
Distinct Influence of Hypercaloric Diets Predominant with Fat or Fat and Sucrose on Adipose Tissue and Liver Inflammation in Mice

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
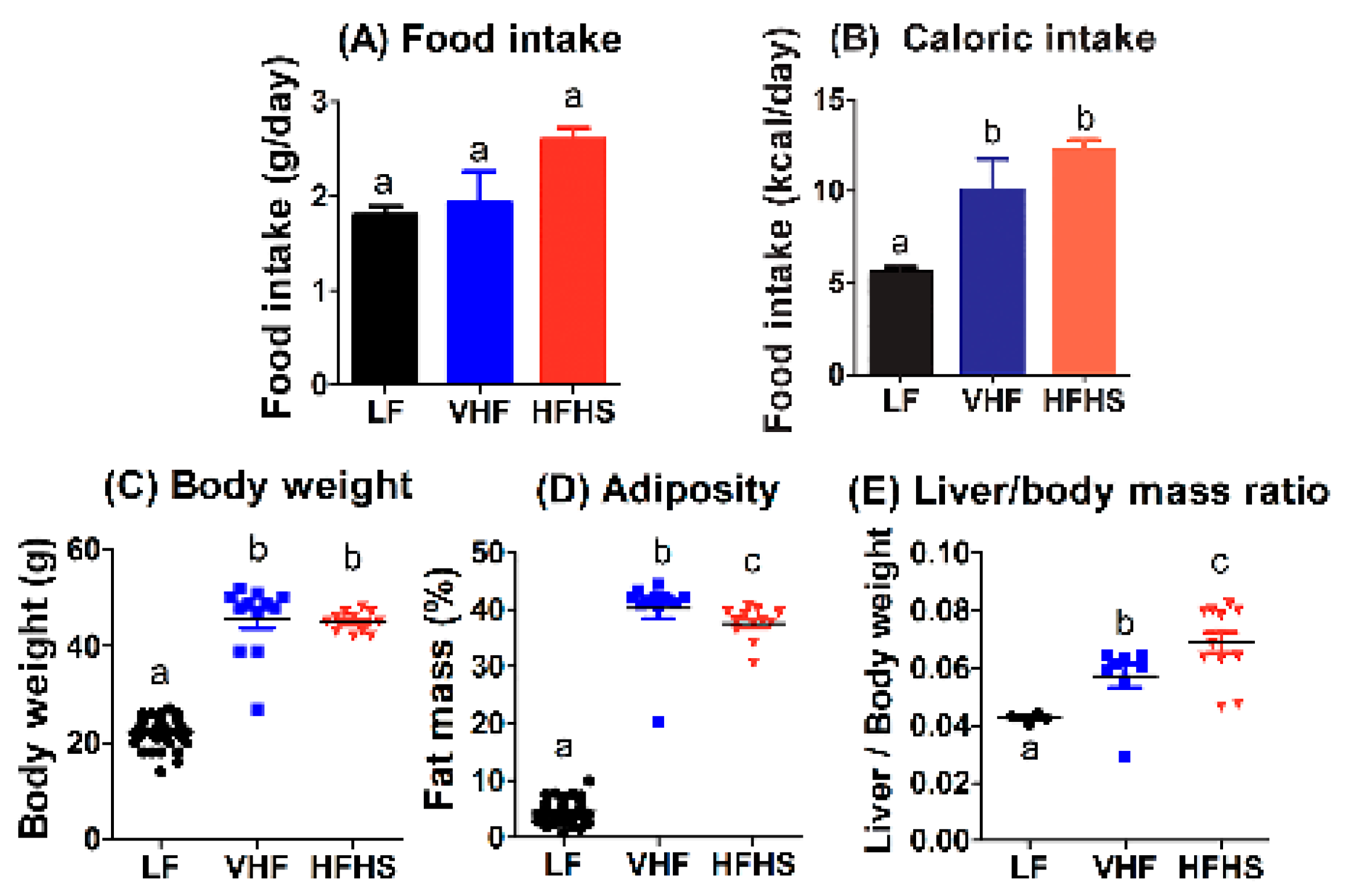
2.1. Diet Effects on Food Intake, Body Weight, and Adiposity
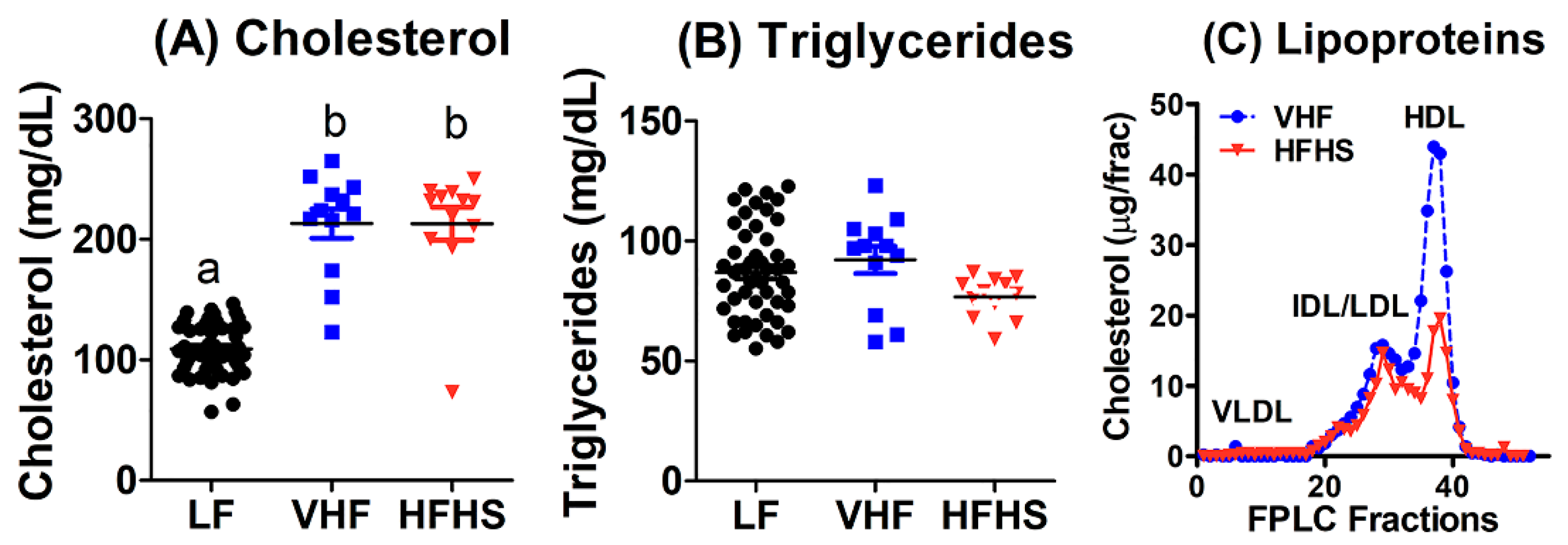
2.2. Diet Effects on Plasma Lipid and Lipoproteins
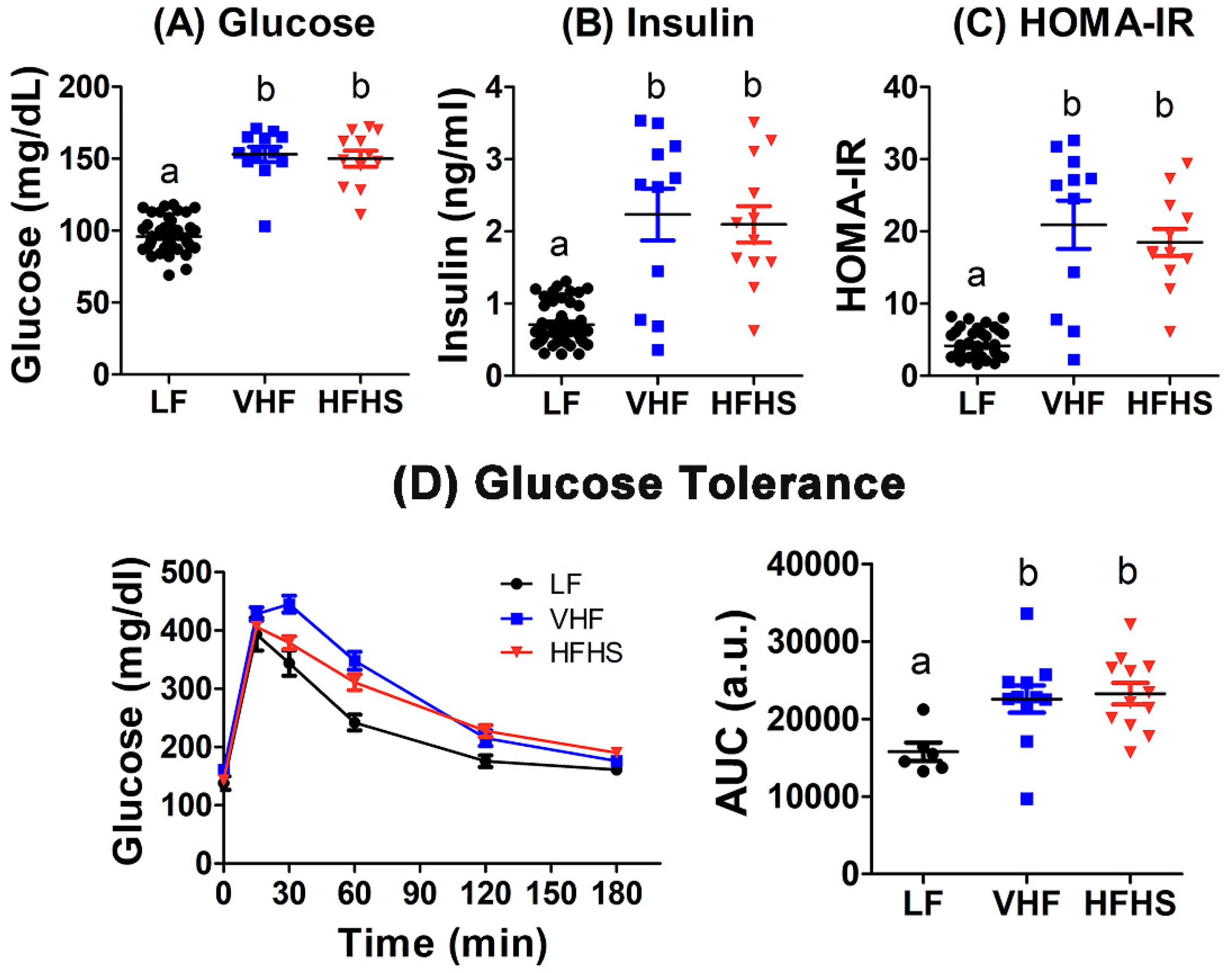
2.3. Diet Effects on Fasting Glucose and Insulin Levels and Insulin Resistance
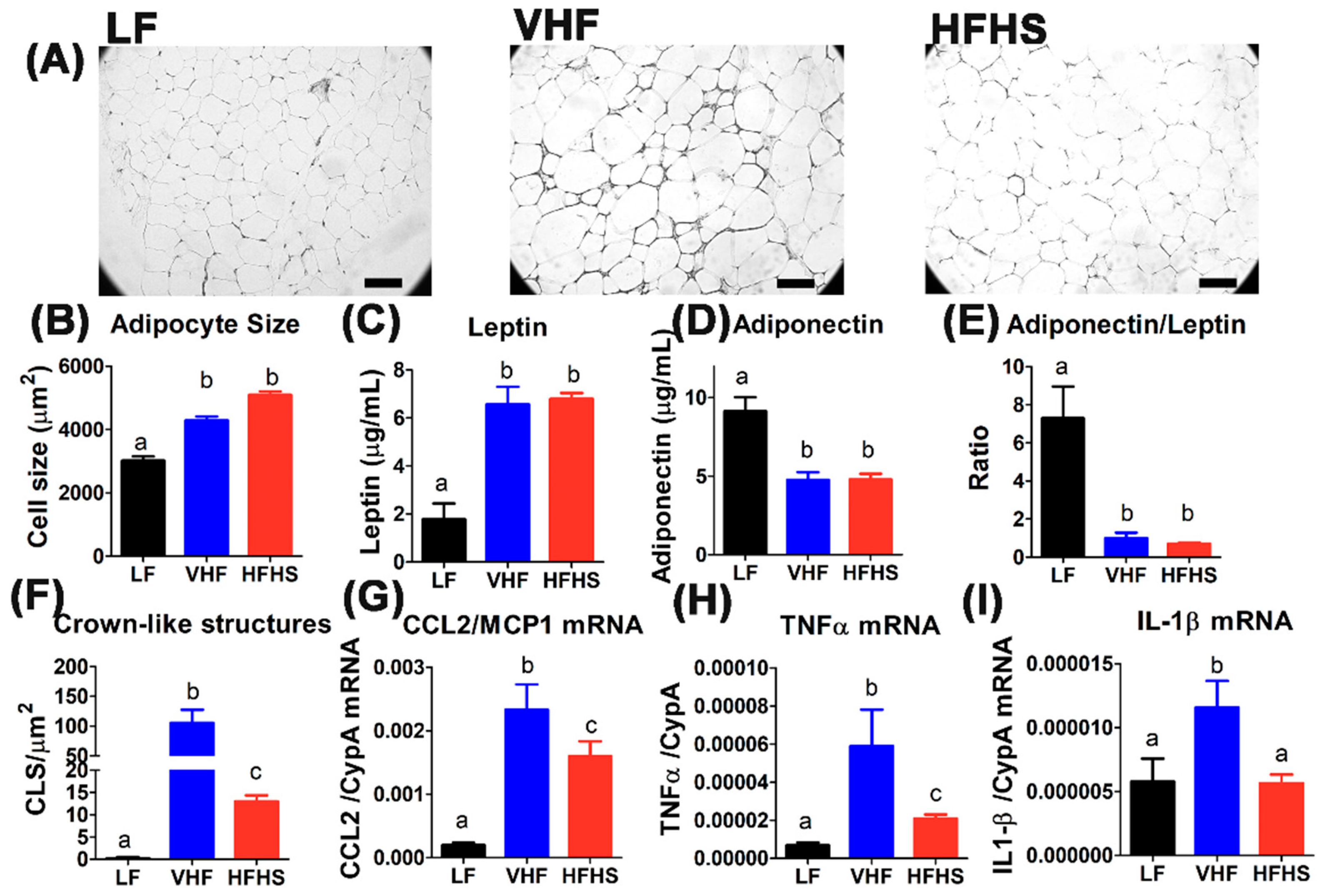
2.4. Hypercaloric Diet with Excess Fat Promotes Adipose Tissue Inflammation
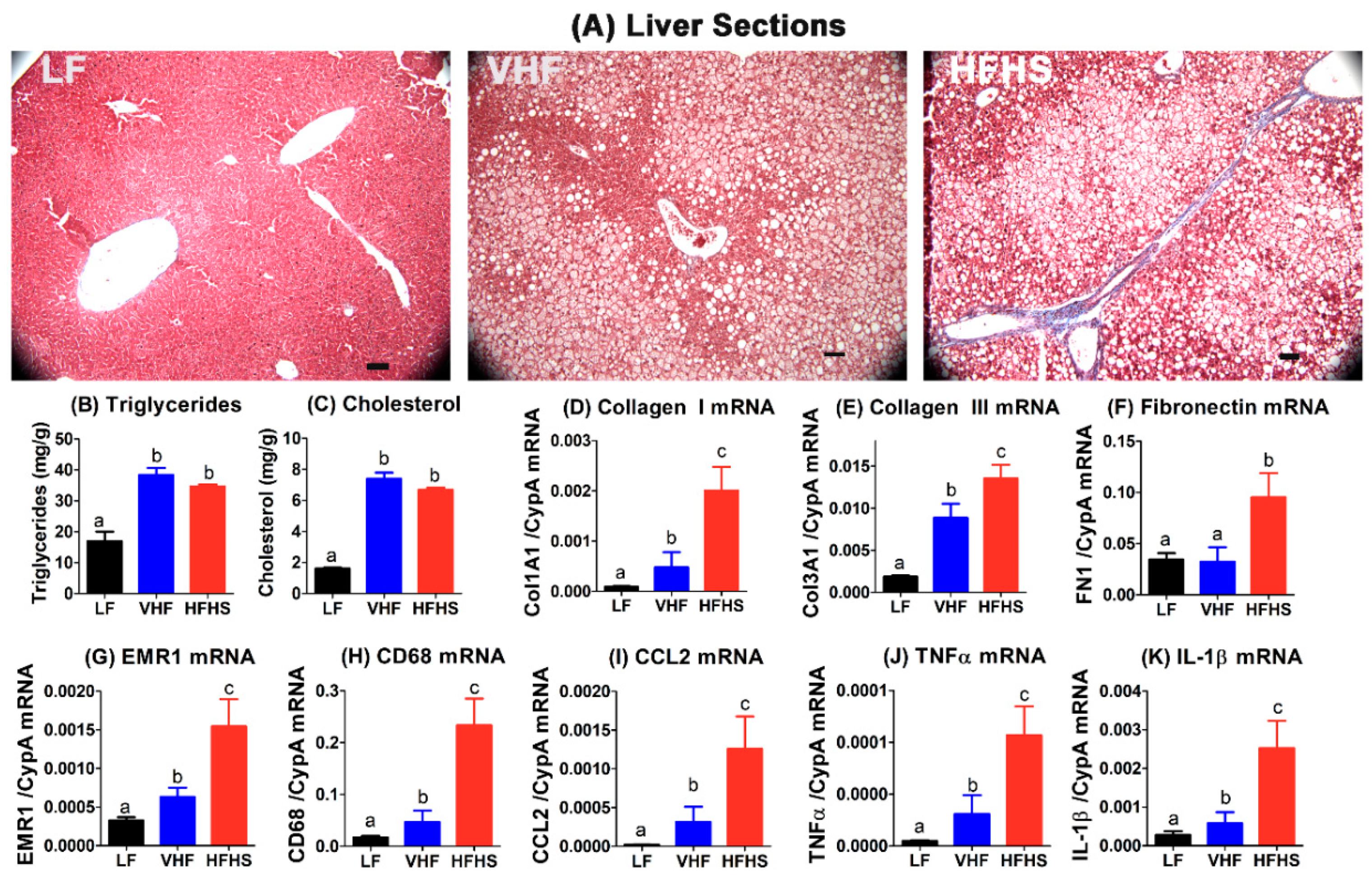
2.5. High-Fat High-Sucrose Diet Accelerates Hepatosteatosis Transition to Steatohepatitis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Analytical Procedures
4.3. Glucose Tolerance Test
4.4. Tissue Histology
4.5. Real-Time Quantitative Polymerase Chain Reaction Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Magno, C.P.; Lane, K.T.; Hinojosa, M.W.; Lane, J.S. Association of hypertension, diabetes, dyslipidemia, and metabolic syndrome with obesity: Findings from the National Health and Nutrition Examination Survey, 1999 to 2004. J. Am. Coll. Surg. 2008, 207, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Corey, K.E.; Kaplan, L.M. Obesity and liver disease. The epidemic of the twenty-first century. Clin. Liver Dis. 2014, 18, 1–18. [Google Scholar] [CrossRef]
