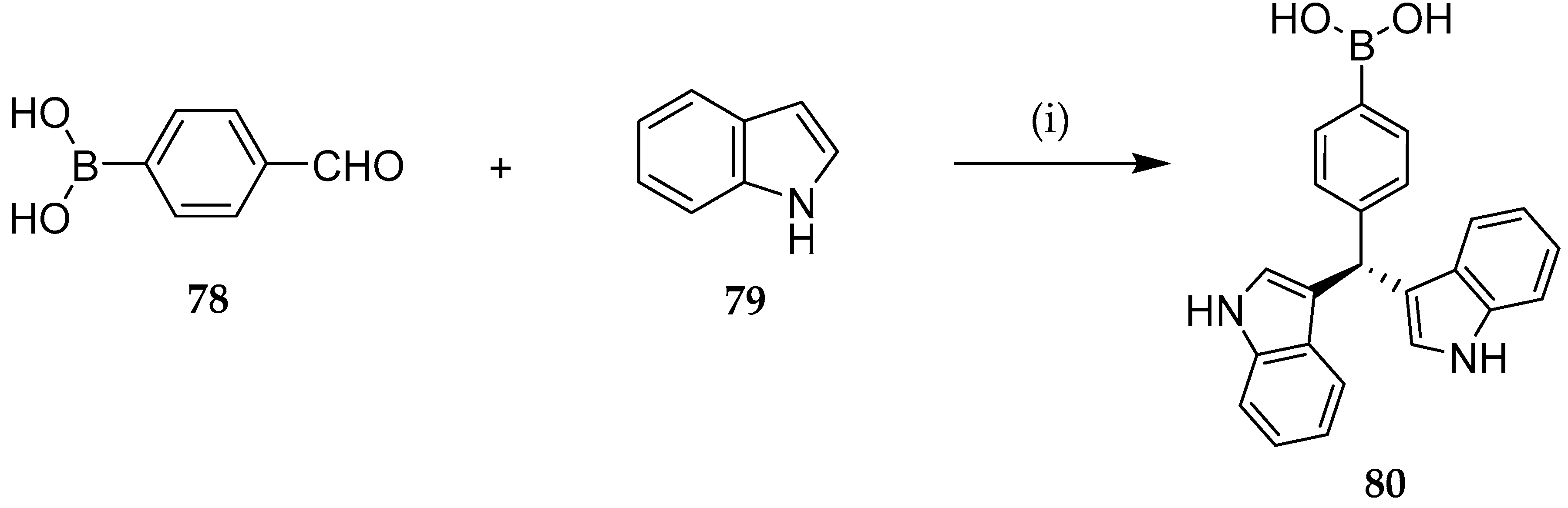{kind=link}
{kind=link}
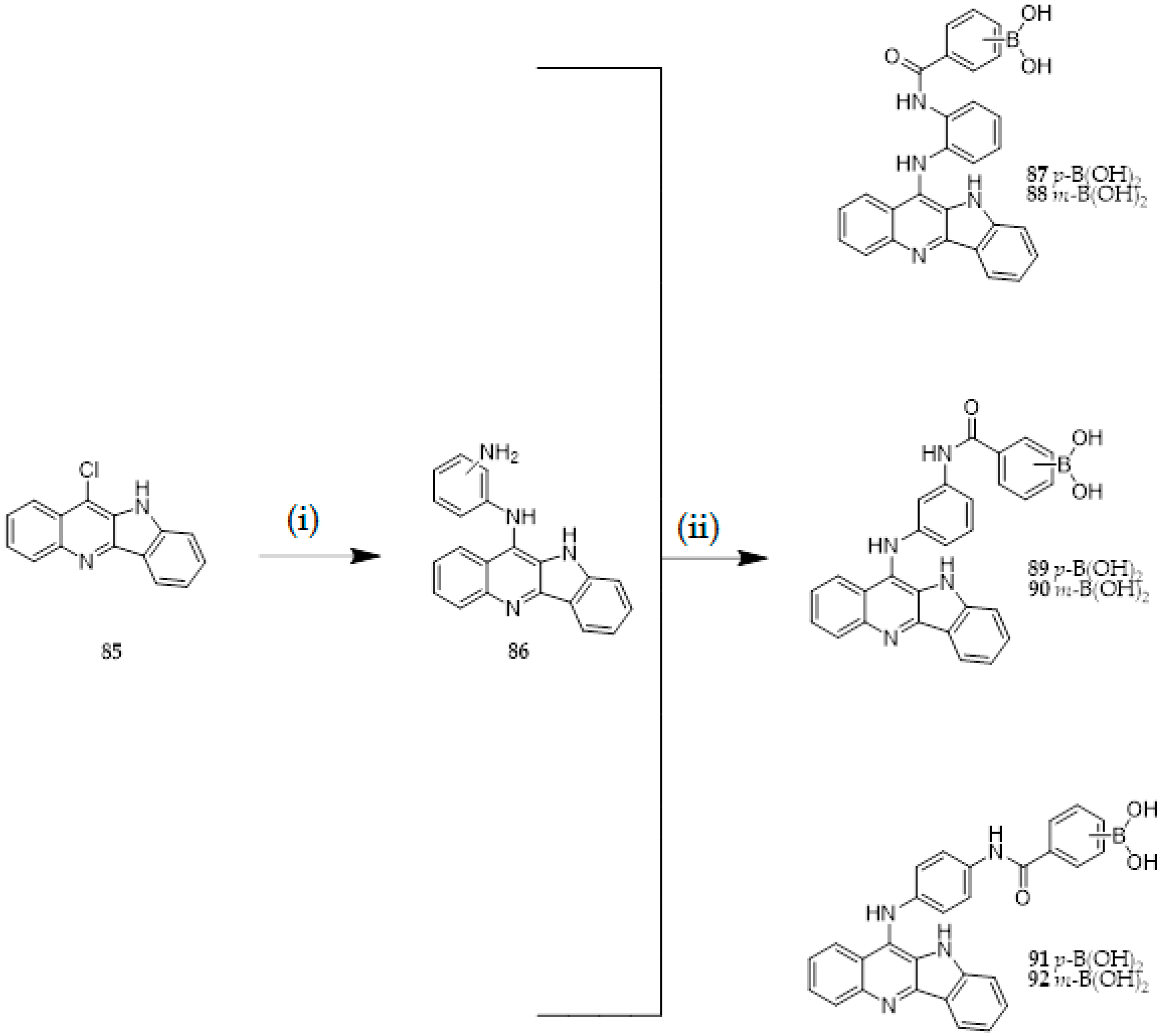{kind=link}
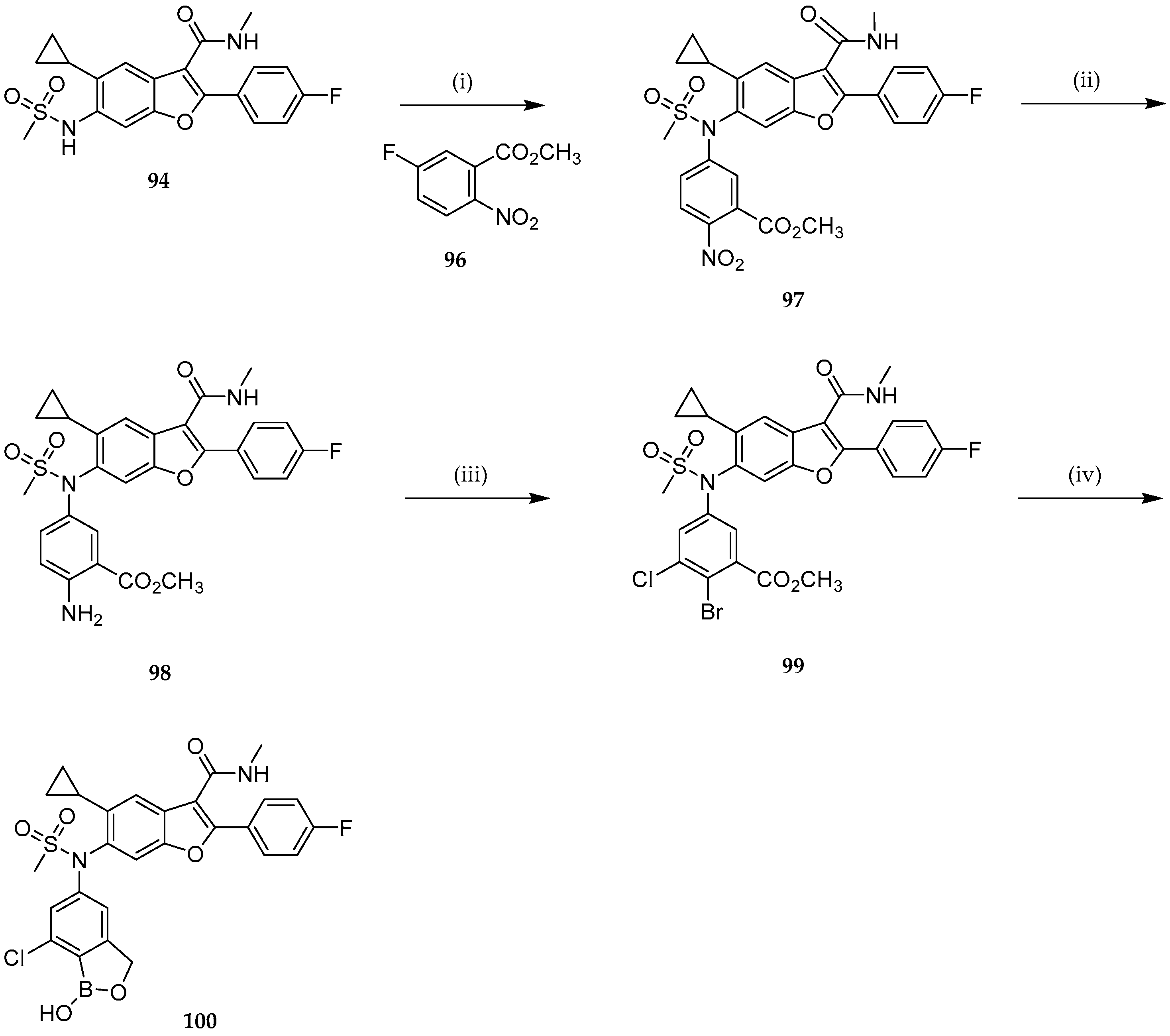{kind=link}
{kind=link}
| Study | Purpose | Phase | Status |
|---|---|---|---|
| Combination therapy with bortezomib in advanced stage aggressive lymphomas | To study whether the combination of CHOP 1 with bortezomib to overcome drug resistance induced by pro-apoptotic proteins BAX and BCL-XS 2 | Phase 1 Phase 2 | Completed (October 2009) |
| Combination therapy with ixazomib for the treatment of sarcoma | To establish a safe and tolerable dose combination of selinexor and ixazomib when used together for the treatment of certain types of advanced sarcoma | Phase 1 | Ongoing |
| Lymphoma study in sombination with bortezomib or vorinostat | To determine the safety, tolerability and MTD 3 of AMG-655 4 when combined with bortezomib or vorinostat | Phase 1 | Completed (August 2011) |
| Bortezomib in relapsed or refractory AIDS 5-related sarcoma | To evaluate the MTD of bortezomib in patients with AIDS 5-related Kaposi sarcoma | Phase 1 | Completed (January 2015) |
| Combination therapy with bortezomib in ovarian epithelial cancer, fallopian tube cancer, or primary peritoneal cancer | To determine the MTD and dose-limiting toxicities of intraperitoneal bortezomib when combined with intraperitoneal carboplatin | Phase 1 | Completed (January 2018) |
| Combination therapy with bortezomib in acute myeloid leukemia | To identify the effective and tolerable dose of the bortezomib/sorafenib combination in acute myeloid leukemia; to recommend a dose level for phase 2 and define specific toxicities | Phase 1 | Ongoing |
| Combination therapy with bortezomib in lymphoma | To determine the RD 6 for phase 2 dose of alisertib and bortezomib when combined with rituximab in patients with relapsed/refractory mantle cell and B-cell low grade non-Hodgkin lymphoma | Phase 1 | Ongoing |
| First time in human safety and pharmacokinetics study of GSK3036656 in healthy subjects | To evaluate the safety, tolerability and pharmacokinetics of single ascending and repeat oral doses of GSK3036656 in healthy adults | Phase 1 | Completed (August 2017) |
| Intravenous ixazomib in pediatric participants with relapsed or refractory acute lymphoblastic leukemia or lymphoblastic lymphoma | To determine the MTD and/or phase 2 RD, safety and toxicity, and pharmacokinetics | Phase 1 | Ongoing |
