
Glucosinolates: Natural Occurrence, Biosynthesis, Accessibility, Isolation, Structures, and Biological Activities




Abstract
:1. Introduction
2. Natural Occurrence of Glucosinolates
3. Glucosinolates: Biosynthetic and Chemical Synthetic Pathways
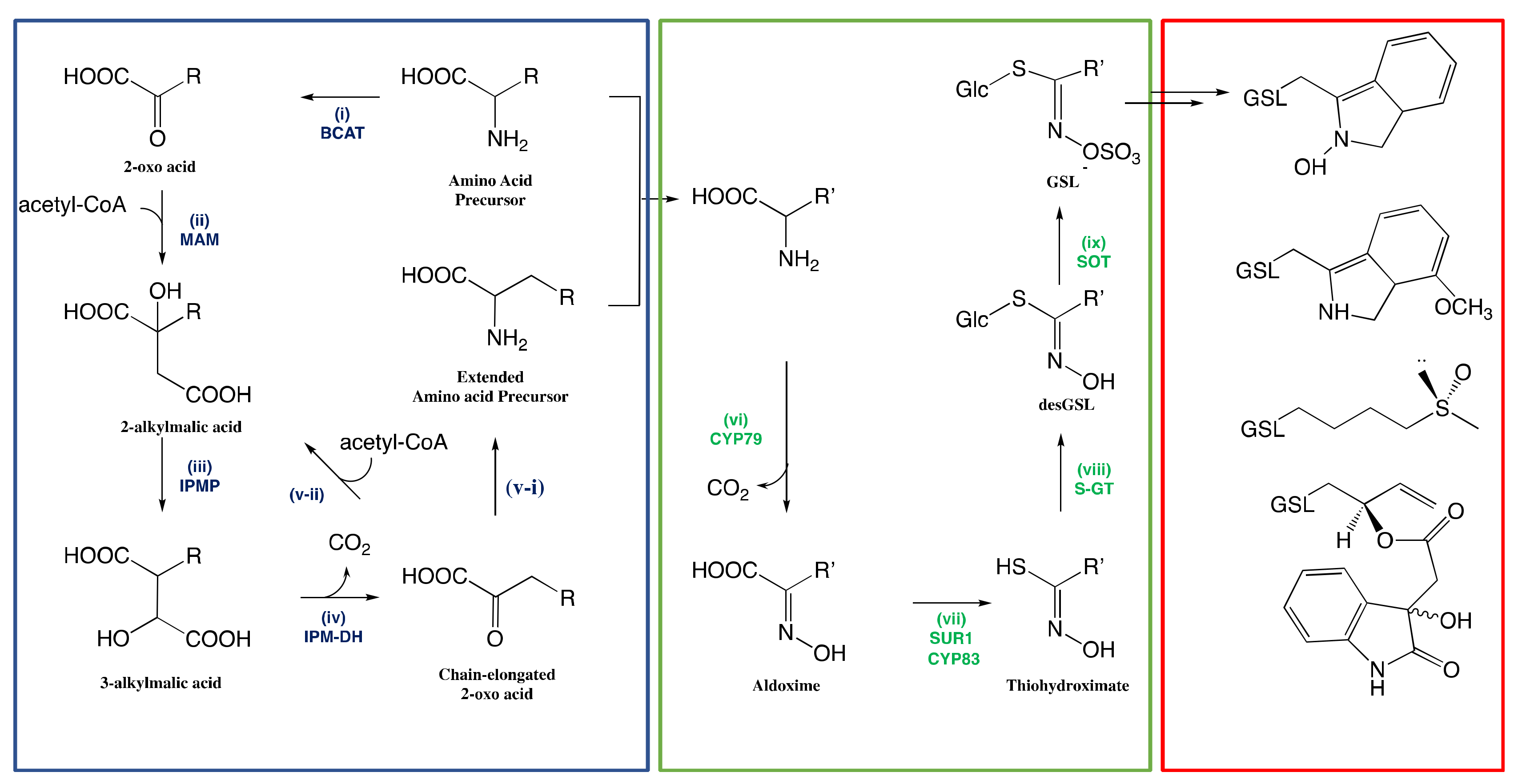
3.1. Biosynthesis of Glucosinolates in Plants
3.1.1. Side-Chain Elongation of Amino Acid
3.1.2. Reconfiguration of Amino Acid to Glucosinolate Core
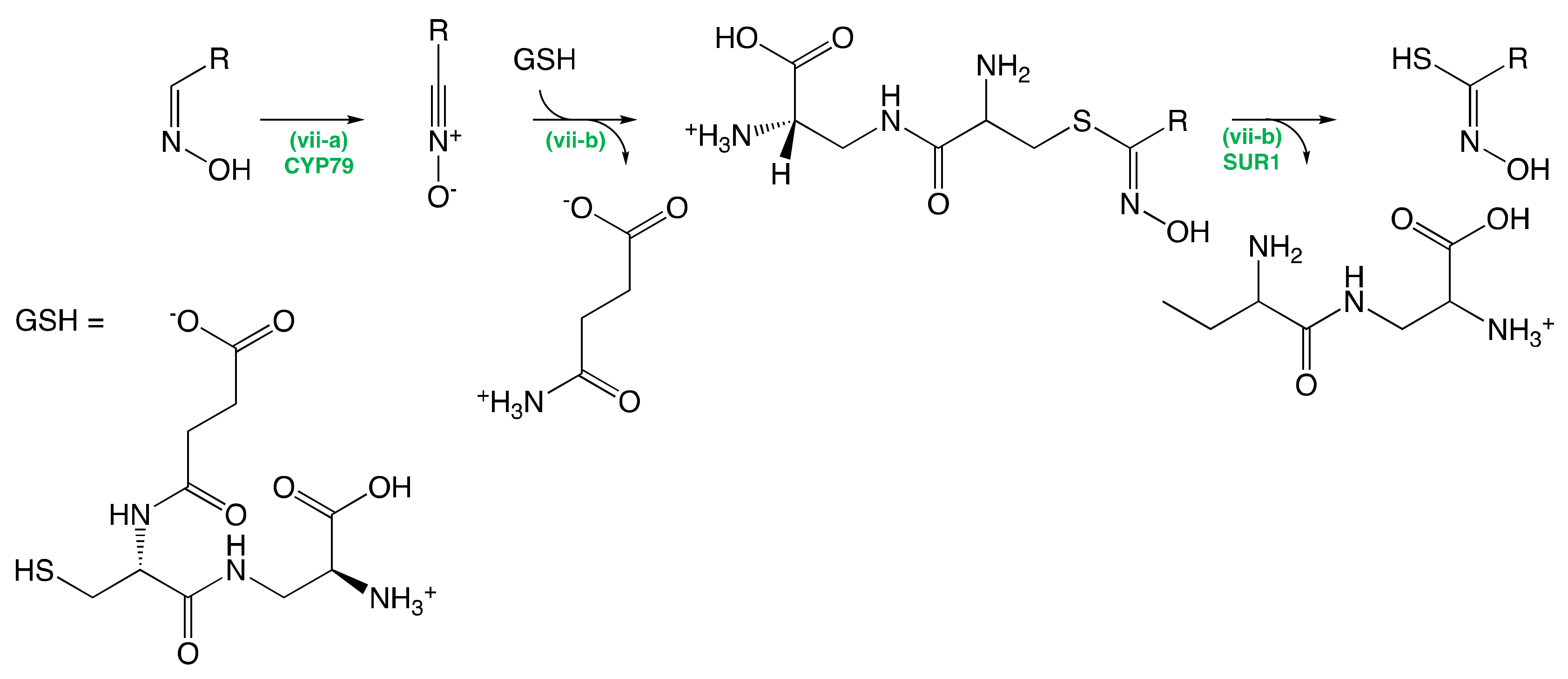
The Conversion of Amino Acid to Aldoximes
The Conversion of Aldoximes to Thiohydroximic Acids
The Formation of Glucosinolate Core
3.1.3. Natural Side-Chain Modification of Glucosinolates
3.1.4. Regulation of Glucosinolate Biosynthesis
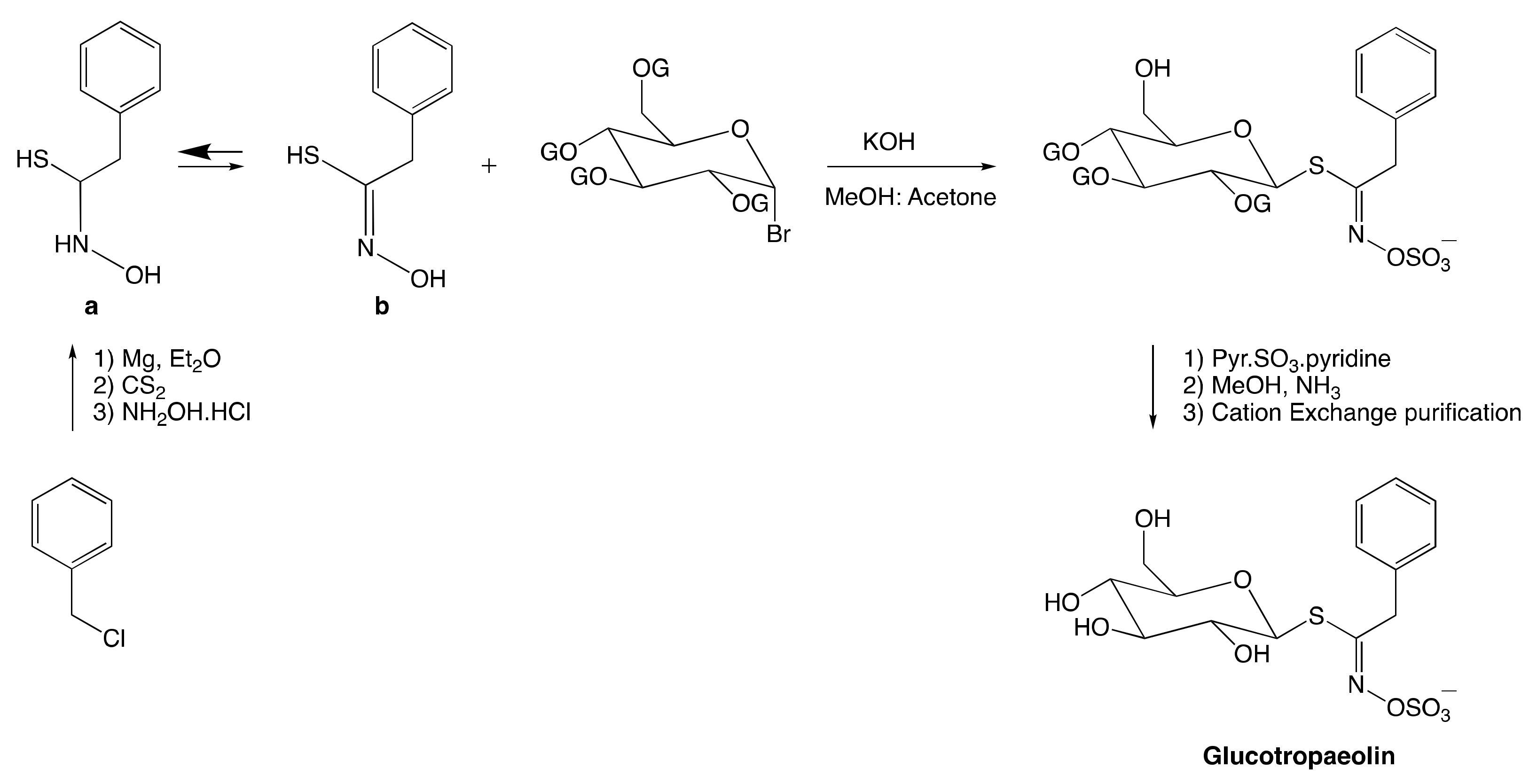
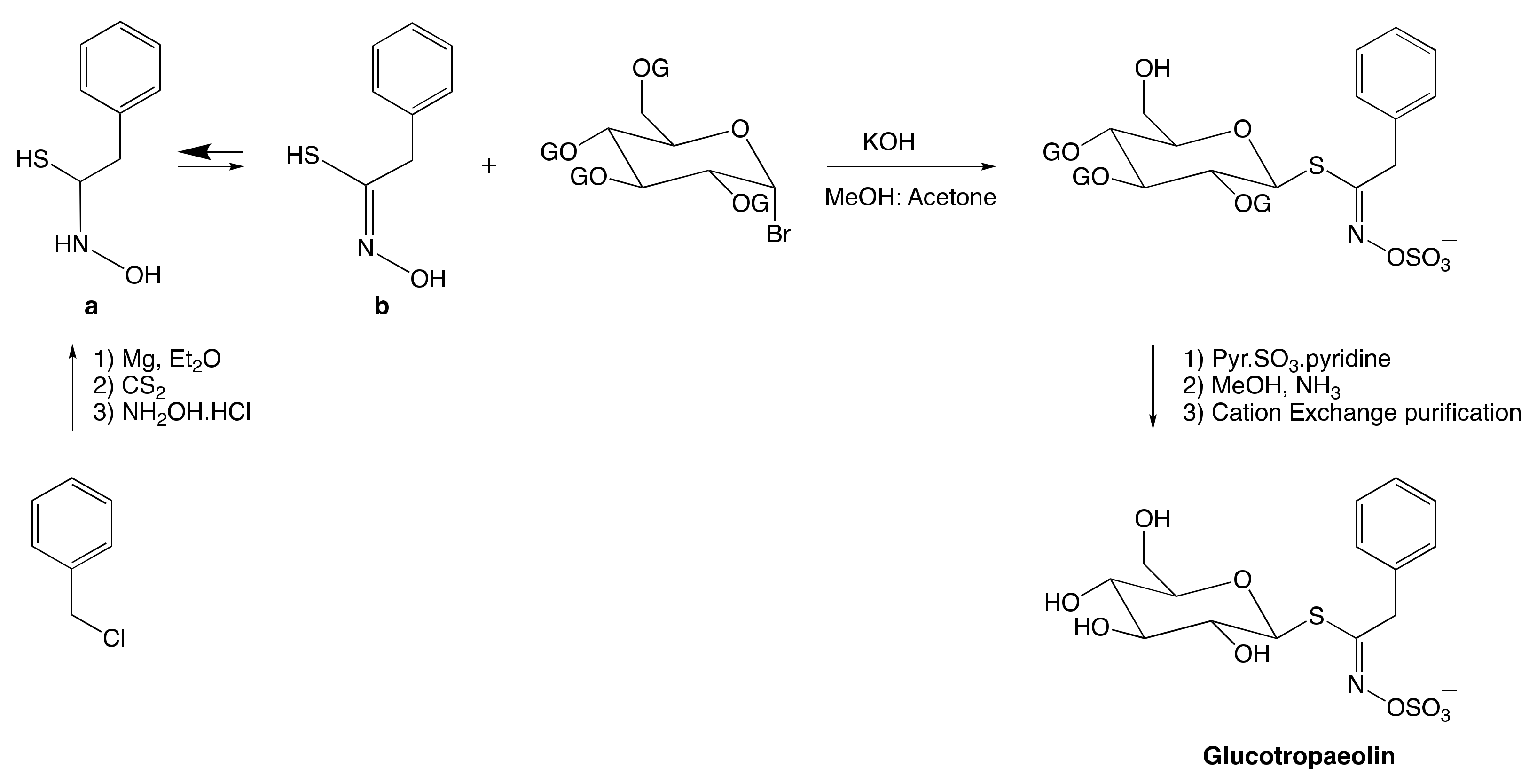
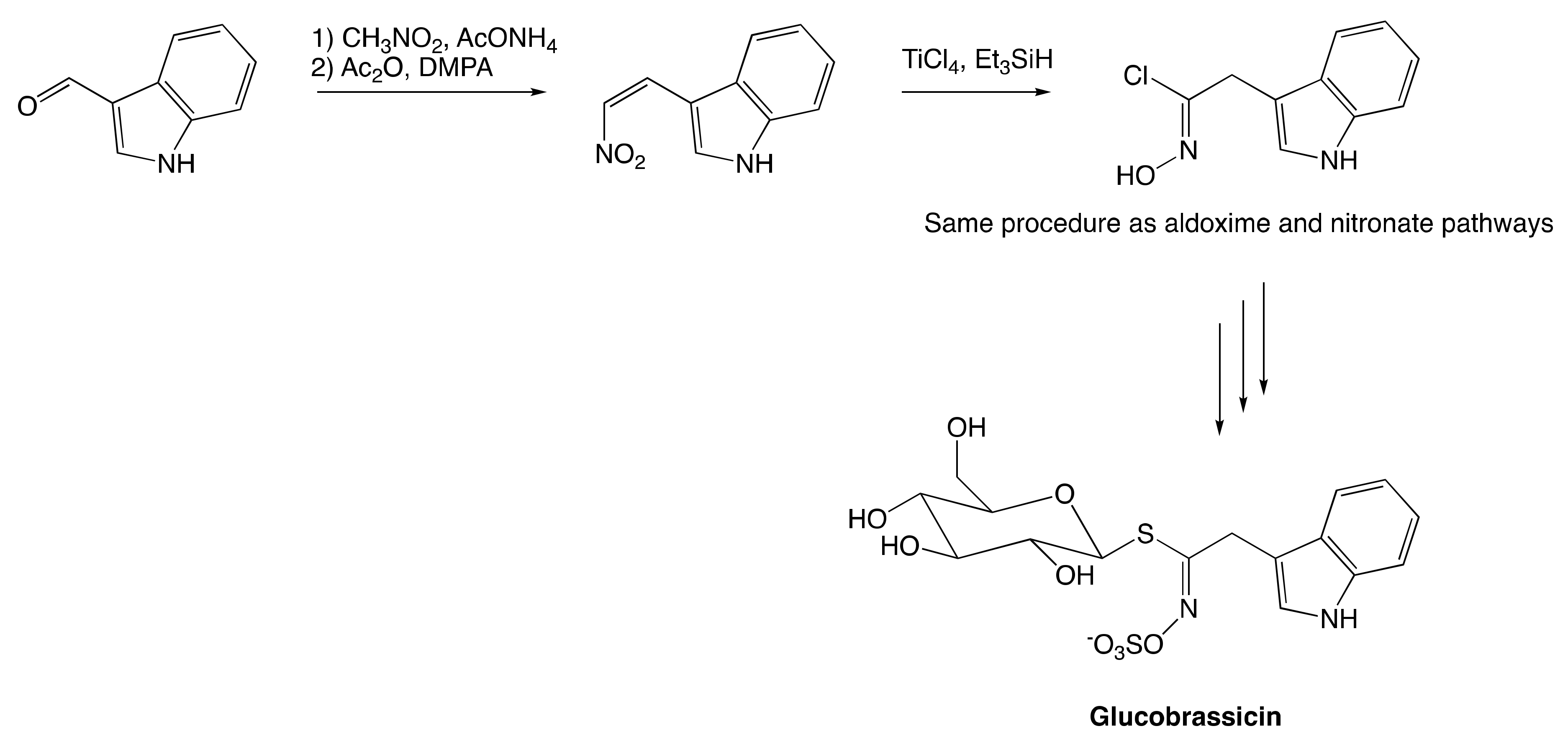
3.2. Chemical Synthesis of Glucosinolates
3.2.1. Anomeric Disconnection
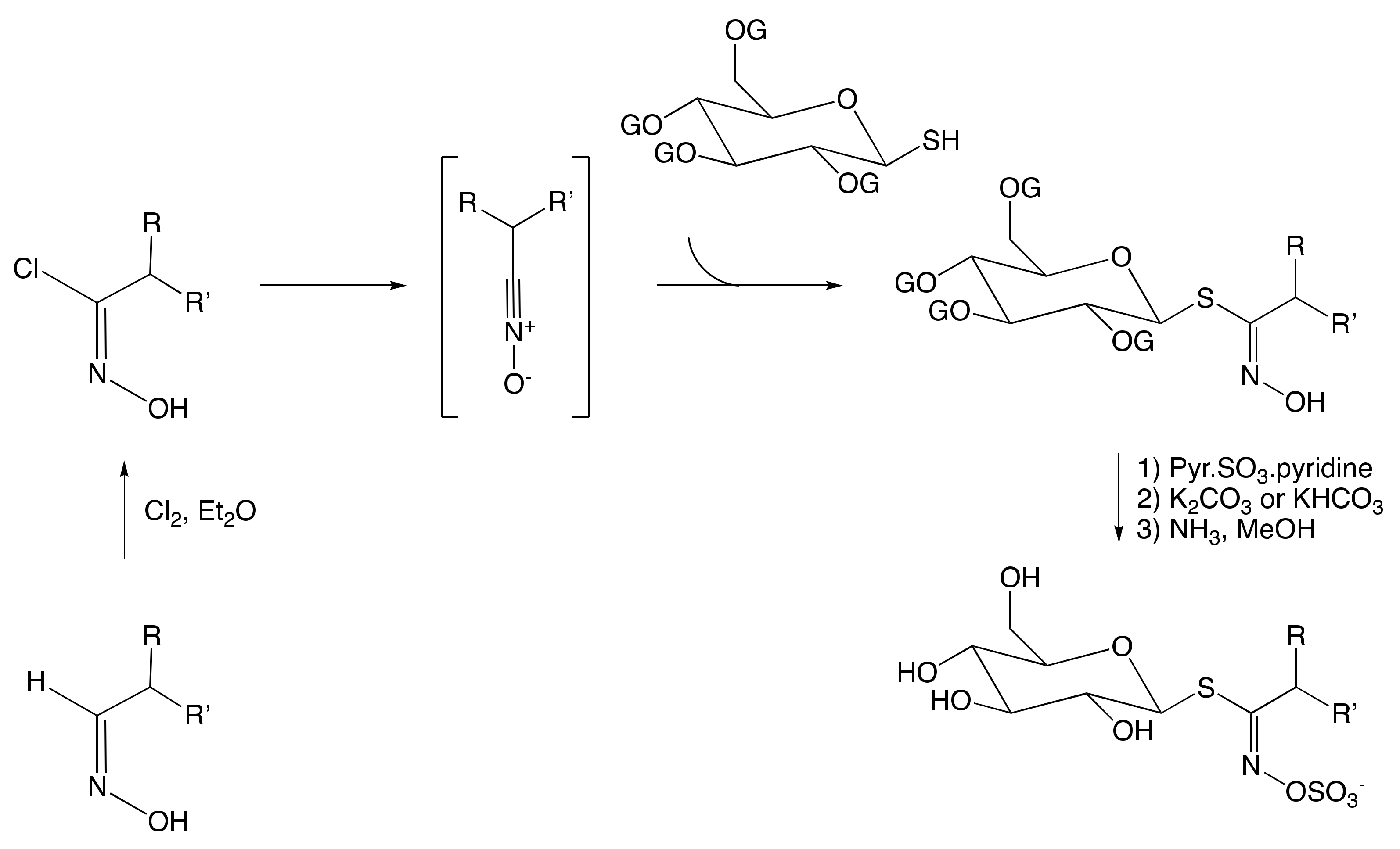
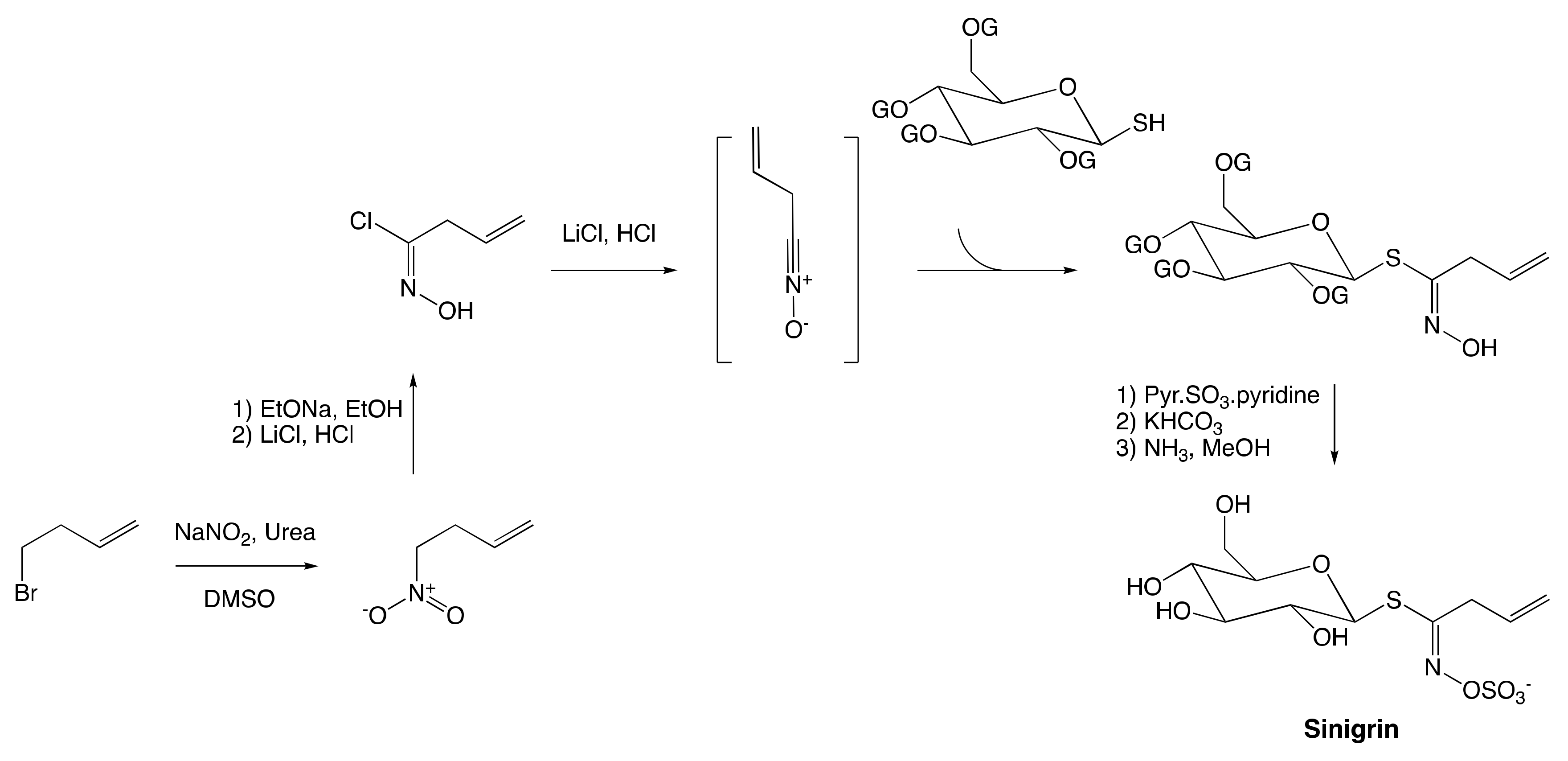
3.2.2. Hydroximate Disconnection
The Aldoxime Pathway
The Nitronate and Nitrovinyl Pathway
4. Extraction, Purification, and Characterization of Glucosinolates
4.1. Extraction of Glucosinolates
4.2. Purification and Separation of Glucosinolates
4.3. Characterization of Glucosinolates
5. Structure and Classification of Glucosinolates
6. Stability of Glucosinolates
6.1. Effects of Processing Methods on Glucosinolate Profile
6.2. Degradation of Glucosinolates in Solution
7. Biological Activities of Glucosinolates
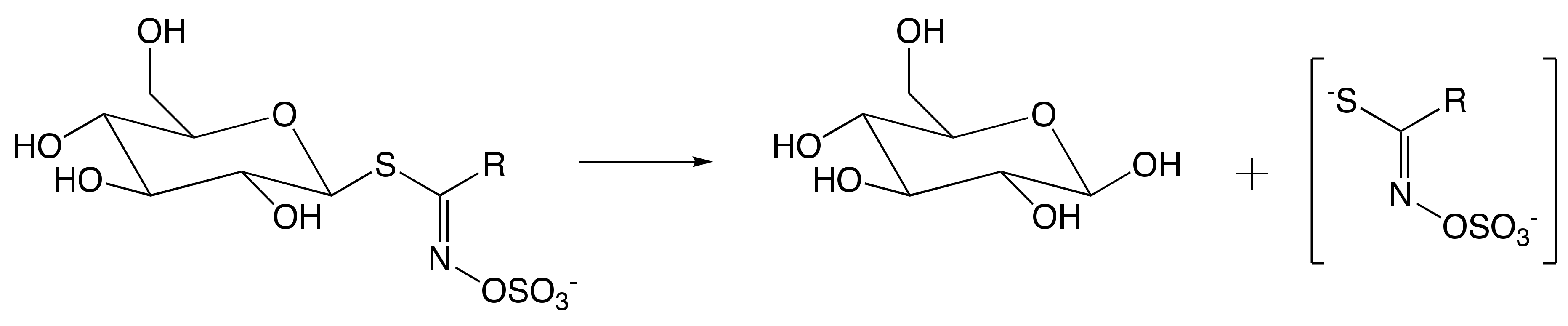
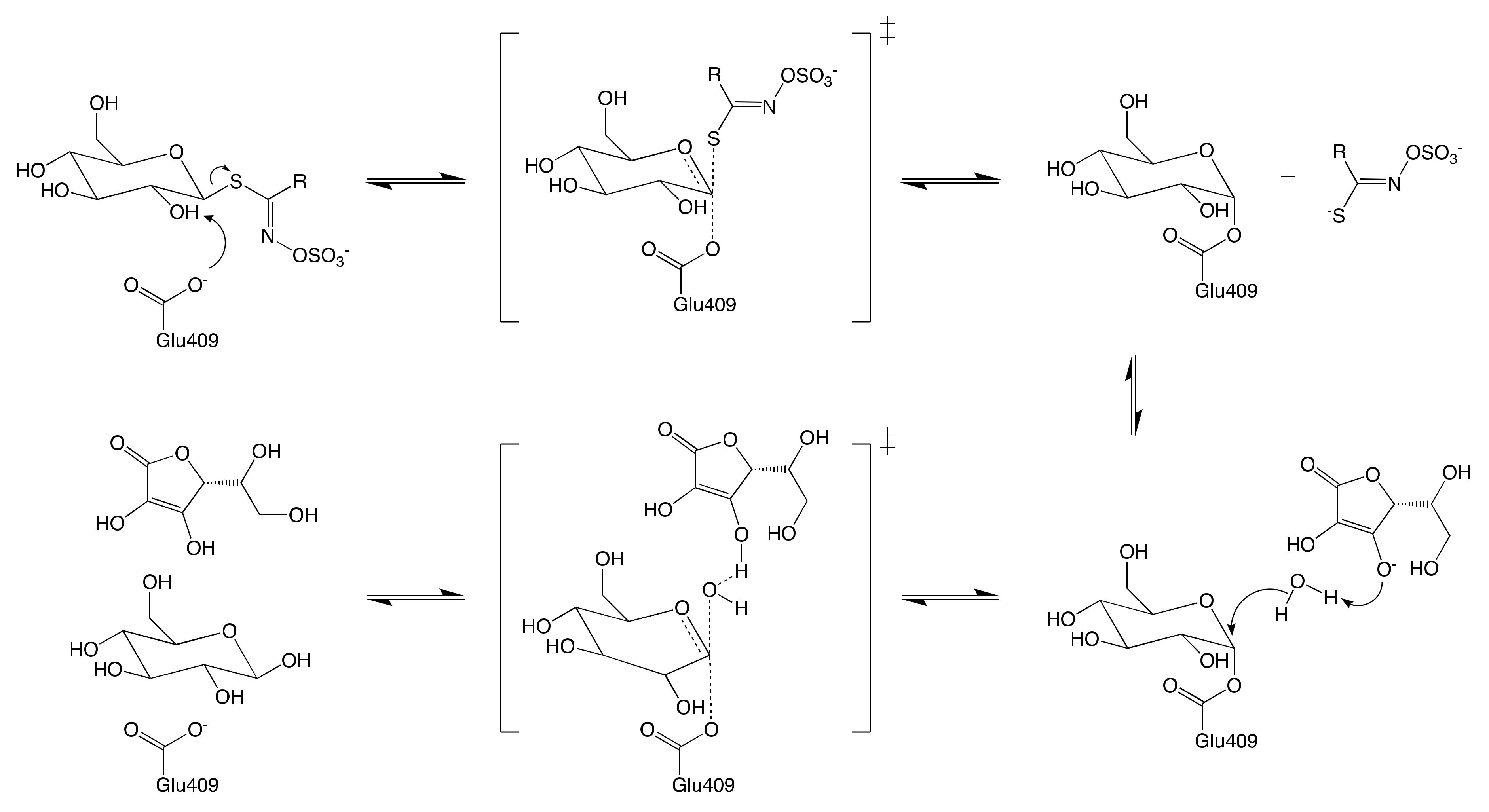
7.1. Mechanism of Myrosinase
7.1.1. Hypothetical Recognition Role of Sulfate Group
7.1.2. Reconfiguration of Unstable Aglucone
7.2. Biological Activities of Glucosinolates and Their Catabolites
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wittstock, U.; Halkier, B.A. Cytochrome P450 CYP79A2 from Arabidopsis thaliana L. catalyzes the conversion of L-phenylalanine to phenylacetaldoxime in the biosynthesis of benzylglucosinolate. J. Biol. Chem. 2000, 275, 14659–14666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgen, B.H.; Thangstad, O.P.; Ahuja, I.; Rossiter, J.T.; Bones, A.M. Removing the mustard oil bomb from seeds: Transgenic ablation of myrosin cells in oilseed rape (Brassica napus) produces MINELESS seeds. J. Exp. Bot. 2010, 61, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ballesta, M.; Moreno-Fernández, D.A.; Castejón, D.; Ochando, C.; Morandini, P.A.; Carvajal, M. The impact of the absence of aliphatic glucosinolates on water transport under salt stress in Arabidopsis thaliana. Front. Plant Sci. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaževic, I.; Montaut, S.; Burcul, F.; Olsen, C.E.; Burow, M.; Rollin, P.; Agerbirk, N. Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. Phytochemistry 2020, 169, 112100. [Google Scholar] [CrossRef]
- Borek, V.; Morra, M.J.; Brown, P.D.; McCaffrey, J.P. Allelochemicals Produced during Sinigrin Decomposition in Soil. J. Agric. Food Chem. 1994, 42, 1030–1034. [Google Scholar] [CrossRef]
- Bednarek, P.; Pislewska-Bednarek, M.; Svatos, A.; Schneider, B.; Doubsky, J.; Mansurova, M.; Humphry, M.; Consonni, C.; Panstruga, R.; Sanchez-Vallet, A.; et al. A Glucosinolate Metabolism Pathway in Living Plant Cells Mediates Broad-Spectrum Antifungal Defense. Science 2009, 323, 101–106. [Google Scholar] [CrossRef]
- Cartea, M.E.; Velasco, P. Glucosinolates in Brassica foods: Bioavailability in food and significance for human health. Phytochem. Rev. 2008, 7, 213–229. [Google Scholar] [CrossRef]
- Mazumder, A.; Dwivedi, A.; Plessis, J.D. Sinigrin and its therapeutic benefits. Molecules 2016, 21, 416. [Google Scholar] [CrossRef] [Green Version]
- Benn, M.H.; Ettlinger, M.G. The synthesis of sinigrin. Chem. Commun. (Lond.) 1965, 445–447. [Google Scholar] [CrossRef]
- Cassel, S.; Casenave, B.; Déléris, G.; Latxague, L.; Rollin, P. Exploring an alternative approach to the synthesis of arylalkyl and indolylmethyl glucosinolates. Tetrahedron 1998, 54, 8515–8524. [Google Scholar] [CrossRef]
- Rollin, P.; Tatibouët, A. Glucosinolates: The synthetic approach. Comptes Rendus Chim. 2011, 14, 194–210. [Google Scholar] [CrossRef]
- Keller, T.H.; Yelland, L.J.; Benn, M.H. A new glucosinolate synthesis. Can. J. Chem. 1984, 62, 437–440. [Google Scholar] [CrossRef]
- Kushad, M.M.; Brown, A.F.; Kurilich, A.C.; Juvik, J.A.; Klein, B.P.; Wallig, M.A.; Jeffery, E.H. Variation of glucosinolates in vegetable crops of Brassica oleracea. J. Agric. Food Chem. 1999, 47, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Barba, F.J.; Nikmaram, N.; Roohinejad, S.; Khelfa, A.; Zhu, Z.; Koubaa, M. Bioavailability of Glucosinolates and Their Breakdown Products: Impact of Processing. Front. Nutr. 2016, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanschen, F.S. Domestic boiling and salad preparation habits affect glucosinolate degradation in red cabbage (Brassica oleracea var. capitata f. rubra). Food Chem. 2020, 321, 126694. [Google Scholar] [CrossRef] [PubMed]
- Crocoll, C.; Halkier, B.A.; Burow, M. Analysis and Quantification of Glucosinolates. Curr. Protoc. Plant Biol. 2016, 1, 385–409. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E. Glucosinolate structures in evolution. Phytochemistry 2012, 77, 16–45. [Google Scholar] [CrossRef]
- Tripathi, M.; Mishra, A. Glucosinolates in animal nutrition: A review. Anim. Feed Sci. Technol. 2007, 132, 1–27. [Google Scholar] [CrossRef]
- Munday, R.; Munday, C.M. Induction of Phase II Detoxification Enzymes in Rats by Plant-Derived Isothiocyanates: Comparison of Allyl Isothiocyanate with Sulforaphane and Related Compounds. J. Agric. Food Chem. 2004, 52, 1867–1871. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, H.; Yi, X.; Zhang, Y.; Yang, Q.; Zhang, C.; Fan, C.; Zhou, Y. Dissection of genetic architecture for glucosinolate accumulations in leaves and seeds of Brassica napus by genome-wide association study. Plant Biotechnol. J. 2020, 18, 1472–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, P.; Soengas, P.; Vilar, M.; Cartea, M.E.; del Rio, M. Comparison of Glucosinolate Profiles in Leaf and Seed Tissues of Different Brassica napus Crops. J. Am. Soc. Hortic. Sci. 2008, 133, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Troufflard, S.; Mullen, W.; Larson, T.R.; Graham, I.A.; Crozier, A.; Amtmann, A.; Armengaud, P. Potassium deficiency induces the biosynthesis of oxylipins and glucosinolates in Arabidopsis thaliana. BMC Plant Biol. 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Ballesta, M.d.C.; Moreno, D.A.; Carvajal, M. The physiological importance of glucosinolates on plant response to abiotic stress in Brassica. Int. J. Mol. Sci. 2013, 14, 11607–11625. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Lema, A.; Fernández-Martínez, J.M.; Pérez-Vich, B.; Velasco, L. Transgressive segregation for reduced glucosinolate content in Brassica carinata A. Braun. Plant Breed. 2006, 125, 400–402. [Google Scholar] [CrossRef]
- Márquez-Lema, A.; Fernández-Martínez, J.M.; Pérez-Vich, B.; Velasco, L. Inheritance of very high glucosinolate content in Ethiopian mustard seeds. Plant Breed. 2009, 128, 278–281. [Google Scholar] [CrossRef]
- Chodur, G.M.; Olson, M.E.; Wade, K.L.; Stephenson, K.K.; Nouman, W.; Garima; Fahey, J.W. Wild and domesticated Moringa oleifera differ in taste, glucosinolate composition, and antioxidant potential, but not myrosinase activity or protein content. Sci. Rep. 2018, 8, 7995. [Google Scholar] [CrossRef]