- Bertola, A.; Bonnafous, S.; Anty, R.; Patouraux, S.; Saint-Paul, M.C.; Iannelli, A.; Gugenheim, J.; Barr, J.; Mato, J.M.; Le Marchand-Brustel, Y.; et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS ONE 2010, 5, e13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummins, T.D.; Holden, C.R.; Sansbury, B.E.; Gibb, A.A.; Shah, J.; Zafar, N.; Tang, Y.; Hellmann, J.; Rai, S.N.; Spite, M.; et al. Metabolic remodeling of white adipose tissue in obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E262–E277. [Google Scholar] [CrossRef]
- Li, L.; Liu, D.W.; Yan, H.Y.; Wang, Z.Y.; Zhao, S.H.; Wang, B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: Evidence from a meta-analysis of 21 cohort studies. Obes. Rev. 2016, 17, 510–519. [Google Scholar] [CrossRef]
- Aby, E.; Saab, S. Nonobese nonalcoholic fatty liver disease. Clin. Liver Dis. 2017, 10, 130–133. [Google Scholar] [CrossRef]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [Green Version]
- Gholam, P.M.; Kotler, D.P.; Flancbaum, L.J. Liver pathology in morbidly obese patients undergoing Roux-en-Y gastric bypass surgery. Obes. Surg. 2002, 12, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Mai, B.H.; Yan, L.-J. The negative and detrimental effects of high fructose on the liver, with special reference to metabolic disorders. Diabetes Metab. Syndr. Obes. 2019, 12, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Smethers, A.D.; Rolls, B.J. Dietary management of obesity: Cornerstones of healthy eating patterns. Med. Clin. N. Am. 2018, 102, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Abdelhamid, A.; Moore, H.J.; Douthwaite, W.; Skeaff, C.M.; Summerbell, C.D. Effect