
ATP-Binding and Hydrolysis in Inflammasome Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The NLR Inflammasomes
1.1. Inflammasomes and Innate Immunity
1.2. Selective Tissue Expression Profiles
1.3. Epigenetic Programming and Innate Immune Memory
1.4. NLR Links between the Innate and Adaptive Immune Systems
2. The ATP-Dependency of NLR Activation
2.1. NLRs are STAND ATPases
2.2. NLR Phylogeny
2.3. The Importance of ATP in NLR Activation
3. ATP-Dependency for the Assembly and Activation of Selected NLR Inflammasomes
3.1. NLRC4
3.2. NLRP1
3.3. NLRP2
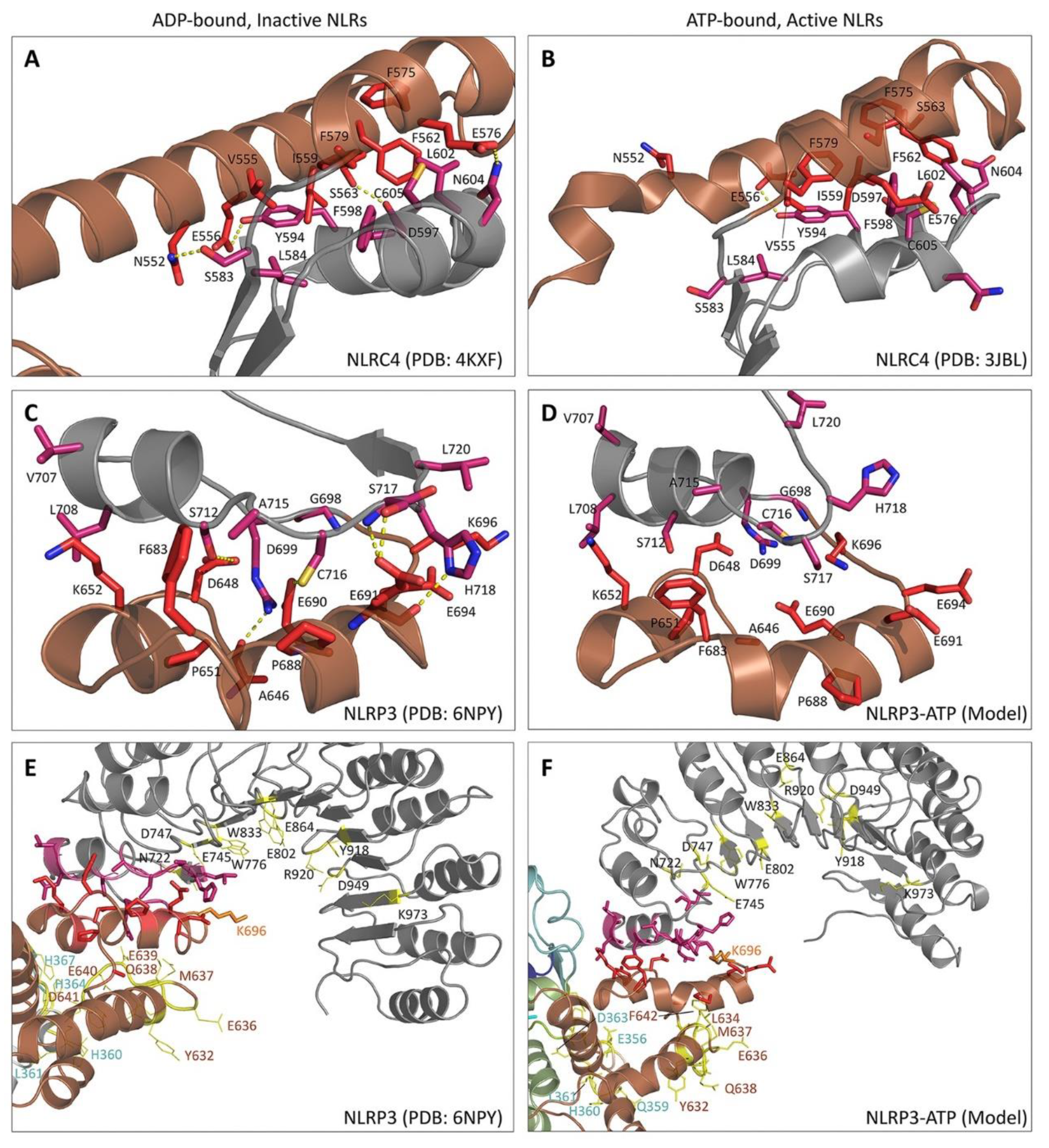
3.4. NLRP3
3.5. NLRP6
3.6. NLRP7
3.7. NLRP9
3.8. NLRP12
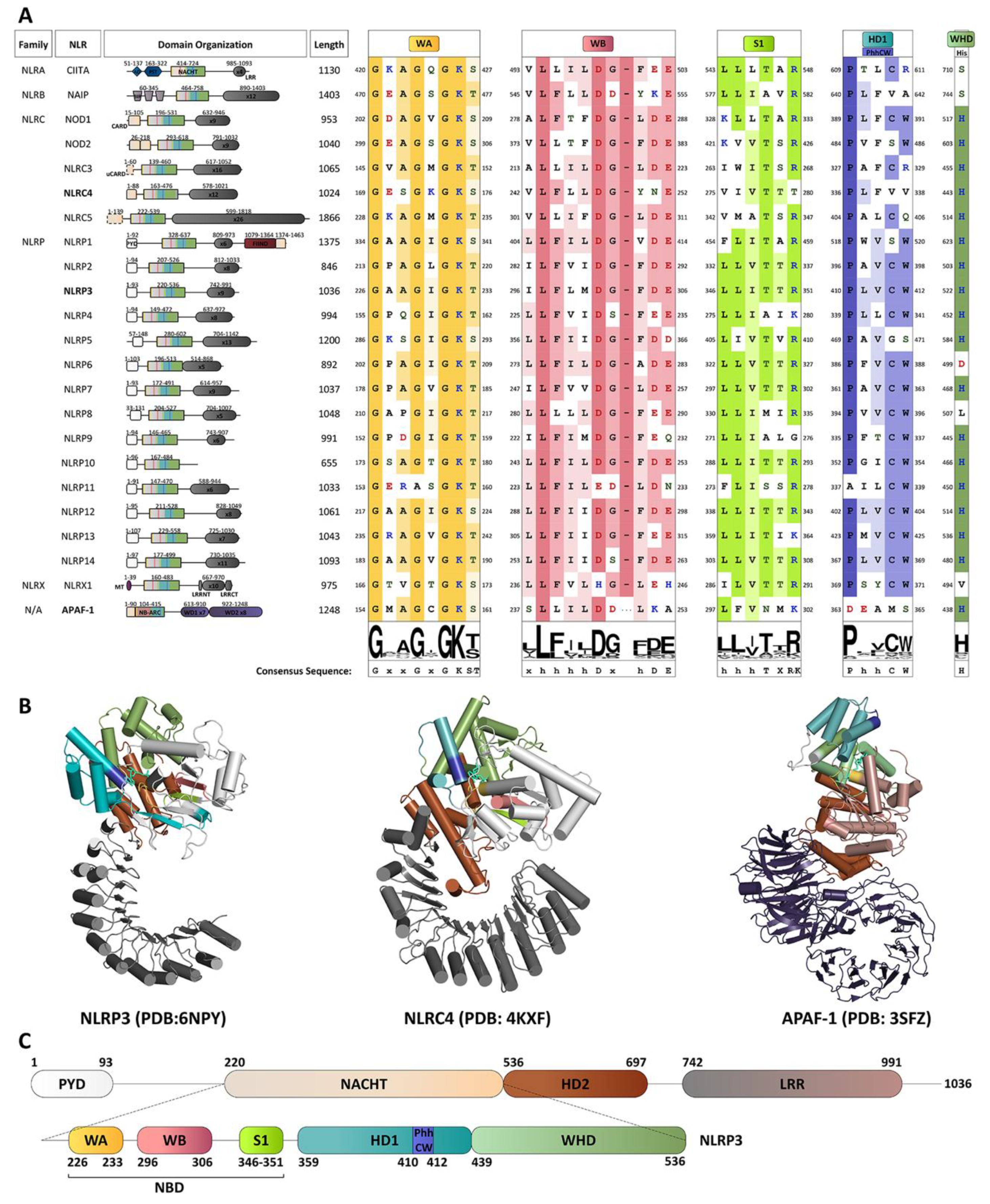
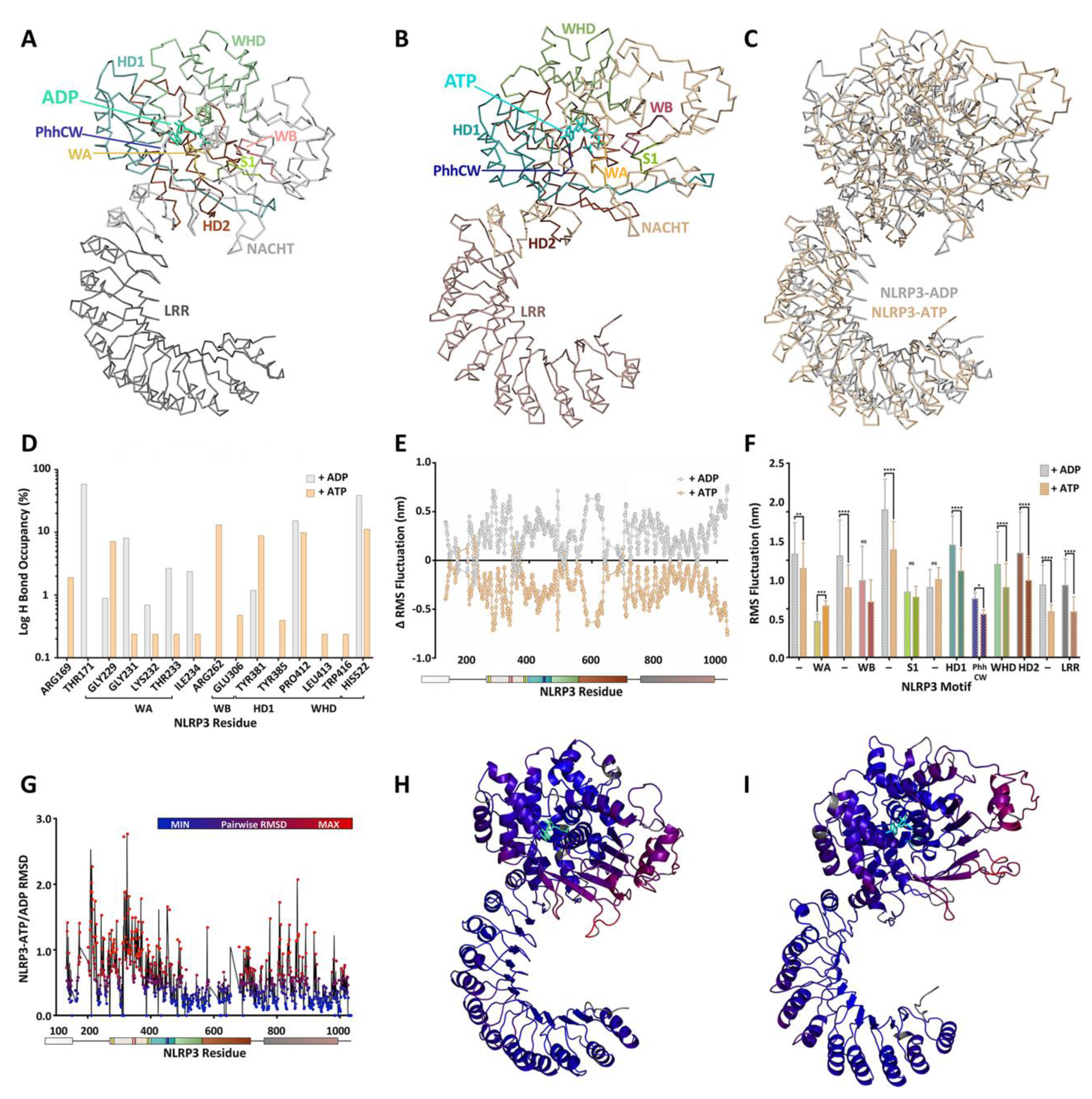
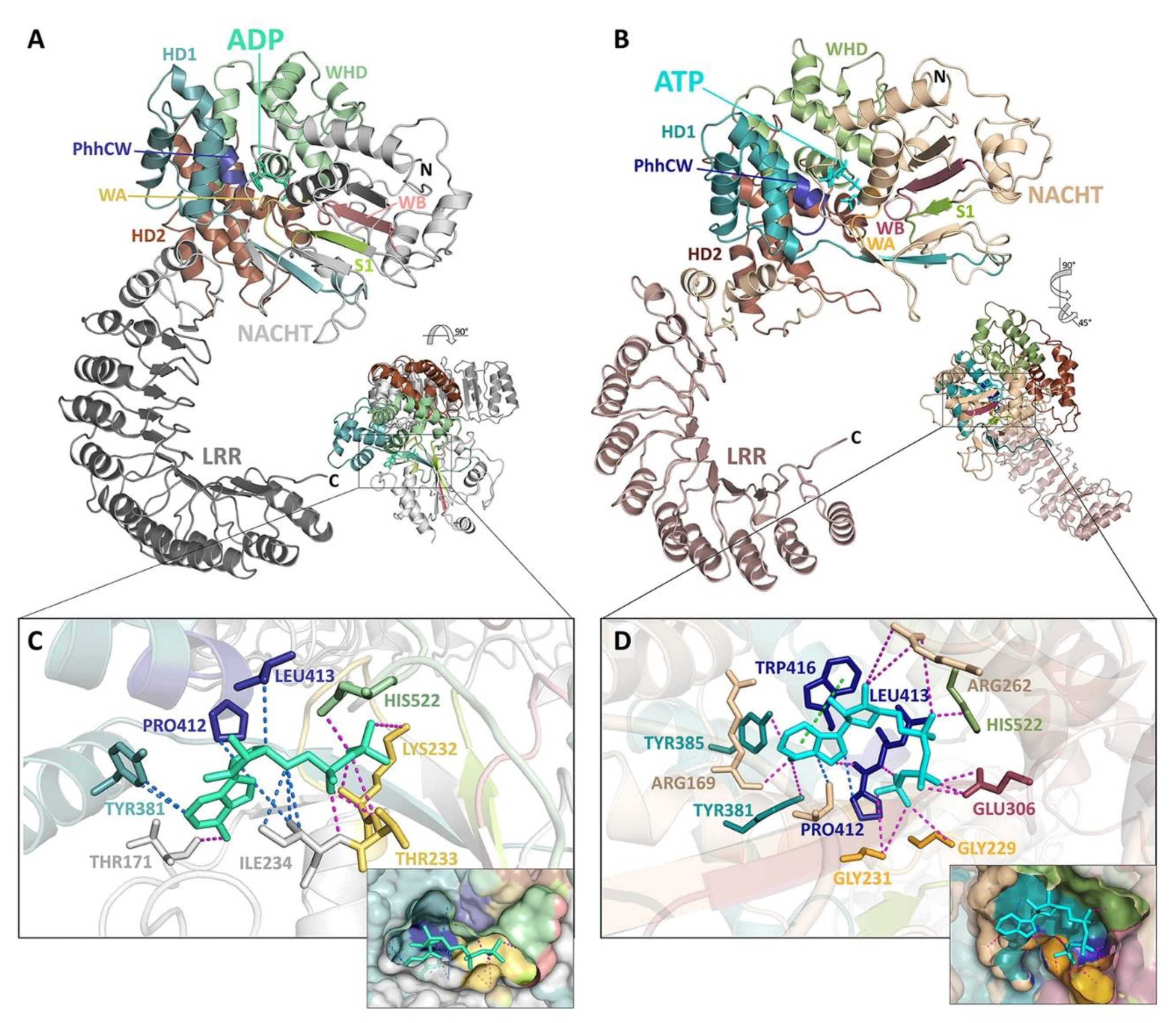
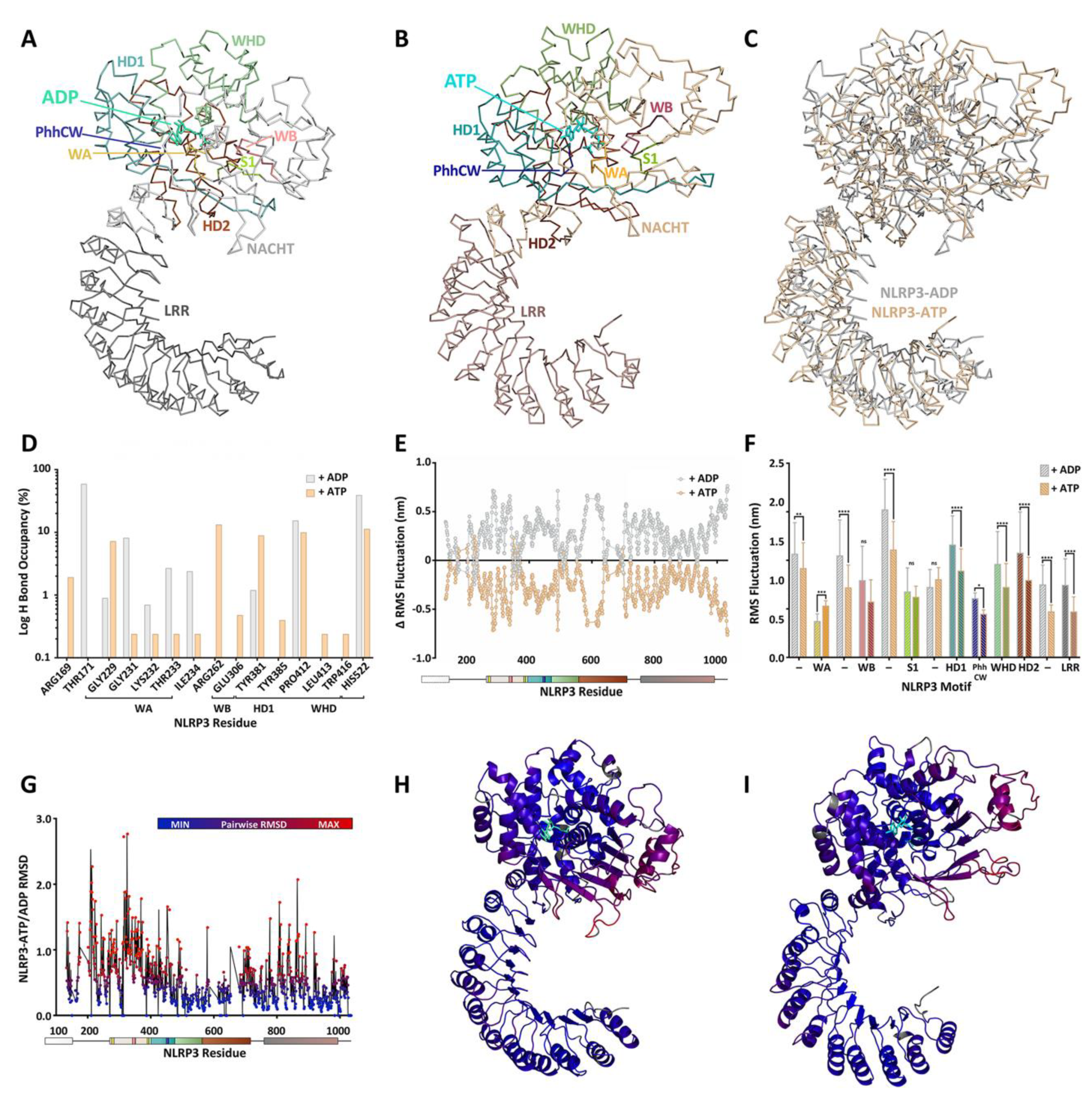
4. A Molecular Description of the NACHT Domain: Key Functional Motifs
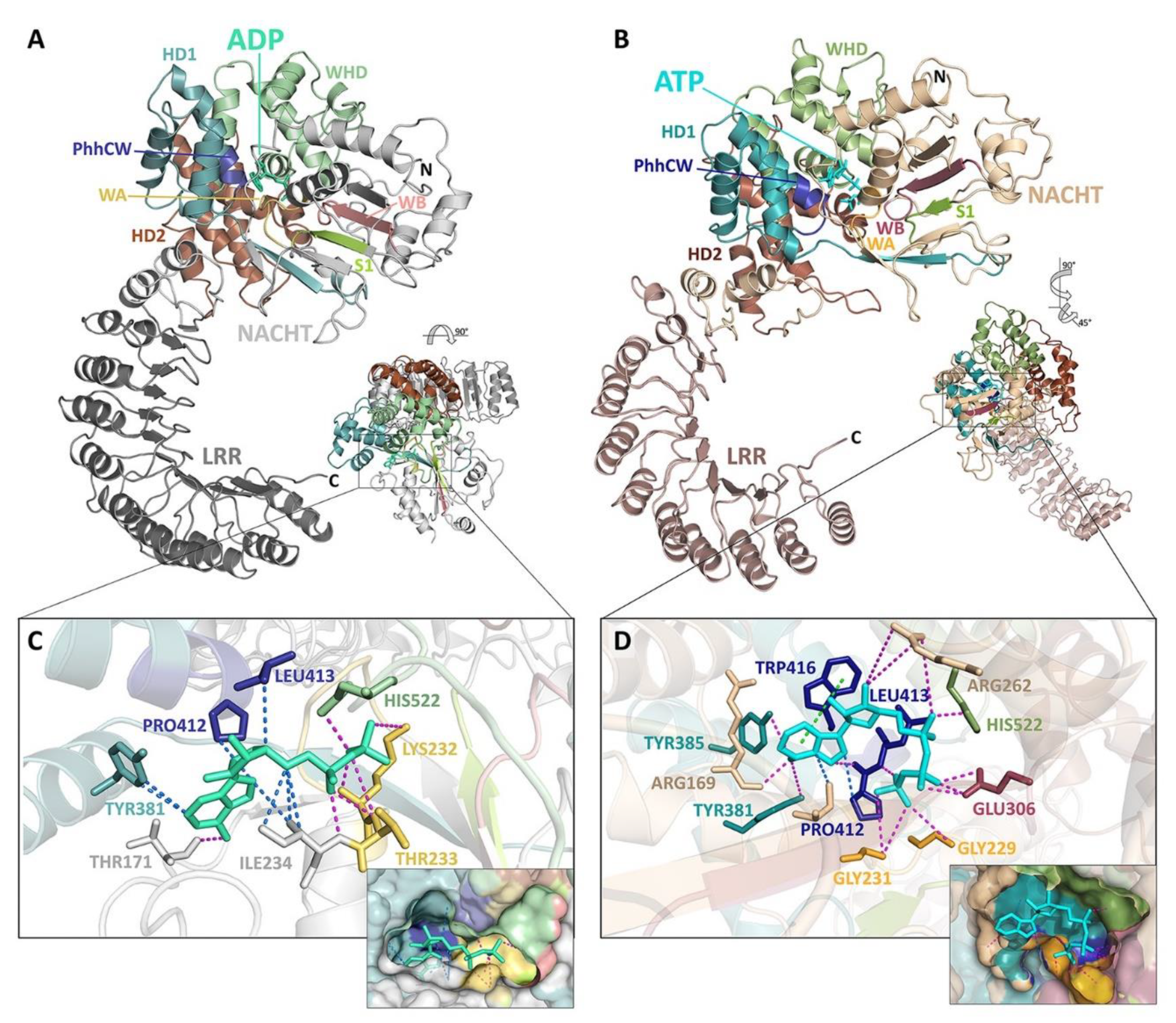
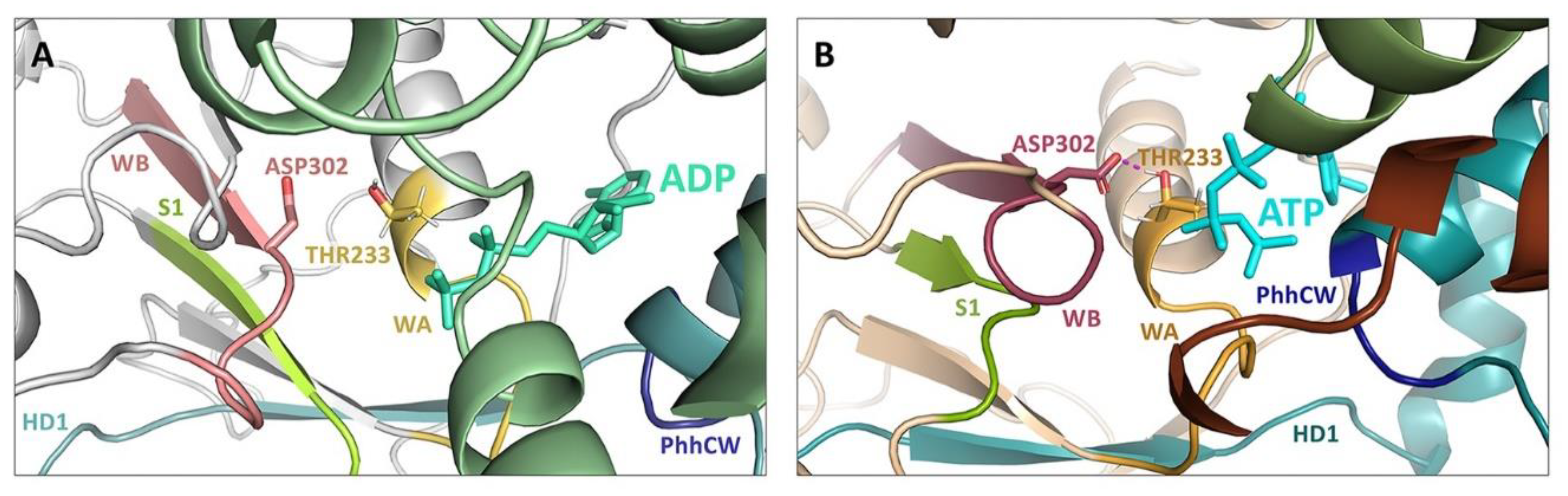
5. Structural Basis for Inflammasome Assembly Mechanisms
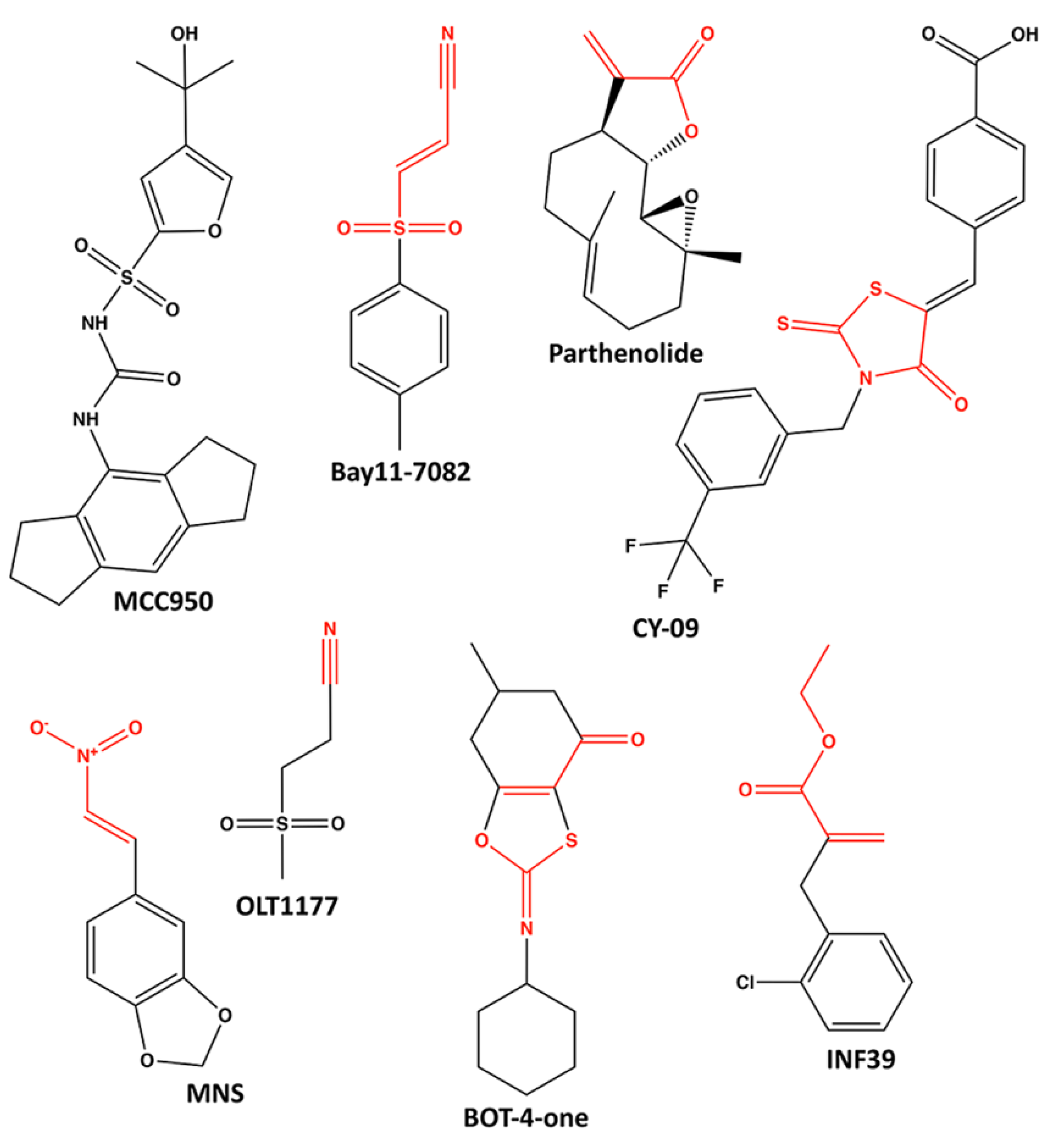
6. Pharmacological Inhibitors of NLRP3 ATPase Activity
6.1. MCC950
6.2. Parthenolide and Bay11-7082
6.3. CY-09
6.4. 3,4-Methylenedioxy-β-Nitrostyrene (MNS)
6.5. OLT1177 (Dapansutrile)
6.6. BOT-4-One
6.7. INF39
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vajjhala, P.R.; Ve, T.; Bentham, A.; Stacey, K.J.; Kobe, B. The molecular mechanisms of signaling by cooperative assembly formation in innate immunity pathways. Mol. Immunol. 2017, 86, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Franz, K.M.; Kagan, J.C. Innate immune receptors as competitive determinants of cell fate. Mol. Cell 2017, 66, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Ann. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef]