- Ishida, M.; Nagata, M.; Ohara, T.; Kakizaki, T.; Hatakeyama, K.; Nishio, T. Small variation of glucosinolate composition in Japanese cultivars of radish (Raphanus sativus L.) requires simple quantitative analysis for breeding of glucosinolate component. Breed. Sci. 2012, 62, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Amyot, L.; McDowell, T.; Martin, S.L.; Renaud, J.; Gruber, M.Y.; Hannoufa, A. Assessment of Antinutritional Compounds and Chemotaxonomic Relationships between Camelina sativa and Its Wild Relatives. J. Agric. Food Chem. 2019, 67, 796–806. [Google Scholar] [CrossRef]
- Moshgani, M.; Kolvoort, E.; de Jong, T. Pronounced effects of slug herbivory on seedling recruitment of Brassica cultivars and accessions, especially those with low levels of aliphatic glucosinolates. Basic Appl. Ecol. 2014, 15, 607–615. [Google Scholar] [CrossRef]
- Baaij, B.M.; Kim, H.K.; Grosser, K.; Worrich, A.; de Jong, T.J. Slug herbivory on hybrids of the crop Brassica napus and its wild relative B. rapa. Basic Appl. Ecol. 2018, 31, 52–60. [Google Scholar] [CrossRef]
- Gupta, S.; Sangha, M.; Kaur, G.; Atwal, A.; Banga, S.; Banga, S. Variability for Leaf and Seed Glucosinolate Contents and Profiles in a Germplasm Collection of the Brassica juncea. Biochem. Anal. Biochem. 2012, 1, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yue, Z.; Zhong, X.; Lei, J.; Tao, P.; Li, B. Distribution of primary and secondary metabolites among the leaf layers of headed cabbage (Brassica oleracea var. capitata). Food Chem. 2020, 312, 126028. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, C.; Zheng, H.; Zheng, L. Melatonin treatment affects the glucoraphanin-sulforaphane system in postharvest fresh-cut broccoli (Brassica oleracea L.). Food Chem. 2020, 307, 125562. [Google Scholar] [CrossRef]
- Döring, A.; Ulber, B. Performance of cabbage stem flea beetle larvae (Psylliodes chrysocephala) in brassicaceous plants and the effect of glucosinolate profiles. Entomol. Exp. Appl. 2020, 168, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Badenes-Perez, F.R.; Reichelt, M.; Gershenzon, J.; Heckel, D.G. Interaction of glucosinolate content of Arabidopsis thaliana mutant lines and feeding and oviposition by generalist and specialist lepidopterans. Phytochemistry 2013, 86, 36–43. [Google Scholar] [CrossRef]
- Yi, G.; Lim, S.; Chae, W.B.; Park, J.E.; Park, H.R.; Lee, E.J.; Huh, J.H. Root Glucosinolate Profiles for Screening of Radish (Raphanus sativus L.) Genetic Resources. J. Agric. Food Chem. 2016, 64, 61–70. [Google Scholar] [CrossRef]
- Tetteh, O.N.A.; Ulrichs, C.; Huyskens-Keil, S.; Mewis, I.; Amaglo, N.K.; Oduro, I.N.; Adarkwah, C.; Obeng-Ofori, D.; Förster, N. Effects of harvest techniques and drying methods on the stability of glucosinolates in Moringa oleifera leaves during post-harvest. Sci. Hortic. 2019, 246, 998–1004. [Google Scholar] [CrossRef]
- Förster, N.; Ulrichs, C.; Schreiner, M.; Müller, C.T.; Mewis, I. Development of a reliable extraction and quantification method for glucosinolates in Moringa oleifera. Food Chem. 2015, 166, 456–464. [Google Scholar] [CrossRef]
- Chen, R.; Wang, X.J.; Zhang, Y.Y.; Xing, Y.; Yang, L.; Ni, H.; Li, H.H. Simultaneous extraction and separation of oil, proteins, and glucosinolates from Moringa oleifera seeds. Food Chem. 2019, 300, 125162. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, Y.; Dräger, B. Glucosinolates in Moringa stenopetala. Planta Med. 2003, 69, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.N.; Mellon, F.A.; Foidl, N.; Pratt, J.H.; Dupont, M.S.; Perkins, L.; Kroon, P.A. Profiling glucosinolates and phenolics in vegetative and reproductive tissues of the multi-purpose trees Moringa oleifera L. (Horseradish tree) and Moringa stenopetala L. J. Agric. Food Chem. 2003, 51, 3546–3553. [Google Scholar] [CrossRef] [PubMed]
- Halkier, B.A.; Du, L. The biosynthesis of glucosinolates. Trends Plant Sci. 1997, 2, 425–431. [Google Scholar] [CrossRef]
- Halkier, B.A.; Gershenzon, J. Biology and Biochemistry of Glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [Green Version]
- Graser, G.; Oldham, N.J.; Brown, P.D.; Temp, U.; Gershenzon, J. The biosynthesis of benzoic acid glucosinolate esters in Arabidopsis thaliana. Phytochemistry 2001, 57, 23–32. [Google Scholar] [CrossRef]
- Adam Sakine, M.N.; Yaya, M.; Dijoux-Franca, M.-G.; Moudachirou, M. In vitro anti-hyperglycaemic effect of glucocapparin isolated from the seeds of Boscia senegalensis (Pers.) Lam. ex Poiret parasuis. Afr. J. Biotechnol. 2012, 11, 6345–6349. [Google Scholar] [CrossRef]
- Bojesen, G.; Larsen, E. Characterization of five glucosinolates by fast atom bombardment mass spectrometry and collision activation of [M-H]-1. Biol. Mass Spectrom. 1991, 20, 286–288. [Google Scholar] [CrossRef]
- Montaut, S.; Bleeker, R.S.; Jacques, C. Phytochemical constituents of Cardamine diphylla. Can. J. Chem. 2010, 88, 50–55. [Google Scholar] [CrossRef]
- Olsen, C.E.; Huang, X.C.; Hansen, C.I.; Cipollini, D.; Ørgaard, M.; Matthes, A.; Geu-Flores, F.; Koch, M.A.; Agerbirk, N. Glucosinolate diversity within a phylogenetic framework of the tribe Cardamineae (Brassicaceae) unraveled with HPLC-MS/MS and NMR-based analytical distinction of 70 desulfoglucosinolates. Phytochemistry 2016, 132, 33–56. [Google Scholar] [CrossRef]
- Ibrahim, N.; Allart-Simon, I.; De Nicola, G.R.; Iori, R.; Renault, J.H.; Rollin, P.; Nuzillard, J.M. Advanced NMR-Based Structural Investigation of Glucosinolates and Desulfoglucosinolates. J. Nat. Prod. 2018, 81, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Kjær, A.; Christensen, B.W.; Larsen, P.O. isoThiocyanates. XXXIX. Glucobenzosisymbrin, a New Glucoside Present in Seeds of Sisymbrium austriacum Jacq. Acta Chem. Scand. 1961, 15, 1477–1484. [Google Scholar] [CrossRef] [Green Version]
- Radulović, N.; Dekić, M.; Stojanović-Radić, Z. A new antimicrobial glucosinolate autolysis product, 4- isothiocyanatobutanoic acid, from the diffuse wallflower (Erysimum diffusum): Methyl 4-isothiocyanatobutanoate, a long unrecognized artifact of the isolation procedure? Food Chem. 2011, 129, 125–130. [Google Scholar] [CrossRef]
- Chisholm, M.D. Biosynthesis of 3-methoxycarbonylpropylglucosinolate in an Erysimum species. Phytochemistry 1973, 12, 605–608. [Google Scholar] [CrossRef]
- Kjær, A.; Larsen, I.; Tjus, E.; Burris, R.H. isoThiocyanates. IX. The Occurrence of Ethyl isoThiocyanate in Nature. Acta Chem. Scand. 1954. [Google Scholar] [CrossRef]
- Kjær, A.; Schuster, A. Glucosinolates in Capparis flexuosa of jamaican origin. Phytochemistry 1971, 10, 3155–3160. [Google Scholar] [CrossRef]
- Kojima, M.; Uchida, M.; Akahori, Y. Studies on the Volatile Components of Wasabia japonica, Brassica juncea and Cocholearia armoracia by Gas Chromatography-Mass Spectrometry. I. Determination of Low Mass Volatile Components. Yakugaku Zasshi 1973, 93, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Grob, K.; Matile, P. Capillary GC of glucosinolate-derived horseradish constituents. Phytochemistry 1980, 19, 1789–1793. [Google Scholar] [CrossRef]
- Kjær, A.; Thomsen, H.; Hansen, S.E.; Levitin, N.E.; Westin, G. isoThiocyanates XXXVIII. Glucocapangulin, a Novel isoThiocyanate-Producing Glucoside. Acta Chem. Scand. 1960, 14, 1226–1227. [Google Scholar] [CrossRef]
- Kjær, A.; Thomsen, H.; Edman, P.; Kvande, P.C.; Meisingseth, E. Gluconorcappasalin, a Thioglucoside Producing 5-Oxoheptyl Isothiocyanate on Enzymic Hydrolysis. Acta Chem. Scand. 1963, 17, 561–562. [Google Scholar] [CrossRef]
- Kjær, A.; Thomsen, H.; Lipschitz, L.; Nielsen, J.T. Isothiocyanates. XLVI. Glucocappasalin, a New Naturally Occurring Thioglucoside. Acta Chem. Scand. 1962, 16, 2065–2066. [Google Scholar] [CrossRef]
- Gaind, K.; Gandhi, K.; Junega, T.; Kjær, A.; Juhl Nielsen, B. 4,5,6,7-tetrahydroxydecyl isothiocyanate derived from a glucosinolate in Capparis grandis. Phytochemistry 1975, 14, 1415–1418. [Google Scholar] [CrossRef]