| Ixazomib and pevonedistat in treating multiple myeloma that has come back or does not respond to treatment | To study the side effects and best dose of pevonedistat when combined with ixazomib in multiple myeloma that has come or does not respond to treatment | Phase 1 | Ongoing |
| The effect of ixazomib on the latent HIV reservoir | To determine the safety and tolerability of ixazomib in HIV infected patients who are on ART 7 that suppresses HIV replication; to determine the effect of ixazomib on the size of the HIV reservoir | Phase 1 | Ongoing |
| Dose escalation study of nelfinavir and ixazomib in patients with advanced solid tumors or lymphoma | To determine the MTD of the combination in advanced solid tumors or lymphoma | Phase 1 | Ongoing |
| Safety, tolerability and pharmacokinetics of multiple rising doses of ixazomib in lupus nephritis | To characterize the safety and tolerability of ixazomib when administered as multiple oral doses at escalating dose levels in participants with lupus nephritis | Phase 1 | Completed (January 2018) |
| Study of the combination of ixazomib and fulvestrant | To determine the proper dose and side effects of ixazomib when combined with fulvestrant | Phase 1 | Completed (June 2018) |
| Dose-finding, pharmacokinetics, and safety of meropenem-vaborbactam in pediatric patients with bacterial infections | To determine the dose-finding and evaluate pharmacokinetics, safety, and tolerability in pediatric patients (˂18 years) with serious bacterial infections | Phase 1 | Ongoing |
| Cumulative irritation test of tavaborole | To determine the cumulative irritation potential of tavaborole | Phase 1 | Completed (February 2007) |
| Study to assess local tolerability of crisaborole 2% ointment in healthy participants | To estimate observed application site adverse events following topical applications of crisaborole and vehicle in healthy participants | Phase 1 | Completed (April 2019) |
| Combination therapy with bortezomib with/without venetoclax in multiple myeloma | To study side effects and best dose of venetoclax combined with daratumumab, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma | Phase 1 Phase 2 | Ongoing |
| Combination therapy with bortezomib in plasma cell leukemia | To determine the MTD/RD of bortezomib and pegylated liposomal doxorubicin hydrochloride when in combination with a fixed dose of daratumumab, lenalidomide, and dexamethasone (phase 2) | Phase 1 Phase 2 | Completed (December 2018) |