- Ting, J.P.Y.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Aravind, L. The NACHT family-A new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem. Sci. 2000, 25, 223–224. [Google Scholar] [CrossRef]
- Danot, O.; Marquenet, E.; Vidal-Ingigliardi, D.; Richet, E. Wheel of life, wheel of death: A mechanistic insight into signaling by STAND proteins. Structure 2009, 17, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Leipe, D.D.; Koonin, E.V.; Aravind, L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: Multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 2004, 343, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, P.; Acharya, V. Computational identification raises a riddle for distribution of putative NACHT NTPases in the genome of early green plants. PLoS ONE 2016, 11, e0150634. [Google Scholar] [CrossRef]
- Hu, Z.; Yan, C.; Liu, P.; Huang, Z.; Ma, R.; Zhang, C.; Wang, R.; Zhang, Y.; Martinon, F.; Miao, D.; et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 2013, 341, 172–175. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Mathur, A.; Hayward, J.A.; Man, S.M. Molecular mechanisms of inflammasome signaling. J. Leukoc. Biol. 2018, 103, 233–257. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Mace, P.D.; Riedl, S.J. Structural mechanisms in NLR inflammasome signaling. Curr. Opin. Struct. Biol. 2014, 29, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Hauenstein, A.V.; Zhang, L.; Wu, H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr. Opin. Struct. Biol. 2015, 31, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Yang, L.; Li, H. The innate immune signaling in cancer and cardiometabolic diseases: Friends or foes? Cancer Lett. 2017, 387, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Conforti-Andreoni, C.; Ricciardi-Castagnoli, P.; Mortellaro, A. The inflammasomes in health and disease: From genetics to molecular mechanisms of autoinflammation and beyond. Cell. Mol. Immunol. 