- El-Migirab, S.; Berger, Y.; Jadot, J. Isothiocyanates, thiourees et thiocarbamates isoles de Pentadiplandra brazzeana. Phytochemistry 1977, 16, 1719–1721. [Google Scholar] [CrossRef]
- Sirinut, P.; Petchkongkeaw, A.; Romsaiyud, J.; Prateeptongkum, S.; Thongyoo, P. Phytochemical constituents from the root of luvunga scandens and biological activity evaluation. Nat. Prod. Commun. 2017, 12, 1483–1484. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.N.; Mellon, F.A.; Kroon, P.A. Screening Crucifer Seeds as Sources of Specific Intact Glucosinolates Using Ion-Pair High-Performance Liquid Chromatography Negative Ion Electrospray Mass Spectrometry. J. Agric. Food Chem. 2004, 52, 428–438. [Google Scholar] [CrossRef]
- Kjær, A.; Wagnières, W. 3-Methyl-3-butenylglucosinolate, a New Isothiocyanate-Producing Thioglucoside. Acta Chem. Scand. 1965, 19, 1989–1991. [Google Scholar] [CrossRef] [Green Version]
- Blažević, I.; Mastelić, J. Glucosinolate degradation products and other bound and free volatiles in the leaves and roots of radish (Raphanus sativus L.). Food Chem. 2009, 113, 96–102. [Google Scholar] [CrossRef]
- Dauvergne, X.; Cérantola, S.; Salaün, S.; Magné, C.; Kervarec, N.; Bessières, M.A.; Deslandes, E. General occurrence of the glucosinolate glucocochlearin within the Cochlearia genus. Carbohydr. Res. 2006, 341, 2166–2169. [Google Scholar] [CrossRef]
- Kjær, A.; Christensen, B.W.; Krohn, C.; Motzfeldt, K.; Theander, O.; Flood, H. Isothiocyanates. XLI. Glucobenzsisaustricin, a New Glucoside Present in Seeds of Sisymbrium austriacum Jacq. Acta Chem. Scand. 1962, 16, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Agerbirk, N.; Olsen, C.E.; Chew, F.S.; Ørgaard, M. Variable glucosinolate profiles of cardamine pratensis (Brassicaceae) with equal chromosome numbers. J. Agric. Food Chem. 2010, 58, 4693–4700. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Zheng, Q.A.; Gadagi, R.S. The first naturally occurring aromatic isothiocyanates, rapalexins A and B, are cruciferous phytoalexins. Chem. Commun. 2007, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Pfalz, M.; Mukhaimar, M.; Perreau, F.; Kirk, J.; Hansen, C.I.C.; Olsen, C.E.; Agerbirk, N.; Kroymann, J. Methyl transfer in glucosinolate biosynthesis mediated by indole glucosinolate O-methyltransferase 5. Plant Physiol. 2016, 172, 2190–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millán, S.; Sampedro, M.C.; Gallejones, P.; Castellón, A.; Ibargoitia, M.L.; Goicolea, M.A.; Barrio, R.J. Identification and quantification of glucosinolates in rapeseed using liquid chromatography-ion trap mass spectrometry. Anal. Bioanal. Chem. 2009, 394, 1661–1669. [Google Scholar] [CrossRef]
- Agerbirk, N.; Petersen, B.L.; Olsen, C.E.; Halkier, B.A.; Nielsen, J.K. 1,4-Dimethoxyglucobrassicin in Barbarea and 4-hydroxyglucobrassicin in Arabidopsis and Brassica. J. Agric. Food Chem. 2001, 49, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Schraudolf, H.; Bäuerle, R. 1N-acetyl-3-indolylmethylglucosinolate in Seedlings of Tovaria pendula Ruiz et Pav. Z. Fur Nat. Sect. C J. Biosci. 1986, 41, 526–528. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.C.; Stowe, B.B. A novel sulphonated natural indole. Phytochemistry 1970, 9, 1629–1632. [Google Scholar] [CrossRef]
- Bianco, G.; Agerbirk, N.; Losito, I.; Cataldi, T.R. Acylated glucosinolates with diverse acyl groups investigated by high resolution mass spectrometry and infrared multiphoton dissociation. Phytochemistry 2014, 100, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Agerbirk, N.; Olsen, C.E. Isoferuloyl derivatives of five seed glucosinolates in the crucifer genus Barbarea. Phytochemistry 2011, 72, 610–623. [Google Scholar] [CrossRef]
- Kusznierewicz, B.; Iori, R.; Piekarska, A.; Namieśnik, J.; Bartoszek, A. Convenient identification of desulfoglucosinolates on the basis of mass spectra obtained during liquid chromatography-diode array-electrospray ionisation mass spectrometry analysis: Method verification for sprouts of different Brassicaceae species extract. J. Chromatogr. A 2013, 1278, 108–115. [Google Scholar] [CrossRef]
- Maldini, M.; Foddai, M.; Natella, F.; Petretto, G.L.; Rourke, J.P.; Chessa, M.; Pintore, G. Identification and quantification of glucosinolates in different tissues of Raphanus raphanistrum by liquid chromatography tandem-mass spectrometry. J. Food Compos. Anal. 2017, 61, 20–27. [Google Scholar] [CrossRef]
- Pagnotta, E.; Agerbirk, N.; Olsen, C.E.; Ugolini, L.; Cinti, S.; Lazzeri, L. Hydroxyl and Methoxyl Derivatives of Benzylglucosinolate in Lepidium densiflorum with Hydrolysis to Isothiocyanates and non-Isothiocyanate Products: Substitution Governs Product Type and Mass Spectral Fragmentation. J. Agric. Food Chem. 2017, 65, 3167–3178. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, G.R.; Nyegue, M.; Montaut, S.; Iori, R.; Menut, C.; Tatibouët, A.; Rollin, P.; Ndoyé, C.; Zollo, P.H.A. Profile and quantification of glucosinolates in Pentadiplandra brazzeana Baillon. Phytochemistry 2012, 73, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Doheny-Adams, T.; Redeker, K.; Kittipol, V.; Bancroft, I.; Hartley, S.E. Development of an efficient glucosinolate extraction method. Plant Methods 2017, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agerbirk, N.; Warwick, S.I.; Hansen, P.R.; Olsen, C.E. Sinapis phylogeny and evolution of glucosinolates and specific nitrile degrading enzymes. Phytochemistry 2008, 69, 2937–2949. [Google Scholar] [CrossRef]
- Montaut, S.; Zhang, W.D.; Nuzillard, J.M.; De Nicola, G.R.; Rollin, P. Glucosinolate Diversity in Bretschneidera sinensis of Chinese Origin. J. Nat. Prod. 2015, 78, 2001–2006. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E. Glucosinolate hydrolysis products in the crucifer Barbarea vulgaris include a thiazolidine-2-one from a specific phenolic isomer as well as oxazolidine-2-thiones. Phytochemistry 2015, 115, 143–151. [Google Scholar] [CrossRef]
- Agerbirk, N.; Matthes, A.; Erthmann, P.; Ugolini, L.; Cinti, S.; Lazaridi, E.; Nuzillard, J.M.; Müller, C.; Bak, S.; Rollin, P.; et al. Glucosinolate turnover in Brassicales species to an oxazolidin-2-one, formed via the 2-thione and without formation of thioamide. Phytochemistry 2018, 153, 79–93. [Google Scholar] [CrossRef]
- Dekić, M.S.; Radulović, N.S.; Stojanović, N.M.; Randjelović, P.J.; Stojanović-Radić, Z.Z.; Najman, S.; Stojanović, S. Spasmolytic, antimicrobial and cytotoxic activities of 5-phenylpentyl isothiocyanate, a new glucosinolate autolysis product from horseradish (Armoracia rusticana P. Gaertn., B. Mey. & Scherb., Brassicaceae). Food Chem. 2017, 232, 329–339. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E.; Nielsen, J.K. Seasonal variation in leaf glucosinolates and insect resistance in two types of Barbarea vulgaris ssp. arcuata. Phytochemistry 2001, 58, 91–100. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E.; Poulsen, E.; Jacobsen, N.; Hansen, P.R. Complex metabolism of aromatic glucosinolates in Pieris rapae caterpillars involving nitrile formation, hydroxylation, demethylation, sulfation, and host plant dependent carboxylic acid formation. Insect Biochem. Mol. Biol. 2010, 40, 126–137. [Google Scholar] [CrossRef]
- Waterman, C.; Cheng, D.M.; Rojas-Silva, P.; Poulev, A.; Dreifus, J.; Lila, M.A.; Raskin, I. Stable, water extractable isothiocyanates from Moringa oleifera leaves attenuate inflammation in vitro. Phytochemistry 2014, 103, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, O.; Rasmussen, K.W.; Sørensen, H. Glucosinolates in Sesamoides canescens and S. pygmaea: Identification of 2-(α-l-arabinopyranosyloxy)-2-phenylethylglucosinolate. Phytochemistry 1981, 20, 1857–1861. [Google Scholar] [CrossRef]