| Combination chemotherapy with bortezomib in lymphoma | To determine the safety and the MTD of bortezomib when combined with rituximab, methotrexate, and cytarabine in lymphoma | Phase 1 Phase 2 | Ongoing |
| Ixazomib in the prophylaxis of chronic graft-versus-host disease | To evaluate the efficacy of ixazomib at the doses stablished in phase 1 | Phase 1 Phase 2 | Ongoing |
| PS-341 8 followed by removal of prostate in prostate cancer | To study the effect of systemic treatment with PS-341 8 doing correlative scientific markers assessing apoptosis; to evaluate protease protein targets and angiogenesis markers | Phase 2 | Completed (December 2013) |
| Bortezomib and vorinostat in patients with multiple myeloma who have undergone stem cell transplant | To evaluate the toxicity of the use of vorinostat and bortezomib as maintenance therapy after autologous transplant | Phase 2 | Completed (March 2017) |
| Fulvestrant with/without bortezomib in inoperable locally or metastatic estrogen receptor positive breast cancer | To determine whether the combination of bortezomib to fulvestrant improves median progression free survival | Phase 2 | Ongoing |
| Bortezomib and lenalidomide in mantle cell lymphoma | To determine the complete and partial response rate of bortezomib and lenalidomide therapy in relapsed or refractory mantle cell lymphoma | Phase 2 | Completed (April 2019) |
| Combination therapy with bortezomib in previously untreated symptomatic multiple myeloma | To evaluate the complete and near-complete response rate of the bortezomib/pegylated liposomal doxorubicin regimen in previously untreated, symptomatic multiple myeloma | Phase 2 | Completed (September 2019) |
| Rituximab, venetoclax, and bortezomib in lymphoma | To determine the overall response rate of rituximab, venetoclax, and bortezomib in relapsed/refractory diffuse large B-cell lymphoma | Phase 2 | Ongoing |
| Combination therapy with/without bortezomib and dexamethasone in immunoglobulin light chain amyloidosis | To evaluate the overall hematologic response rate for ibrutinib with bortezomib and dexamethasone added for lack of response in amyloid light chain | Phase 2 | Completed (April 2019) |
| Combination therapy with bortezomib followed by combination therapy with ixazomib in multiple myeloma | To evaluate the progression-free survival of patients with daratumumab, bortezomib, and dexamethasone treatment followed by daratumumab, ixazomib, and dexamethasone treatment | Phase 2 | Ongoing |