2011, 8, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Christgen, S.; Kanneganti, T.D. Inflammasomes and the fine line between defense and disease. Curr. Opin. Immunol. 2020, 62, 39–44. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- de Alba, E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch. Biochem. Biophys. 2019, 670, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019, 364, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswamis, C.; Bertin, J.J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frew, B.C.; Joag, V.R.; Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 2012, 8, e1002659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef]
- Poyet, J.L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef] [Green Version]
- Nour, A.M.; Yeung, Y.G.; Santambrogio, L.; Boyden, E.D.; Stanley, E.R.; Brojatsch, J. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect. Immun. 2009, 77, 1262–1271. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Manji, G.A.; Wang, L.; Geddes, B.J.; Brown, M.; Merriam, S.; Al-Garawi, A.; Mak, S.; Lora, J.M.; Briskin, M.; Jurman, M.; et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-κB. J. Biol. Chem. 2002, 277, 11570–11575. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lin, G.; Han, Z.; Chai, J. Structural biology of NOD-like receptors. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; pp. 119–141. [Google Scholar]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P.Y. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Lich, J.D.; Moore, C.B.; Duncan, J.A.; Williams, K.L.; Ting, J.P.-Y. ATP binding by Monarch-1/NLRP12 Is critical for its inhibitory function. Mol. Cell. Biol. 2008, 28, 1841–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radian, A.D.; Khare, S.; Chu, L.H.; Dorfleutner, A.; Stehlik, C. ATP binding by NLRP7 is required for inflammasome activation in response to bacterial lipopeptides. Mol. Immunol. 2015, 67, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, L.; Holland, L.; Christodoulou, E.; Kunzelmann, S.; Esposito, D.; Rittinger, K. The biophysical characterisation and SAXS analysis of human NLRP1 uncover a new level of complexity of NLR proteins. PLoS ONE 2016, 11, e0164662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurek, B.; Proell, M.; Wagner, R.N.; Schwarzenbacher, R.; Kufer, T.A. Mutational analysis of human NOD1 and NOD2 NACHT domains reveals different modes of activation. Innate Immun. 2012, 18, 100–111. [Google Scholar] [CrossRef]
- Maharana, J.; Sahoo, B.R.; Bej, A.; Jena, I.; Parida, A.; Sahoo, J.R.; Dehury, B.; Patra, M.C.; Martha, S.R.; Balabantray, S.; et al. Structural models of zebrafish (Danio rerio) NOD1 and NOD2 NACHT domains suggest differential ATP binding orientations: Insights from computational modeling, docking and molecular dynamics simulations. PLoS ONE 2015, 10, e01215415. [Google Scholar] [CrossRef]
- Maharana, J.; Panda, D.; De, S. Deciphering the ATP-binding mechanism(s) in NLRP-NACHT 3D models using structural bioinformatics approaches. PLoS ONE 2018, 13, e0209420. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ardlie, K.G.; DeLuca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; De Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [Green Version]
- Bahar Halpern, K.; Caspi, I.; Lemze, D.; Levy, M.; Landen, S.; Elinav, E.; Ulitsky, I.; Itzkovitz, S. Nuclear Retention of mRNA in Mammalian Tissues. Cell Rep. 2015, 13, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, C.D.C.C.; Noz, M.P.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Keating, S.T. Epigenetics and trained immunity. Antioxid. Redox Signal. 