of reducing total fat intake on body weight: Systematic review and meta-analysis of randomised controlled trials and cohort studies. BMJ 2012, 345, e7666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, F.M.; Bray, G.A.; Carey, V.J.; Smith, S.R.; Ryan, D.H.; Anton, S.D.; McManus, K.; Champagne, C.M.; Bishop, L.M.; Laranjo, N.; et al. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. N. Engl. J. Med. 2009, 360, 859–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogelholm, M.; Anderssen, S.; Gunnarsdottir, I.; Lahti-Koski, M. Dietary macronutrients and food consumption as determinants of long-term weight change in adult populations: A systematic literature review. Food Nutr. Res. 2012, 56. [Google Scholar] [CrossRef] [Green Version]
- Bray, G.A.; Siri-Tarino, P.W. The role of macronutrient content in the diet for weight management. Endocrinol. Metab. Clin. N. Am. 2016, 45, 581–604. [Google Scholar] [CrossRef]
- Makris, A.; Foster, G.D. Dietary approaches to the treatment of obesity. Psychiatr. Clin. N. Am. 2011, 34, 813–827. [Google Scholar] [CrossRef] [Green Version]
- Bueno, N.B.; de Melo, I.S.V.; de Oliveira, S.L.; da Rocha Ataide, T. Very low carbohydrate ketogenic diet v. low fat diet for long term weight loss: A meta-analysis of randomised controlled trials. Br. J. Nutr 2013, 110, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Mansoor, N.; Vinknes, K.J.; Veierod, M.B.; Retterstol, K. Effects of low carbohydrate diets v. low fat diets on body weight and cardiovascular risk