| Melphalan and bortezomib as conditioning regimen for autologous and allogeneic stem cell transplants in multiple myeloma | To evaluate the effectiveness of bortezomib when combined to standard chemotherapy medicine(s) for treatment of multiple myeloma | Phase 2 | Completed (May 2019) |
| Study of ixazomib and erlotinib in solid tumors | To find the MTD of the combination of ixazomib and erlotinib in advanced solid tumors; to study the safety of these drugs | Phase 2 | Ongoing |
| Efficacy and safety of ixazomib and dexamethasone refractory autoimmune cytopenia | To evaluate the safety and efficacy of this combination in refractory autoimmune cytopenia | Phase 2 | Completed (October 2019) |
| Ibrutinib and ixazomib citrate in relapsed or refractory Waldenstrom macroglobulinemia | To study the side effects of ibrutinib citrate when combined with ixazomib; to determine the effect in refractory Waldenstrom macroglobulinemia | Phase 2 | Ongoing |
| Ixazomib for Desensitization | To study the effect of ixazomib to desensitize highly sensitized kidney transplant recipients | Phase 2 | Ongoing |
| Absorption and systemic study of tavaborole in patients with moderate to severe onychomycosis (ADME 9 I) | To determine the absorption, systemic pharmacokinetics and accumulation in the nail of tavaborole | Phase 2 | Completed (May 2007) |
| Absorption and systemic study of tavaborole in patients with moderate to severe onychomycosis (ADME II) | To determine the absorption and systemic pharmacokinetics of tavaborole | Phase 2 | Completed (May 2007) |
| Safety and efficacy study of crisaborole ointment to treat plaque type psoriasis | To determine the safety and efficacy of crisaborole ointment 5% in the treatment of plaque type psoriasis | Phase 2 | Completed (March 2008) |
| Crisaborole ointment 2% skin biomarker biopsy study in atopic dermatitis | To characterize the mechanism of action of crisaborole ointment 2% through evaluation of efficacy and changes in key skin biomarkers in atopic dermatitis lesions treated with crisaborole ointment 2% | Phase 2 | Completed (May 2018) |
| Combination therapy with/without bortezomib in previously untreated multiple myeloma | To study the effect of lenalidomide, dexamethasone, and bortezomib compared to dexamethasone and lenalidomide alone in treating patients with previously untreated multiple myeloma | Phase 3 | Ongoing |
| Combination therapy with bortezomib in newly diagnosed acute myeloid leukemia | To study the effect of bortezomib and sorafenib tosylate in newly diagnosed acute myeloid leukemia | Phase 3 | Ongoing |