- Gueyrard, D.; Barillari, J.; Iori, R.; Palmieri, S.; Rollin, P. First synthesis of an O-glycosylated glucosinolate isolated from Moringa oleifera. Tetrahedron Lett. 2000, 41, 8307–8309. [Google Scholar] [CrossRef]
- Maldini, M.; Maksoud, S.A.; Natella, F.; Montoro, P.; Petretto, G.L.; Foddai, M.; De Nicola, G.R.; Chessa, M.; Pintore, G. Moringa oleifera: Study of phenolics and glucosinolates by mass spectrometry. J. Mass Spectrom. 2014, 49, 900–910. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, R.M.; Krosse, S.; Swolfs, A.E.; te Brinke, E.; Prill, N.; Leimu, R.; van Galen, P.M.; Wang, Y.; Aarts, M.G.; van Dam, N.M. Isolation and identification of 4-α-rhamnosyloxy benzyl glucosinolate in Noccaea caerulescens showing intraspecific variation. Phytochemistry 2015, 110, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Chidewe, C.; Castillo, U.F.; Sem, D.S. Structural Analysis and Antimicrobial Activity of Chromatographically Separated Fractions of Leaves of Sesamum angustifolium (Oliv.) Engl. J. Biol. Act. Prod. Nat. 2017, 7, 463–474. [Google Scholar] [CrossRef]
- Montaut, S.; Grandbois, J.; Righetti, L.; Barillari, J.; Iori, R.; Rollin, P. Updated glucosinolate profile of Dithyrea wislizenii. J. Nat. Prod. 2009, 72, 889–893. [Google Scholar] [CrossRef]
- Montaut, S.; Montagut-Romans, A.; Chiari, L.; Benson, H.J. Glucosinolates in Draba borealis DC. (Brassicaceae) in a taxonomic perspective. Biochem. Syst. Ecol. 2018, 78, 31–34. [Google Scholar] [CrossRef]
- Denicola, G.R.; Blažević, I.; Montaut, S.; Rollin, P.; Mastelić, J.; Iori, R.; Tatibouët, A. Glucosinolate distribution in aerial parts of degenia velebitica. Chem. Biodivers. 2011, 8, 2090–2096. [Google Scholar] [CrossRef]
- Daxenbichler, M.E.; Spencer, G.F.; Carlson, D.G.; Rose, G.B.; Brinker, A.M.; Powell, R.G. Glucosinolate composition of seeds from 297 species of wild plants. Phytochemistry 1991, 30, 2623–2638. [Google Scholar] [CrossRef]
- Reichelt, M.; Brown, P.D.; Schneider, B.; Oldham, N.J.; Stauber, E.; Tokuhisa, J.; Kliebenstein, D.J.; Mitchell-Olds, T.; Gershenzon, J. Benzoic acid glucosinolate esters and other glucosinolates from Arabidopsis thaliana. Phytochemistry 2002, 59, 663–671. [Google Scholar] [CrossRef]
- Jaki, B.; Sticher, O.; Veit, M.; Fröhlich, R.; Pauli, G.F. Evaluation of glucoiberin reference material from Iberis amara by spectroscopic fingerprinting. J. Nat. Prod. 2002, 65, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, S.; Caridi, D.; Stinton, M.; Trenerry, V.C.; Jones, R. The isolation and purification of glucoraphanin from broccoli seeds by solid phase extraction and preparative high performance liquid chromatography. J. Chromatogr. A 2006, 1120, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, N.S.; Gerendás, J.; Krumbein, A. Identification of desulphoglucosinolates in Brassicaceae by LC/MS/MS: Comparison of ESI and atmospheric pressure chemical ionisation-MS. Mol. Nutr. Food Res. 2007, 51, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Agerbirk, N.; Olsen, C.E.; Cipollini, D.; Ørgaard, M.; Linde-Laursen, I.; Chew, F.S. Specific glucosinolate analysis reveals variable levels of epimeric glucobarbarins, dietary precursors of 5-phenyloxazolidine-2-thiones, in watercress types with contrasting chromosome numbers. J. Agric. Food Chem. 2014, 62, 9586–9596. [Google Scholar] [CrossRef]
- Yamane, A.; Fujikura, J.; Ogawa, H.; Mizutani, J. Isothiocyanates as alleopathic compounds from Rorippa indica Hiern. (Cruciferae) roots. J. Chem. Ecol. 1992, 18, 1941–1954. [Google Scholar] [CrossRef]
- Berhow, M.A.; Polat, U.; Glinski, J.A.; Glensk, M.; Vaughn, S.F.; Isbell, T.; Ayala-Diaz, I.; Marek, L.; Gardner, C. Optimized analysis and quantification of glucosinolates from Camelina sativa seeds by reverse-phase liquid chromatography. Ind. Crop Prod. 2013, 43, 119–125. [Google Scholar] [CrossRef]
- Huang, X.; Renwick, J.A.; Sachdev-Gupta, K. A chemical basis for differential acceptance of Erysimum cheiranthoides by two Pieris species. J. Chem. Ecol. 1993, 19, 195–210. [Google Scholar] [CrossRef]
- Fabre, N.; Bon, M.; Moulis, C.; Fouraste, I.; Stanislas, E. Three glucosinolates from seeds of Brassica juncea. Phytochemistry 1997, 45, 525–527. [Google Scholar] [CrossRef]
- Iori, R.; Barillari, J.; Gallienne, E.; Bilardo, C.; Tatibouët, A.; Rollin, P. Thio-functionalised glucosinolates: Unexpected transformation of desulfoglucoraphenin. Tetrahedron Lett. 2008, 49, 292–295. [Google Scholar] [CrossRef]
- Kjær, A.; Schuster, A.; Miksche, G.E.; Liaaen-Jensen, S.; Lamvik, A.; Sunde, E.; Sørensen, N.A. Glucosinolates in Erysimum hieracifolium L.; Three New, Naturally Occurring Glucosinolates. Acta Chem. Scand. 1970, 24, 1631–1638. [Google Scholar] [CrossRef]
- Kjaer, A.; Schuster, A. ω-Methylthioalkylglucosinolates and some oxidized congeners in seeds of Erysimum rhaeticum. Phytochemistry 1973, 12, 929–933. [Google Scholar] [CrossRef]
- Kjær, A.; Schuster, A.; Vestersjø, E.; Andresen, A.F.; Pearson, W.B.; Meisalo, V. Glucosinolates in Seeds of Arabis hirsuta (L.) Scop.: Some New, Naturally Derived Isothiocyanates. Acta Chem. Scand. 1972, 26, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.N.; Mellon, F.A.; Botting, N.P.; Eagles, J.; Rosa, E.A.; Williamson, G. Identification of the major glucosinolate (4-mercaptobutyl glucosinolate) in leaves of Eruca sativa L. (salad rocket). Phytochemistry 2002, 61, 25–30. [Google Scholar] [CrossRef]
- Cataldi, T.R.I.; Rubino, A.; Lelario, F.; Bufo, S.A. Naturally occurring glucosinolates in plant extracts of rocket salad (Eruca sativa L.) identified by liquid chromatography coupled with negative ion electrospray ionization and quadrupole ion-trap mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2374–2388. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jander, G. Myzus persicae (green peach aphid) feeding on Arabidopsis induces the formation of a deterrent indole glucosinolate. Plant J. 2007, 49, 1008–1019. [Google Scholar] [CrossRef]
- Kim, S.J.; Jin, S.; Ishii, G. Isolation and Structural Elucidation of 4-(β-D-Glucopyranosyldisulfanyl)butyl Glucosinolate from Leaves of Rocket Salad Eruca sativa L.) and Its Antioxidative Activity. Biosci. Biotechnol. Biochem. 2004, 68, 2444–2450. [Google Scholar] [CrossRef] [Green Version]
- Lelario, F.; Bianco, G.; Bufo, S.A.; Cataldi, T.R. Establishing the occurrence of major and minor glucosinolates in Brassicaceae by LC-ESI-hybrid linear ion-trap and Fourier-transform ion cyclotron resonance mass spectrometry. Phytochemistry 2012, 73, 74–83. [Google Scholar] [CrossRef]
- Kim, K.H.; Moon, E.; Kim, S.Y.; Choi, S.U.; Lee, J.H.; Lee, K.R. 4-Methylthio-butanyl derivatives from the seeds of Raphanus sativus and their biological evaluation on anti-inflammatory and antitumor activities. J. Ethnopharmacol. 2014, 151, 503–508. [Google Scholar] [CrossRef]
- Matich, A.J.; McKenzie, M.J.; Lill, R.E.; Brummell, D.A.; McGhie, T.K.; Chen, R.K.; Rowan, D.D. Selenoglucosinolates and their metabolites produced in Brassica spp. fertilised with sodium selenate. Phytochemistry 2012, 75, 140–152. [Google Scholar] [CrossRef]
- Fréchard, A.; Fabre, N.; Péan, C.; Montaut, S.; Fauvel, M.T.; Rollin, P.; Fourasté, I. Novel indole-type glucosinolates from woad (Isatis tinctoria L.). Tetrahedron Lett. 2001, 42, 9015–9017. [Google Scholar] [CrossRef]
- Mohn, T.; Hamburger, M. Glucosinolate pattern in Isatis tinctoria and I. indigotica seeds. Planta Med. 2008, 74, 885–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Lewis, J.A.; Hanley, A.B.; Fenwick, G.R. 2-Hydroxyethyl glucosinolate from Capparis masaikai of chinese origin. Phytochemistry 1989, 28, 1252–1254. [Google Scholar] [CrossRef]