2018, 29, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Schlitzer, A.; Placek, K.; Joosten, L.A.B.; Schultze, J.L. Innate and adaptive immune memory: Aan evolutionary continuum in the host’s response to pathogens. Cell Host Microbe 2019, 25, 13–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, S.; Jeffrey, K.L. Beyond receptors and signaling: Epigenetic factors in the regulation of innate immunity. Immunol. Cell Biol. 2015, 93, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Fu, J. Novel Insights into the NLRP3 Inflammasome in atherosclerosis. J. Am. Heart Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [Green Version]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018, 172, 162–175.e14. [Google Scholar] [CrossRef] [Green Version]
- Ciraci, C.; Janczy, J.R.; Sutterwala, F.S.; Cassel, S.L. Control of innate and adaptive immunity by the inflammasome. Microbes Infect. 2012, 14, 1263–1270. [Google Scholar] [CrossRef] [Green Version]
- Evavold, C.L.; Kagan, J.C. How inflammasomes inform adaptive immunity. J. Mol. Biol. 2018, 430, 217–237. [Google Scholar] [CrossRef]
- Trunk, G.; Oxenius, A. Innate instruction of CD4+ T cell immunity in respiratory bacterial infection. J. Immunol. 2012, 189, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Pedra, J.H.F.; Sutterwala, F.S.; Sukumaran, B.; Ogura, Y.; Qian, F.; Montgomery, R.R.; Flavell, R.A.; Fikrig, E. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-γ axis during Anaplasma phagocytophilum infection. J. Immunol. 2007, 179, 4783–4791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1Β-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Felley, L.E.; Sharma, A.; Theisen, E.; Romero-Masters, J.C.; Sauer, J.-D.; Gumperz, J.E. Human invariant NKT cells induce IL-1β secretion by peripheral blood monocytes via a P2X7-independent pathway. J. Immunol. 