| Ixazomib citrate, lenalidomide, dexamethasone, and zoledronic acid or zoledronic acid alone after radiation therapy in solitary plasmacytoma of bone | To evaluate the impact of combination of ixazomib, lenalidomide, and dexamethasone to zoledronic acid in multiple myeloma progression rate | Phase 3 | Ongoing |
| Study of dexamethasone plus ixazomib or physician’s choice in relapsed or refractory systemic light chain amyloidosis | To determine if this combination therapy improves hematologic response, heart and kidney deterioration and mortality rate comparing to a physician’s choice of a chemotherapy regimen in relapsed or refractory systemic light chain amyloidosis | Phase 3 | Ongoing |
| Study of oral ixazomib maintenance therapy after initial therapy in newly diagnosed multiple myeloma not treated with stem cell transplantation | To determine the effect of ixazomib maintenance therapy on progression-free survival in newly diagnosed multiple myeloma who have had a major or partial response to initial therapy and who have not undergone stem-cell transplantation | Phase 3 | Ongoing |
| Efficacy, safety, tolerability of meropenem-vaborbactam compared to best available therapy in treating serious infections in adults | To compare the efficacy, safety and tolerability of meropenem-vaborbactam with the best available therapy for infections caused by carbapenem-resistant Enterobacteriaceae | Phase 3 | Completed (July 2017) |
| Efficacy and safety evaluation of tavaborole topical solution to treat onychomycosis of the toenail | To determine whether tavaborole topical solution is a safe and effective treatment for onychomycosis of the toenail | Phase 3 | Completed (January 2013) |
| Evaluation of the safety and pharmacokinetics of tavaborole topical solution for the treatment of onychomycosis in children and adolescents | To evaluate the safety and pharmacokinetics of tavaborole 5% topical solution in treating distal subungual onychomycosis of the toenail in children and adolescents | Phase 4 | Completed (July 2017) |
| Compounds | MDA-MB-435 | MDA-MB-231 |
|---|---|---|
| 2 | 10 | 8.8 |
| 4 | 16 | 8.5 |
| 5 | 8.8 | 8.8 |
 | 18 | 44 |
 | 13 | 18 |
| Cell Line | GI50 1 (µM) |
|---|---|
| MCF7 (breast cancer) | 0.034 |
| SNB-19 (CNS 2 cancer) | ≤0.01 |
| NCI-H460 (lung cancer) | 0.028 |
| SNB-19 (colon cancer) | ≤0.01 |
| UACC-62 (melanoma) | 0.034 |
| SK-OV-3 (ovarian cancer) | ≤0.01 |
| DU-145 (prostate cancer) | 0.050 |
| Compound | MCF-7 | T47D |
|---|---|---|