- Daxenbichler, M.E.; Spencer, G.F.; Schroeder, W.P. 3-Hydroxypropylglucosinolate, a new glucosinolate in seeds of Erysimum hieracifolium and Malcolmia maritima. Phytochemistry 1980, 19, 813–815. [Google Scholar] [CrossRef]
- Bringmann, G.; Kajahn, I.; Nuesuüß, C.; Pelzing, M.; Laug, S.; Unger, M.; Holzgrabe, U. Analysis of the glucosinolate pattern of Arabidopsis thaliana seeds by capillary zone electrophoresis coupled to electrospray ionization-mass spectrometry. Electrophoresis 2005, 26, 1513–1522. [Google Scholar] [CrossRef]
- Hogge, L.R.; Reed, D.W.; Underhill, E.W.; Haughn, G.W. Hplc separation of glucosinolates from leaves and seeds of Arabidopsls thaliana and their identification using thermospray liquid chromatography/mass spectrometry. J. Chromatogr. Sci. 1988, 26, 551–556. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; D’Auria, J.C.; Behere, A.S.; Kim, J.H.; Gunderson, K.L.; Breen, J.N.; Lee, G.; Gershenzon, J.; Last, R.L.; Jander, G. Characterization of seed-specific benzoyloxyglucosinolate mutations in Arabidopsis thaliana. Plant J. 2007, 51, 1062–1076. [Google Scholar] [CrossRef]
- Chisholm, M.D.; Wetter, L.R. Biosynthesis of Mustard Oil Glucosides: IV. The Administration of Methionine-C14 and Related Compounds to Horseradish. Can. J. Biochem. 1964, 42, 1033–1040. [Google Scholar] [CrossRef]
- Graser, G.; Schneider, B.; Oldham, N.J.; Gershenzon, J. The methionine chain elongation pathway in the biosynthesis of glucosinolates in Eruca sativa (Brassicaceae). Arch. Biochem. Biophys. 2000, 378, 411–419. [Google Scholar] [CrossRef]
- Sønderby, I.E.; Geu-Flores, F.; Halkier, B.A. Biosynthesis of glucosinolates—Gene discovery and beyond. Trends Plant Sci. 2010, 15, 283–290. [Google Scholar] [CrossRef]
- Mikkelsen, M.D.; Naur, P.; Halkier, B.A. Arabidopsis mutants in the C-S lyase of glucosinolate biosynthesis establish a critical role for indole-3-acetaldoxime in auxin homeostasis. Plant J. 2004, 37, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Czerniawski, P.; Bednarek, P. Glutathione S-transferases in the biosynthesis of sulfur-containing secondary metabolites in brassicaceae plants. Front. Plant Sci. 2018, 871, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, M.; Schemenewitz, A.; Lopukhina, A.; Müller, A.; Janowitz, T.; Weiler, E.W.; Oecking, C. Desulfoglucosinolate sulfotransferases from Arabidopsis thaliana catalyze the final step in the biosynthesis of the glucosinolate core structure. J. Biol. Chem. 2004, 279, 50717–50725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubb, C.D.; Zipp, B.J.; Ludwig-Müller, J.; Masuno, M.N.; Molinski, T.F.; Abel, S. Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J. 2004, 40, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Grubb, C.D.; Abel, S. Glucosinolate metabolism and its control. Trends Plant Sci. 2006, 11, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.D.; Petersen, B.L.; Olsen, C.E.; Halkier, B.A. Biosynthesis and metabolic engineering of glucosinolates. Amino Acids 2002, 22, 279–295. [Google Scholar] [CrossRef]
- Wittstock, U.; Halkier, B.A. Glucosinolate research in the Arabidopsis era. Trends Plant Sci. 2002, 7, 263–270. [Google Scholar] [CrossRef]
- Mackay, T.F.C. Quantitative trait loci in Drosophila. Nat. Rev. Genet. 2001, 2, 11–20. [Google Scholar] [CrossRef]
- Kliebenstein, D.J.; Kroymann, J.; Brown, P.; Figuth, A.; Pedersen, D.; Gershenzon, J.; Mitchell-Olds, T. Genetic control of natural variation in arabidopsis glucosinolate accumulation. Plant Physiol. 2001, 126, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Pfalz, M.; Vogel, H.; Kroymann, J. The gene controlling the Indole Glucosinolate Modifier1 quantitative trait locus alters indole glucosinolate structures and aphid resistance in Arabidopsis. Plant Cell 2009, 21, 985–999. [Google Scholar] [CrossRef] [Green Version]
- Wentzell, A.M.; Rowe, H.C.; Hansen, B.G.; Ticconi, C.; Halkier, B.A.; Kliebenstein, D.J. Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet. 2007, 3, 1687–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroymann, J.; Textor, S.; Tokuhisa, J.G.; Falk, K.L.; Bartram, S.; Gershenzon, J.; Mitchell-Olds, T. A gene controlling variation in arabidopsis glucosinolate composition is part of the methionine chain elongation pathway. Plant Physiol. 2001, 127, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hansen, B.G.; Ober, J.A.; Kliebenstein, D.J.; Halkier, B.A. Subclade of flavin-monooxygenases involved in aliphatic glucosinolate biosynthesis. Plant Physiol. 2008, 148, 1721–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirani, A.H.; Zelmer, C.D.; McVetty, P.B.; Daayf, F.; Li, G. Homoeologous GSL-ELONG gene replacement for manipulation of aliphatic glucosinolates in Brassica rapa L. by marker assisted selection. Front. Plant Sci. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettlinger, M.G.; Lundeen, A.J. First Synthesis of a Mustard Oil Glucoside; The Enzymatic Lossen Rearrangement. J. Am. Chem. Soc. 1957, 79, 1764–1765. [Google Scholar] [CrossRef]
- Benn, M.H. a New Mustard Oil Glucoside Synthesis: The Synthesis of Glucotropaeolin. Can. J. Chem. 1963, 41, 2836–2838. [Google Scholar] [CrossRef]
- Kumaran, G.; Kulkarni, G.H. Synthesis of α-Functionalized and Nonfunctionalized Hydroximoyl Chlorides from Conjugated Nitroalkenes and Nitroalkanes. J. Org. Chem. 1997, 62, 1516–1520. [Google Scholar] [CrossRef]
- MacLeod, A.J.; Rossiter, J.T. Synthesis of 2-hydroxybut-3-enylglucosinolate (progoitrin). J. Chem. Soc. Perkin Trans. 1 1983, 53, 717. [Google Scholar] [CrossRef]
- Szmigielska, A.M.; Schoenau, J.J.; Levers, V. Determination of glucosinolates in canola seeds using anion exchange membrane extraction combined with the high-pressure liquid chromatography detection. J. Agric. Food Chem. 2000, 48, 4487–4491. [Google Scholar] [CrossRef]
- Mohn, T.; Cutting, B.; Ernst, B.; Hamburger, M. Extraction and analysis of intact glucosinolates—A validated pressurized liquid extraction/liquid chromatography-mass spectrometry protocol for Isatis tinctoria, and qualitative analysis of other cruciferous plants. J. Chromatogr. A 2007, 1166, 142–151. [Google Scholar] [CrossRef]
- Grosser, K.; van Dam, N.M. A Straightforward Method for Glucosinolate Extraction and Analysis with High-pressure Liquid Chromatography (HPLC). J. Vis. Exp. 2017, 55425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blažević, I.; Đulović, A.; Čikeš Čulić, V.; Popović, M.; Guillot, X.; Burćul, F.; Rollin, P. Microwave-Assisted versus Conventional Isolation of Glucosinolate Degradation Products from Lunaria annua L. and Their Cytotoxic Activity. Biomolecules 2020, 10, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popović, M.; Maravić, A.; Čikeš Čulić, V.; Đulović, A.; Burćul, F.; Blažević, I. Biological effects of glucosinolate degradation products from horseradish: A horse that wins the race. Biomolecules 2020, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Thies, W. Isolation of Sinigrin and Glucotropaeolin from Cruciferous Seeds. Fett Wiss. Technol. Fat Sci. Technol. 1988, 90, 311–314. [Google Scholar] [CrossRef]
- Wang, T.; Liang, H.; Yuan, Q. Optimization of ultrasonic-stimulated solvent extraction of sinigrin from Indian mustard seed (Brassica juncea L.) using response surface methodology. Phytochem. Anal. 2011, 22, 205–213. [Google Scholar] [CrossRef]