2016, 197, 2455–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarda, G.; Dostert, C.; Staehli, F.; Cabalzar, K.; Castillo, R.; Tardivel, A.; Schneider, P.; Tschopp, J. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 2009, 460, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Dickerman, A.W.; Michelmore, R.W.; Sivaramakrishnan, S.; Sobral, B.W.; Young, N.D. Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J. 1999, 20, 317–332. [Google Scholar] [CrossRef]
- Tameling, W.I.L.; Elzinga, S.D.J.; Darmin, P.S.; Vossen, J.H.; Takken, F.L.W.; Haring, M.A.; Cornelissen, B.J.C. The tomato R gene products i-2 and Mi-1 are functional ATP binding proteins with ATPase activity. Plant Cell 2002, 14, 2929–2939. [Google Scholar] [CrossRef]
- Chinnaiyan, A.; Chaudhary, D.; O’Rourke, K.; Koonin, E.V.; Dixit, V.M. Role of CED-4 in the activation of CED-3. Nature 1997, 388, 728–729. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAf-1 · cytochrome C multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J. Biol. Chem. 2000, 275, 31199–31203. [Google Scholar] [CrossRef] [Green Version]
- van der Biezen, E.A.; Jones, J.D. The NB-ARC domain: A novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. Curr. Biol. 1998, 8, R226–R227. [Google Scholar] [CrossRef] [Green Version]
- Ammelburg, M.; Frickey, T.; Lupas, A.N. Classification of AAA+ proteins. J. Struct. Biol. 2006, 156, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New insights into apoptosome structure and function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Fusco, W.G.; Duncan, J.A. Novel aspects of the assembly and activation of inflammasomes with focus on the NLRC4 inflammasome. Int. Immunol. 2018, 30, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef]