factors: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2016, 115, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Sackner-Bernstein, J.; Kanter, D.; Kaul, S. Dietary intervention for overweight and obese adults: Comparison of low carbohydrate and low fat diets. A meta-analysis. PLoS ONE 2015, 10, e0139817. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.U.; Dethlefsen, C.; Joensen, A.M.; Stegger, J.; Tjonneland, A.; Schmidt, E.B.; Overvad, K. Intake of carbohydrates compared with intake of saturated fatty acids and risk of myocardial infarction: Importance of the glycemic index. Am. J. Clin. Nutr 2010, 91, 1764–1768. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.D.; Wyatt, H.R.; Hill, J.O.; Makris, A.P.; Rosenbaum, D.L.; Brill, C.; Stein, R.I.; Mohammed, B.S.; Miller, B.; Rader, D.J.; et al. Weight and metabolic outcomes after 2 years on a low-carbohydrate versus low-fat diet: A randomized trial. Ann. Intern. Med. 2010, 153, 147–157. [Google Scholar] [CrossRef]
- Hjorth, M.F.; Astrup, A.; Zohar, Y.; Urban, L.E.; Sayer, R.D.; Patterson, B.W.; Herring, S.J.; Klein, S.; Zemel, B.S.; Foster, G.D.; et al. Personalized nutrition: Pretreatment glucose metabolism determines individual long-term weight loss responsiveness in individuals with obesity on low carbohydrate versus low fat diet. Int. J. Obes. 2019, 43, 2037–2044. [Google Scholar] [CrossRef]