- Sut, S.; Boschiero, I.; Solana, M.; Malagoli, M.; Bertucco, A.; Dall’Acqua, S. Supercritical CO2 extraction of eruca sativa using cosolvents: Phytochemical composition by LC-MS analysis. Molecules 2018, 23, 3240. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.E.; Jones, B.A.; Ezzell, J.L.; Porter, N.L.; Avdalovic, N.; Pohl, C. Accelerated Solvent Extraction: A Technique for Sample Preparation. Anal. Chem. 1996, 68, 1033–1039. [Google Scholar] [CrossRef]
- Rafińska, K.; Pomastowski, P.; Rudnicka, J.; Krakowska, A.; Maruśka, A.; Narkute, M.; Buszewski, B. Effect of solvent and extraction technique on composition and biological activity of Lepidium sativum extracts. Food Chem. 2019, 289, 16–25. [Google Scholar] [CrossRef]
- Wang, T.; Liang, H.; Yuan, Q. Separation of sinigrin from Indian mustard (Brassica juncea L.) seed using macroporous ion-exchange resin. Korean J. Chem. Eng. 2012, 29, 396–403. [Google Scholar] [CrossRef]
- Charpentier, N.; Bostyn, S.; Coïc, J.P. Isolation of a rich glucosinolate fraction by liquid chromatography from an aqueous extract obtained by leaching dehulled rapeseed meal (Brassica napus L.). Ind. Crop Prod. 1998, 8, 151–158. [Google Scholar] [CrossRef]
- Ito, Y. High-speed countercurrent chromatography. Nature 1987, 326, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Chou, F.E. Separation and purification of glucosinolates from crude plant homogenates by high-speed counter-current chromatography. J. Chromatogr. A 2003, 996, 85–93. [Google Scholar] [CrossRef]
- Marsh, R.E.; Waser, J. Refinement of the crystal structure of sinigrin. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Jing, B.; Guo, R.; Wang, M.; Zhang, L.; Yu, X. Influence of seed roasting on the quality of glucosinolate content and flavor in virgin rapeseed oil. Lwt 2020, 126, 109301. [Google Scholar] [CrossRef]
- Oerlemans, K.; Barrett, D.M.; Suades, C.B.; Verkerk, R.; Dekker, M. Thermal degradation of glucosinolates in red cabbage. Food Chem. 2006, 95, 19–29. [Google Scholar] [CrossRef]
- Song, L.; Thornalley, P.J. Effect of storage, processing and cooking on glucosinolate content of Brassica vegetables. Food Chem. Toxicol. 2007, 45, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, F.S.; Lamy, E.; Schreiner, M.; Rohn, S. Reactivity and Stability of Glucosinolates and Their Breakdown Products in Foods. Angew. Chem. Int. Ed. 2014, 53, 11430–11450. [Google Scholar] [CrossRef]
- Lazzeri, L.; Curto, G.; Leoni, O.; Dallavalle, E. Effects of Glucosinolates and Their Enzymatic Hydrolysis Products via Myrosinase on the Root-knot Nematode Meloidogyne incognita (Kofoid et White) Chitw. J. Agric. Food Chem. 2004, 52, 6703–6707. [Google Scholar] [CrossRef]
- Buskov, S.; Serra, B.; Rosa, E.; Sørensen, H.; Sørensen, J.C. Effects of intact glucosinolates and products produced from glucosinolates in myrosinase-catalyzed hydrolysis on the potato cyst nematode (Globodera rostochiensis cv. Woll). J. Agric. Food Chem. 2002, 50, 690–695. [Google Scholar] [CrossRef]
- Burmeister, W.P.; Cottaz, S.; Driguez, H.; Iori, R.; Palmieri, S.; Henrissat, B. The crystal structures of Sinapis alba myrosinase and a covalent glycosyl–enzyme intermediate provide insights into the substrate recognition and active-site machinery of an S-glycosidase. Structure 1997, 5, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Cottaz, S.; Henrissat, B.; Driguez, H. Mechanism-based inhibition and stereochemistry of glucosinolate hydrolysis by myrosinase. Biochemistry 1996, 35, 15256–15259. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, W.P.; Cottaz, S.; Rollin, P.; Vasella, A.; Henrissat, B. High resolution X-ray crystallography shows that ascorbate is a cofactor for myrosinase and substitutes for the function of the catalytic base. J. Biol. Chem. 2000, 275, 39385–39393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettlinger, M.G.; Dateo, G.P.; Harrison, B.W.; Mabry, T.J.; Thompson, C.P. Vitamin C As A Coenzyme: The Hydrolysis of Mustard Oil Glucosides. Proc. Natl. Acad. Sci. USA 1961, 47, 1875–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratzka, A.; Vogel, H.; Kliebenstein, D.J.; Mitchell-Olds, T.; Kroymann, J. Disarming the mustard oil bomb. Proc. Natl. Acad. Sci. USA 2002, 99, 11223–11228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, K.L.; Gershenzon, J. The desert locust, Schistocerca gregaria, detoxifies the glucosinolates of Schouwia purpurea by desulfation. J. Chem. Ecol. 2007, 33, 1542–1555. [Google Scholar] [CrossRef]
- Heidel-Fischer, H.M.; Kirsch, R.; Reichelt, M.; Ahn, S.J.; Wielsch, N.; Baxter, S.W.; Heckel, D.G.; Vogel, H.; Kroymann, J. An Insect Counteradaptation against Host Plant Defenses Evolved through Concerted Neofunctionalization. Mol. Biol. Evol. 2019, 36, 930–941. [Google Scholar] [CrossRef]
- Ahn, S.J.; Betzin, F.; Gikonyo, M.W.; Yang, Z.L.; Köllner, T.G.; Beran, F. Identification and evolution of glucosinolate sulfatases in a specialist flea beetle. Sci. Rep. 2019, 9, 15725. [Google Scholar] [CrossRef]
- Wittstock, U.; Burow, M. Glucosinolate Breakdown in Arabidopsis: Mechanism, Regulation and Biological Significance. Arab. Book 2010, 8, e0134. [Google Scholar] [CrossRef] [Green Version]
- Eisenschmidt-Bönn, D.; Schneegans, N.; Backenköhler, A.; Wittstock, U.; Brandt, W. Structural diversification during glucosinolate breakdown: Mechanisms of thiocyanate, epithionitrile and simple nitrile formation. Plant J. 2019, 99, 329–343. [Google Scholar] [CrossRef]
- Aumaitre, A.; Bourdon, D.; Peiniau, J.; Freire, J.B. Effect of graded levels of raw and processed rapeseed on feed digestibility and nutrient utilization in young pigs. Anim. Feed Sci. Technol. 1989, 24, 275–287. [Google Scholar] [CrossRef]
- Mabon, N.; Mandiki, S.N.; Derycke, G.; Bister, J.L.; Wathelet, J.P.; Marlier, M.; Paquay, R. Chemical changes and influences of rapeseed antinutritional factors on lamb physiology and performance. 3. Antinutritional factors in plasma and organs. Anim. Feed Sci. Technol. 2000, 85, 111–120. [Google Scholar] [CrossRef]
- Kensler, T.W. Chemoprevention by inducers of carcinogen detoxication enzymes. Environ. Health Perspect. 1997, 105, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Aires, A.; Saavedra, M. Antimicrobial Activity of Isothiocyanates from Cruciferous Plants against Methicillin-Resistant Staphylococcus aureus (MRSA). Int. J. Mol. Sci. 2014, 15, 19552–19561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanschen, F.S.; Yim, B.; Winkelmann, T.; Smalla, K.; Schreiner, M. Degradation of biofumigant isothiocyanates and allyl glucosinolate in soil and their effects on the microbial community composition. PLoS ONE 2015, 10, e0132931. [Google Scholar] [CrossRef] [Green Version]
- Kuchernig, J.C.; Burow, M.; Wittstock, U. Evolution of specifier proteins in glucosinolate-containing plants. BMC Evol. Biol. 2012, 12. [Google Scholar] [CrossRef] [Green Version]
- Mumm, R.; Burow, M.; Bukovinszkine’Kiss, G.; Kazantzidou, E.; Wittstock, U.; Dicke, M.; Gershenzon, J. Formation of simple nitriles upon glucosinolate hydrolysis affects direct and indirect defense against the specialist herbivore, Pieris rapae. J. Chem. Ecol. 2008, 34, 1311–1321. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Tissue | GSL Content | Reference |
|---|---|---|---|---|
| Brassicaceae | Camelina sativa | Seed | 15.8–19.4 | [30] |
| Camelina rumelica subsp. rumelica | Seed | 18.6–21.7 | ||
| Camelina macrocarpa | Seed | 8.0–19.1 | ||
| Brassica napus | Leaf | 0.6–6.9 | [22,31,32] | |
| Seed | 10.8–57.9 | |||
| Brassica carinata A Braun | Seed | 35–170 | [26,27] | |
| Brassica juncea | Leaf | 4.3–129.9 | [33] | |
| Seed | 15.7–127.6 | |||
| Brassica oleracea L. var capitata | Leaf | 2.3–11.5 | [34] | |
| Brassica oleracea L. var italica | Floret | 8.2–19.5 | [35] | |
| Brassica oleracea L. convar capitata var alba | Petiole | 0.5–31.7 | [36] | |
| Brassica rapa | Leaf | 17.3 | [31] | |