- Hughes, A.L. Evolutionary relationships of vertebrate NACHT domain-containing proteins. Immunogenetics 2006, 58, 785–791. [Google Scholar] [CrossRef]
- Proell, M.; Riedl, S.J.; Fritz, J.H.; Rojas, A.M.; Schwarzenbacher, R. The Nod-Like Receptor (NLR) family: A tale of similarities and differences. PLoS ONE 2008, 3, e2119. [Google Scholar] [CrossRef]
- Tian, X.; Pascal, G.; Monget, P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol. Biol. 2009, 9, 202. [Google Scholar] [CrossRef] [Green Version]
- Liao, K.C.; Sandall, C.F.; Carlson, D.A.; Ulke-Lemée, A.; Platnich, J.M.; Hughes, P.F.; Muruve, D.A.; Haystead, T.A.J.; MacDonald, J.A. Application of immobilized ATP to the study of NLRP inflammasomes. Arch. Biochem. Biophys. 2019, 670, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Yan, N.; Chai, J.; Eui, S.L.; Gu, L.; Liu, Q.; He, J.; Wu, J.W.; Kokel, D.; Li, H.; Hao, Q.; et al. Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature 2005, 437, 831–837. [Google Scholar] [CrossRef]
- Murphy, J.M.; Farhan, H.; Eyers, P.A. Bio-Zombie: The rise of pseudoenzymes in biology. Biochem. Soc. Trans. 2017, 45, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Mace, P.D.; Eyers, P.A. Live and let die: Insights into pseudoenzyme mechanisms from structure. Curr. Opin. Struct. Biol. 2017, 47, 95–104. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.A.; Wijekoon, C.P.; Liao, K.C.; Muruve, D.A. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life 2013, 65, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Sandall, C.F.; MacDonald, J.A. Effects of phosphorylation on the NLRP3 inflammasome. Arch. Biochem. Biophys. 2019, 670, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011, 477, 592–597. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–602. [Google Scholar] [CrossRef]