| Fulvestrant 43 | 0.0015 0.0032 | 0.0012 0.0061 |
| CPM 1 | CPM + 75 | CTLZN 2 | CTLZN + 75 | MP 3 | MP + 35 |
|---|---|---|---|---|---|
| 32 | ≤0.06 | 32 | 0.125 | 1 | ≤0.015 |
| CIPR 1 | CIPR + 76 | CIPR + 77 | NOR 2 | NOR + 76 | NOR + 77 |
|---|---|---|---|---|---|
| 16 | 4 | 4 | 64 | 8 | 8 |
| Dengue Virus Protease | West Nile Virus Protease | Zika Virus Protease |
|---|---|---|
| 0.051 | 0.082 | 0.04 |
| Compound | Biological Activity | References |
|---|---|---|
 | Drug for treatment of onychomycosis—leucyl-tRNA synthetase inhibitor | [88,90] |
 | Selective leucyl-tRNA synthetase inhibitor for Mycobacterium tuberculosis (M. tuberculosis, IC50 1 of 0.2 μM) | [91] |
 | Selective inhibitor of human carbonic anhydrases (CAs) (Ki 2 of 69 -414 nM) | [92] |
 | Selective inhibitor of β-CA (Cryptococcus neoformans, Ki of 78 nM; Candida glabrata, Ki of 75 nM) | [93] |
 | Anti-Plasmodium agent (P. falciparum, ED90 3 of 0.85 mg/kg) - promising for the treatment of malaria | [94] |
 | Oral ectoparasiticide agent for Dermacentor variabilis (American dog ticks) and Ctenocephalides felis (cat fleas) (EC50 4 of 96.6 μM) | [95] |
 | Topical agent for treatment of cutaneous leishmaniasis (Leishmania major, EC50 of 4.26 μM; L.tropica, EC50 of 2.01 μM) | [96] |
 | Serine β- lactamase inhibitor (Class A, IC50 ˂ 0.005–0.026 μΜ; Class C, IC50 of 0.012–0.067 μM; Class D, IC50 of 0.088–0.12 μM) of pathogens such as E. coli (MIC 5 of 0.25–1 μg/ml) | [97] |
 | Antiproliferative and proapoptotic agent for ovarian cancer line cell A2780 (IC50 of 14.8 μM) | [98] |
 | Topical anti-inflammatory drug for atopic dermatitis—phosphodiesterase type 4 (PDE4) inhibitor promising for psoriasis | [99] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, M.P.; Saraiva, L.; Pinto, M.; Sousa, M.E. Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications. Molecules 2020, 25, 4323. https://doi.org/10.3390/molecules25184323
Silva MP, Saraiva L, Pinto M, Sousa ME. Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications. Molecules. 2020; 25(18):4323. https://doi.org/10.3390/molecules25184323
Chicago/Turabian StyleSilva, Mariana Pereira, Lucília Saraiva, Madalena Pinto, and Maria Emília Sousa. 2020. "Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications" Molecules 25, no. 18: 4323. https://doi.org/10.3390/molecules25184323
APA StyleSilva, M. P., Saraiva, L., Pinto, M., & Sousa, M. E. (2020). Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications. Molecules, 25(18), 4323. https://doi.org/10.3390/molecules25184323