| Seed | 39.4–81.3 | |||
| Arabidopsis thaliana | Leaf | 5.0–30.7 | [37] | |
| Raphannus sativus L. | Root | 1.0–145.5 | [29,38] | |
| Moringacea | Moringa oleifera Lam. | Leaf | 4.7–217 | [28,39] |
| Seed | 112–354.4 | [40,41] | ||
| Moringa stenopetala L. | Leaf | 33.9–59.4 | [42,43] | |
| Seed | 256–282 |
| No | Class | Index | Semi Systematic Name | Trivial Name | Characterization Methods | Reference |
|---|---|---|---|---|---|---|
| 1 | Ala | A | Methyl GSL | Glucocapparin | MS, NMR of GSL; MS of desGSL | [47,48] |
| 2 | Val | A | 1-Methylethyl GSL | Glucoputranjivin | UV, IR, MS, NMR of GSL | [49] |
| 3 | Val | A | (1R)-Methyl-2-hydroxyethyl GSL | Glucosisymbrin | MS, NMR of desGSL | [50,51] |
| 4 | Val | B | (1R)-2-Bezoyloxt-1-methylethyl GSL | Glucobenzosisymbrin | UV, IR of ITC | [52] |
| 5 | Glu | A | 3-Carboxypropyl GSL | Deducted from ITC structure | [53] | |
| 6 | Glu | A | 3-Methoxycarbonyl- propyl GSL | Glucoerypestrin | Partial NMR of GSL | [54] |
| 7 | ? | A | Ethyl GSL | Glucolepidiin | Thiourea-type, IR compared to GSL structure | [55] |
| 8 | ? | A | n-Butyl GSL | Thiourea-type method compared to GSL | [56,57] | |
| and MS from ITC | ||||||
| 9 | ? | A | n-Pentyl GSL | MS of ITC | [58] | |
| 10 | ? | A | n-Hexyl GSL | MS of ITC | [57] | |
| 11 | ? | A | 4-Oxoheptyl GSL | Glucocapangulin | Deducted from IR and 5-oxooctanoic acid | [59] |
| 12 | ? | A | 5-Oxoheptyl GSL | Gluconorcappasalin | Thiourea-type, IR compared to GSL; | [60] |
| MS from ITC | ||||||
| 13 | ? | A | 5-Oxooctyl GSL | Glucocappasalin | UV, IR of GSL and desGSL; partial NMR of desGSL | [61] |
| 14 | ? | A | 4,5,6,7-Tetrahydroxydecyl GSL | UV, IR, NMR of ITC | [62] | |
| 15 | ? | B | Phenyl GSL | MS of GSL | [57] | |
| 16 | ? | B | 2-(4-Methoxyphenyl)-2,2-dimethyl ethyl GSL | IR, MS, NMR of ITC | [63] | |
| 17 | Leu | A | 2-Methylpropyl GSL | MS, NMR of GSL and desGSL | [50,64] | |
| 18 | Leu | A | 2-Hydroxy-2-methylpropyl GSL | Glucoconringiin | MS, NMR | [65] |
| 19 | Leu | A | 3-Methylbutyl GSL | MS of ITC | [58] | |
| 20 | Leu | A | 3-Methylbut-3-eyl GSL | IR, MS, NMR of | [66] | |
| ITC | ||||||
| 21 | Leu | A | 4-Methylpentyl GSL | MS of ITC | [67] | |
| 22 | Ile | A | (1S)-1-Methylpropyl GSL | Glucocochlearin | MS, NMR of GSL and desGSL | [50,68] |
| 23 | Ile | A | (1R)-1-(Hydroxymethyl)-propyl GSL | Glucosisaustricin | MS, NMR of desGSL | [50] |
| 24 | Ile | B | (1R)-1-(Benzoyloxymethyl)-propylGSL | Glucobenzsisaustricin | Thiourease-type, IR compared to GSL | [69] |
| 25 | Ile | A | (2S)-2-Methylbutyl GSL | Glucojiaputin | UV, IR, MS, NMR of GSL and des GSL | [49,50] |
| 26 | Ile | A | (2S)-2-Hydroxy-2-methylbutyl GSL | Glucocleomin | NMR of desGSL | [51] |
| 27 | Ile | A | 3-Methylpentyl GSL | UV, IR, MS, NMR of GSL; MS, NMR of desGSL | [49,50] | |
| 28 | Ile | A | 3-(Hydroxymethyl)pentyl GSL | NMR of GSL | [70] | |
| 29 | Ile | A | 2-Hydroxy-3-methylpenyl GSL | MS, NMR of desGSL | [50,70] | |
| 30 | Trp | C | 4-Methoxyindol-3-yl GSL | Glucorapassicin A | UV, IR, MS, NMR of synthesized GSL | [71] |
| 31 | Trp | C | Indol-3-ymethyl GSL | Glucobrassicin | UV, IR, MS, NMR of GSL and desGSL | [49,50] |
| 32 | Trp | C | 1-Hydroxyindol-3ylmethyl GSL | MS of GSL; UV, MS of desGSL | [72] | |
| 33 | Trp | C | 4-Hydroxyindol-3-ylmethyl GSL | 4-Hydroxy-glucobrassicin | MS of GSL; UV, MS, NMR of desGSL | [72,73,74] |
| 34 | Trp | C | 4-Methoxyindol-3-ylmethyl GSL | 4-Methoxy-glucobrassicin | UV, MS, MS, NMR of GSL and desGSL | [49,74] |
| 35 | Trp | C | 1-Methoxyindol-3-ylmethyl GSL | Neoglucobrassicin | UV, IR MS, NMR of GSL; MS, NMR of desGSL | [49,50,72] |
| 36 | Trp | C | 1,4-Dimethoxyindol-3-ymethyl GSL | 1,4-Dimethoxy-glucobrassicin | UV, MS, NMR ofdesGSL | [50,70] |
| 37 | Trp | C | 1-Acetylindol-3-ymethyl GSL | N-Acetyl-glucobrassicin | MS of desGSL | [75] |
| 38 | Trp | C | 1-Sulfoindol-3-ylmethyl GSL | N-Sulfo-glucobrassicin | UV, IR, MS, NMR of GSL | [65,76] |
| 39 | Trp | C | 6′-Isoferuloylindol-3-ylmethyl GSL | 6′-Isoferuloyl-glucobrassicin | MS of GSL; UV, MS, NMR of desGSL | [77,78] |
| 40 | Phe | B | Benzyl GSL | Glucotropaeolin | MS, NMR of GSL; UV, MS, NMR of desGSL | [51,79,80] |
| 41 | Phe | B | 3-Hydroxybenzyl GSL | Glucolepigramin | MS of GSL; MS, NMR of desGSL | [65,81] |
| 42 | Phe | B | 3-Methoxybenzyl GSL | Glucolimnanthin | MS, NMR of GSL; UV, MS, NMR of desGSL | [51,82] |
| 43 | Phe/Trp | B | 4-Hydroxybenzyl GSL | Glucosinalbin | UV, MS, NMR of GSL and desGSL | [52,78,83] |
| 44 | Phe/Trp | B | 4-Methoxybenzyl GSL | Glucoaubrietin | MS of GSL; UV, | [50,65,84] |
| MS, NMR of | ||||||
| desGSL | ||||||
| 45 | Phe/Trp | B | 3,4-Dihydroxybenzyl GSL | Glucomatronalin | MS of GSL | [65] |
| 46 | Phe/Tyr | B | 4-Hydroxy-3-methoxybenzyl GSL | 3-Methoxysinalbin | UV, MS, NMR of desGSL | [81] |
| 47 | Phe/Tyr | B | 3-Hydroxy-4-methoxybenzyl GSL | Glucobretschneiderin | UV, IR, MS, NMR of GSL | [85] |
| 48 | Phe/Tyr | B | 3,4-Dimethoxybenzyl GSL | UV, MS, NMR of | [81] | |
| desGSL | ||||||
| 49 | Phe/Tyr | B | 4-Hydroxy-3,5-dimethoxybenzyl GSL | 3,5-Dimethoxy-sinalbin | UV, MS, NMR of desGSL | [81] |
| 50 | Phe/Tyr | B | 3,4,5-Trimethoxybenzyl GSL | MS of GSL; UV, MS, NMR of desGSL | [80,81] | |
| 51 | Phe | B | 2-Phenylethyl GSL | Gluconasturtiin | NMR of GSL; UV, MS, NMR of desGSL | [51,65] |
| 52 | Phe | B | (2S)-2-hydroxy-2-phenylethyl GSL | Glucobarbarin | MS, NMR of GSL and desGSL | [50,65,74] |
| 53 | Phe | B | (2R)-2-Hydroxy-2-phenylethyl GSL | Epiglucobarbarin | MS, NMR of GSL and des GSL | [65,77,86] |
| 54 | Phe | B | 2-(3-Hydroxy-phenyl)ethyl GSL | UV, MS, NMR of desGSL | [86] | |
| 55 | Phe | B | 2-(4-Hydroxy-phenyl)ethyl GSL | Homosinalbin | MS, NMR of GSL; UV, MS, NMR of desGSL | [65,87] |
| 56 | Phe | B | (2R)-2-Hydroxy-2- | m-Hydroxy-epiglucobarbarin | UV, MS, NMR of GSL and desGSL | [18] |
| (3-hydroxyphenyl)ethyl GSL | ||||||
| 57 | Phe | B | 3-Phenylpropyl GSL | MS of ITC | [88] | |
| 58 | Phe | B | 4-Phenylbutyl GSL | MS of ITC | [88] | |
| 59 | Phe | B | 5-Phenylpentyl GSL | Glucoarmoracin | MS of ITC | [88] |
| 60 | Phe/Tyr | B | 2-(4-Methoxy-phenyl)ethyl GSL | NMR of GSL, MS, NMR of desGSL | [65,70,84] | |
| 61 | Phe/Tyr | B | (2R)-2-Hydroxy-2-(4-hydroxyphenyl)ethyl GSL | p-Hydroxy-epiglucobarbarin | MS, NMR of GSL; UV, MS, NMR of desGSL | [70,89] |
| 62 | Phe/Tyr | B | (2S)-2-Hydroxy-2(4-hydroxyphenyl)ethyl GSL | p-Hydroxy-glucobarbarin | UV, MS, NMR of desGSL | [89] |
| 63 | Phe/Tyr | B | (2R)-2-Hydroxy-2(4-methoxyphenyl)ethyl GSL | MS, NMR of GSL | [90] | |
| 64 | Phe | B | 2-(α-l-Rhamnopyranosyloxy)-benzyl GSL | MS of GSL and desGSL | [65,87] | |
| 65 | Phe | B | 4-(4′-O-Acetyl-α-l- 4- rhamnopyranosyloxy)-benzyl GSL | 4-Acetyl-glucomoringin | MS of GSL and ITC | [43,91] |
| 66 | Phe | B | 2-(α-l-Arabinopyranosyloxy)-2phenylethyl GSL | NMR of GSL | [92] | |
| 67 | Phe | B | 6′-Isoferuloyl-2-phenylethyl GSL | 6′-Isoferuloyl-gluconasturtiin | MS of GSL, UV, MS NMR of desGSL | [77,78] |
| 68 | Phe | B | 6′-Isoferuloyl-(2R)-2-hydroxy-2phenylethyl GSL | 6′-Isoferuloyl-epiglucobarbarin | MS, NMR of GSL; UV, MS, NMR of desGSL | [78] |
| 69 | Phe | B | 6′-Isoferuloyl-(2S)-2-hydroxy-2phenylethyl GSL | 6′-Isoferuloyl-glucobarbarin | MS, NMR of GSL; UV, MS, NMR of desGSL | [78] |
| 70 | Phe/Tyr | B | 6′-Isoferuloyl-(R)-2-hydroxy-2(4-hydroxyphenyl)ethyl GSL | MS of GSL; UV, MS NMR of desGSL | [78] | |
| 71 | Phe/Tyr | B | 4-(α-l-Rhamnopyranosyloxy)-benzyl GSL | Glucomorinigin | MS, NMR of GSL and desGSL | [51,93,94,95] |
| 72 | Met | A | 3-(Methylsulfanyl)propyl GSL | Glucoibervirin | MS, NMR of GSL | [96] |
| 73 | Met | A | 4-Oxoheptyl GSL | Glucocapangulin | Deduction from IR, 5-oxooctanoic acid | [59] |