- Fergusson, G.; Etheir, M.; Guevremont, M.; Chretien, C.; Attane, C.; Joly, E.; Fioramonti, X.; Prentki, M.; Poitout, V.; Alquier, T. Defective insulin secretory response to intravenous glucose in C57BL/6J compared to C57BL/6N mice. Mol. Metab. 2014, 3, 848–854. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Toth, L.A. The influence of the cage environment on rodent physiology and behavior: Implications for reproducibility of pre-clinical rodent research. Exp. Neurol. 2015, 270, 72–77. [Google Scholar] [CrossRef]
- Leulier, F.; MacNeil, L.T.; Lee, W.-J.; Rawls, J.F.; Cani, P.D.; Schwarzer, M.; Zhao, L.; Simpson, S.J. Integrative physiology: At the crossroads of nutrition, microbiota, animal physiology, and human health. Cell Metab. 2017, 25, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Hynes, G.R.; Jones, P.J.H. Leptin and its role in lipid metabolism. Curr. Opin. Lipidol. 2001, 12, 321–327. [Google Scholar] [CrossRef]
- Lopez-Jaramillo, P.; Gomez-Arbelaez, D.; Lopez-Lopez, J.; Lopoz-Lopez, C.; Martinez-Ortega, J.; Gomez-Rodriguez, A.; Triana-Cubillos, S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm. Mol. Biol. Clin. Investig. 2014, 18, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iikuni, N.; Lam, Q.L.; Lu, L.; Matarese, G.; La Cava, A. Leptin and inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Walsh, K. Adiponectin as an anti-inflammatory factor. Clin. Chim. Acta 2007, 380, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.M.F.; Olefsky, J.M. The origins and drivers of insulin resistance. Cell 2013, 152, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Heijden, R.A.; Sheedfar, F.; Morrison, M.C.; Hommelberg, P.P.H.; Kor, D.; Kloosterhuis, N.J.; Gruben, N.; Youssef, S.A.; de Bruin, A.; Hofker, M.H.; et al. High fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6J mice. Aging 2015, 7, 256–267. [Google Scholar] [CrossRef] [Green Version]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two hits. Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Wouters, K.; van Gorp, P.J.; Bieghs, V.; Gijbels, M.J.; Duimel, H.; Lutjohann, D.; Kerksiek, A.; van Kruchten, R.; Maeda, N.; Staels, B.; et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology 2008, 48, 474–486. [Google Scholar] [CrossRef]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Stanhope, K.L.; Schwarz, J.-M.; Havel, P.J. Adverse metabolic effects of dietary fructose: Results from the recent epidemiological, clinical, and mechanistic studies. Curr. Opin. Lipidol. 2013, 24, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Perazza, L.R.; Mitchell, P.L.; Jensen, B.A.H.; Daniel, N.; Boyer, M.; Varin, T.V.; Bouchareb, R.; Nachbar, R.T.; Bouchard, M.; Blais, M.; et al. Dietary sucrose induces metabolic inflammation and atherosclerotic cardiovascular diseases more than dietary fat in Ldlr−/−ApoB100/100 mice. Atherosclerosis 2020, 304, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Veniant, M.M.; Beigneux, A.P.; Bensadoun, A.; Fong, L.G.; Young, S.G. Lipoprotein size and susceptibility to atherosclerosis—Insights from genetically modified mouse models. Curr. Drug Targets 2008, 9, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Alogaili, F.; Chinnarasu, S.; Jaeschke, A.; Kranias, E.G.; Hui, D.Y. Hepatic HAX-1 inactivation prevents metabolic diseases by enhancing mitochondrial activity and bile salt export. J. Biol. Chem. 2020, 295, 4631–4646. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples are not available from the authors. All resources and reagents are available commercially. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, C.S.M.; Basford, J.E.; Kuhel, D.G.; Konaniah, E.S.; Cash, J.G.; Lima, V.L.M.; Hui, D.Y. Distinct Influence of Hypercaloric Diets Predominant with Fat or Fat and Sucrose on Adipose Tissue and Liver Inflammation in Mice. Molecules 2020, 25, 4369. https://doi.org/10.3390/molecules25194369
Fonseca CSM, Basford JE, Kuhel DG, Konaniah ES, Cash JG, Lima VLM, Hui DY. Distinct Influence of Hypercaloric Diets Predominant with Fat or Fat and Sucrose on Adipose Tissue and Liver Inflammation in Mice. Molecules. 2020; 25(19):4369. https://doi.org/10.3390/molecules25194369
Chicago/Turabian StyleFonseca, Caíque S. M., Joshua E. Basford, David G. Kuhel, Eddy S. Konaniah, James G. Cash, Vera L. M. Lima, and David Y. Hui. 2020. "Distinct Influence of Hypercaloric Diets Predominant with Fat or Fat and Sucrose on Adipose Tissue and Liver Inflammation in Mice" Molecules 25, no. 19: 4369. https://doi.org/10.3390/molecules25194369
APA StyleFonseca, C. S. M., Basford, J. E., Kuhel, D. G., Konaniah, E. S., Cash, J. G., Lima, V. L. M., & Hui, D. Y. (2020). Distinct Influence of Hypercaloric Diets Predominant with Fat or Fat and Sucrose on Adipose Tissue and Liver Inflammation in Mice. Molecules, 25(19), 4369. https://doi.org/10.3390/molecules25194369