| 74 | Met | A | 4-(Methylsulfanyl)butyl GSL | Glucoerucin | UV, IR, MS NMR of GSL | [51,80] |
| 75 | Met | A | 5-(Methylsulfanyl)pentyl GSL | Glucoberteroin | UV, IR, MS, NMR of GSL; UV, MS, NMR of desGSL | [97,98,99] |
| 76 | Met | A | 6-(Methylsulfanyl)heptyl GSL | UV, IR, MS, NMR of GSL | [97,98] | |
| 77 | Met | A | 6-(Methylsulfanyl)hexyl GSL | Glucolesquerellin | UV, IR, MS, NMR of GSL | [97,98] |
| 78 | Met | A | 8-(Methylsulfanyl)-3-oxooctyl GSL | MS of GSL; MS, NMR of des GSL | [50,65] | |
| 79 | Met | A | 9-(Methylsulfanyl)nonyl GSL | MS of GSL | [65] | |
| 80 | Met | A | 10 -(Methylsulfanyl)decyl GSL | MS of ITC | [100] | |
| 81 | Met | A | 2-Methylsulfinylethyl GSL | UV, MS, NMR of | [101] | |
| desGSL | ||||||
| 82 | Met | A | (R)-11-(Methylsulfinyl)-propyl glucosinolate | Glucoiberin | MS, NMR, X-Ray of GSL; UV, MS, NMR of desGSL | [51,80,102] |
| 83 | Met | A | (R/S)-4-(Methylsulfinyl)-butyl glucosinolate | Glucoraphanin | MS, NMR of GSL, UV, MS NMR of desGSL | [51,80,103] |
| 84 | Met | A | (R/S)-5-(Methylsulfinyl)pentyl GSL | Glucoalyssin | MS, NMR of GSL; MS of desGSL | [80,104] |
| 85 | Met | A | (R/S)-6-(Methylsulfinyl)-hexyl GSL | Glucohesperin | UV, IR, MS, NMR of GSL | [80,97,98] |
| 86 | Met | A | (R/S)-7-(Methylsulfinyl)-heptyl GSL | NMR of GSL; MS, NMR of desGSL | [50,105] | |
| 87 | Met | A | (R/S)-8-(Methylsulfinyl)-octyl GSL | Glucohirsutin | UV, IR, MS, NMR of GSL; MS, NMR of desGSL | [50,98] |
| 88 | Met | A | (R/S)-9-(Methylsulfinyl)-nonyl GSL | Glucoarabin | UV, IR, MS, NMR of GSL; MS, NMR of desGSL | [98,106] |
| 89 | Met | A | (R/S)-10-(Methylsulfinyl)decyl GSL | Glucocamelinin | MS, NMR of GSL; MS of desGSL | [50,107] |
| 90 | Met | A | (R/S)-11-(Methylsulfinyl)undecyl GSL | MS of GSL | [107] | |
| 91 | Met | A | 3-(Methylsulfonyl)-propyl GSL | Glucocheirolin | MS of GSL; NMR of desGSL | [48,108] |
| 92 | Met | A | 4-(Methylsulfonyl)butyl GSL | Glucoerysolin | MS of GSL; MS, NMR of desGSL | [65,84,106] |
| 93 | Met | A | 6-(Methylsulfonyl)hexyl GSL | MS of GSL | [65] | |
| 94 | Met | A | 8-(Methylsulfonyl)octyl GSL | UV, IR, MS, NMR of GSL; MS, NMR of desGSL | [50,106,109] | |
| 95 | Met | A | 9-(Methylsulfonyl)nonyl GSL | UV, IR, MS, NMR of GSL; MS, NMR of desGSL | [50,106,109] | |
| 96 | Met | A | 10-(Methylsulfonyl)decyl GSL | MS, NMR of desGSL | [84,106] | |
| 97 | Met | A | (3E)-4-(Methylsulfanyl)-but-3-enyl GSL | IR, MS, NMR of GSL; NMR of desGSL | [51,80] | |
| 98 | Met | A | (R/S,3E)-4-(Methylsulfinyl)-but-3-enyl GSL | Glucoraphenin | MS, NMR of GSL; UV, NMR of desGSL | [51,80,110] |
| 99 | Met | A | 3-Hydroxy-5-(methylsulfinyl)pentyl GSL | Deducted from tetrahydro-1,3-oxazine-2-thione | [111] | |
| 100 | Met | A | 3-Hydroxy-5-(methylsulfony)pentyl GSL | UV, IR, MS, NMR of ITC | [111] | |
| 101 | Met | A | 3-Hydroxy-6-(methylsulfanyl)hexyl GSL | Deducted from tetrahydro-1,3-oxazine-2-thione | [112] | |
| 102 | Met | A | 3-Hydroxy-6-(methylsufinyl)hexyl GSL | Deducted from ITC | [112] | |
| 103 | Met | A | 3-Hydroxy-5-(methylsulfinyl)pentyl GSL | Deducted from tetrahydro-1,3oxazine-2-thione and ITC | [112] | |
| 104 | Met | A | 8-(Methylsulfanyl)-3-oxooctyl GSL | Deducted from ITC | [113] | |
| 105 | Met | A | (R/S)-8-(Methylsulfinyl)-3-oxooctyl GSL | Deducted from ITC | [113] | |
| 106 | Met | A | 4-Mercaptobutyl GSL | MS, NMR of GSL | [114,115] | |
| 107 | Met | A | (R)-4-(Cystein-S-yl)butyl GSL | Glucorucolamine | MS, NMR of desGSL | [116] |
| 108 | Met | A | Dimeric 4-mercaptobutyl GSL | MS, NMR of GSL; MS of desGSL | [114] | |
| 109 | Met | A | 4-(β-d-Glucopyranosyl- disulfanyl)-butyl GSL | Diglucothiobeinin | MS of GSL; MS, NMR of desGSL | [117,118] |
| 110 | Met | A | 6′-Benzoyl-4(methylsulfanyl)butyl GSL | 6′-Benzoyl-glucoerucin | UV, MS, NMR of desGSL | [101] |
| 111 | Met | A | 6′-Benzoyl-4(methylsulfinyl)butyl- GSL | 6′-Benzoyl -glucopharanin | UV, MS, NMR of desGSL | [101] |
| 112 | Met | A | (R/S, 3E)-6′-Sinapoyl-4-(methylsulfinyl)but-3-enyl GSL | 6′-Sinapoyl-glucoraphenin | UV, IR, MS, NMR of desGSL | [119] |
| 113 | Se-Met | A | 3-(Methylseleno)propyl GSL | Comparing MS with natural S-analog | [120] | |
| 114 | Se-Met | A | 4-(Methylseleno)butyl GSL | Comparing MS with natural S-analog | [120] | |
| 115 | Se-Met | A | 5-(Methylseleno)pentyl GSL | Comparing MS with natural S-analog | [120] | |
| 116 | Met | A | Allyl glucosinolate | Sinigrin | MS, NMR, X-Ray of GSL; UV, MS, NMR of desGSL | [51,65] |
| 117 | Met | A | But-3-enyl GSL | Gluconapin | MS, NMR of GSL; UV, MS, NMR of desGSL | [50,51,80,104] |
| 118 | Met | A | Pent-4-enyl GSL | Glucobrassicanapin | MS of GSL; MS, NMR of desGSL | [50,104] |
| 119 | Met | A | (2S)-2-Hydroxypent-4-enyl GSL | Gluconapoleiferin | MS of GSL | [73] |
| 120 | Met | A | (2R)-2-Hydroxybut-3-enyl GSL | Progoitrin | MS, NMR of GSL; UV, MS, NMR of desGSL | [50,51,79,80,84] |
| 121 | Met | A | (2S)-2-Hydroxybut-3-enyl GSL | Epiprogoitrin | MS, NMR of GSL; UV, MS, NMR of desGSL | [50,51,73,79] |
| 122 | Met | C | 2′,3′-Dihydro-2′-oxoindol-3′-ylacetate ester at 2-OH of (R)-2-hydroxbut-3-enyl GSL | Glucoisatisin | UV, MS, NMR of GSL | [121,122] |
| 123 | Met | C | 2′,3′-Dihydro-2′-oxoindol-3′-ylacetate of ester at 2-OH of (S)-2-hydroxbut-3-enyl GSL | Epiglucoisatisin | UV, MS, NMR of GSL | [121,122] |
| 124 | Met | C | 2′,3′-Dihydro-3′-hydroxy-2′-oxoindol-3′-ylacetate ester at 2-OH of (R)-2-hydroxybut-3-enyl GSL | (S)-3′-Hydroxy-glucoisasitin | UV, MS, NMR of GSL | [121] |
| 125 | Met | C | 2′,3′-Dihydro-3′-hydroxy-2′-oxoindol-3′-ylacetate ester at 2-OH of (S)-2-hydroxybut-3-enyl GSL | (S)-3′-Hydroxy-epiglucoisasitin | UV, MS, NMR of GSL | [121] |
| 126 | Met | B | (2S)-2-Benzoyloxybut-3-enyl GSL | 2-O-Benzoyl-epiprogoitrin | MS of desGSL | [101] |
| 127 | Met | A | 2-Hydroxyethyl GSL | NMR of GSL | [123] | |
| 128 | Met | A | 3-Hydroxypropyl GSL | MS, NMR of ITC | [124] | |
| 129 | Met | A | 4-Hydroxylbutyl GSL | MS of GSL | [125] | |
| 130 | Met | A | 3-Hydroxylbutyl GSL | Deducted from tetrahydro-1,3oxazine-2-thione | [56] | |
| 131 | Met | B | 2-(Benzoyloxy)ethyl GSL | MS of GSL | [125] | |
| 132 | Met | B | 3-(Benzoyloxy)propyl GSL | Glucomalcolmiin | MS of GSL; UV, MS of desGSL | [101,125,126] |
| 133 | Met | B | 4-(Benzoyloxy)butyl GSL | MS of GSL; UV, MS, NMR of desGSL | [101,125] | |
| 134 | Met | B | 5-(Benzoyloxy)pentyl GSL | MS, NMR of desGSL | [101] | |
| 135 | Met | B | 6-(Benzoyloxy)hexyl GSL | Deducted from ITC | [126] | |
| 136 | Met | B | 3-Sinapoyloxypropyl GSL | MS, NMR of desGSL | [127] | |
| 137 | Met | B | 6′-Benzoyl-4-benzoyloxybutyl GSL | UV, MS, NMR of desGSL | [101] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, V.P.T.; Stewart, J.; Lopez, M.; Ioannou, I.; Allais, F. Glucosinolates: Natural Occurrence, Biosynthesis, Accessibility, Isolation, Structures, and Biological Activities. Molecules 2020, 25, 4537. https://doi.org/10.3390/molecules25194537
Nguyen VPT, Stewart J, Lopez M, Ioannou I, Allais F. Glucosinolates: Natural Occurrence, Biosynthesis, Accessibility, Isolation, Structures, and Biological Activities. Molecules. 2020; 25(19):4537. https://doi.org/10.3390/molecules25194537
Chicago/Turabian StyleNguyen, V. P. Thinh, Jon Stewart, Michel Lopez, Irina Ioannou, and Florent Allais. 2020. "Glucosinolates: Natural Occurrence, Biosynthesis, Accessibility, Isolation, Structures, and Biological Activities" Molecules 25, no. 19: 4537. https://doi.org/10.3390/molecules25194537
APA StyleNguyen, V. P. T., Stewart, J., Lopez, M., Ioannou, I., & Allais, F. (2020). Glucosinolates: Natural Occurrence, Biosynthesis, Accessibility, Isolation, Structures, and Biological Activities. Molecules, 25(19), 4537. https://doi.org/10.3390/molecules25194537