- Lightfield, K.L.; Persson, J.; Trinidad, N.J.; Brubaker, S.W.; Kofoed, E.M.; Sauer, J.D.; Dunipace, E.A.; Warren, S.E.; Miao, E.A.; Vance, R.E. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect. Immun. 2011, 79, 1606–1614. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef] [Green Version]
- Tenthorey, J.L.; Kofoed, E.M.; Daugherty, M.D.; Malik, H.S.; Vance, R.E. Molecular Basis for Specific Recognition of Bacterial Ligands by NAIP/NLRC4 Inflammasomes. Mol. Cell 2014, 54, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Diebolder, C.A.; Halff, E.F.; Koster, A.J.; Huizinga, E.G.; Koning, R.I. Cryoelectron Tomography of the NAIP5/NLRC4 Inflammasome: Implications for NLR Activation. Structure 2015, 23, 2349–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Zhou, Q.; Zhang, C.; Fan, S.; Cheng, W.; Zhao, Y.; Shao, F.; Wang, H.W.; Sui, S.F.; Chai, J. Structural and biochemical basis for induced self-propagation of NLRC4. Science 2015, 350, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, S.; Ruan, J.; Wu, J.; Tong, A.B.; Yin, Q.; Li, Y.; David, L.; Lu, A.; Wang, W.L.; et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 2015, 350, 404–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fu, T.M.; Lu, A.; Witt, K.; Ruan, J.; Shen, C.; Wu, H. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl. Acad. Sci. USA 2018, 115, 10845–10852. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Wang, A.; Wang, L.; Dorsch, M.; Ocain, T.D.; Xu, Y. Nucleotide binding to CARD12 and its role in CARD12-mediated caspase-1 activation. Biochem. Biophys. Res. Commun. 2005, 331, 1114–1119. [Google Scholar] [CrossRef]
- Canna, S.W.; De Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; Dimattia, M.A.; Zaal, K.J.M.; Sanchez, G.A.M.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.S.; Sandstrom, A.; Vance, R.E. The NLRP1 inflammasome: New mechanistic insights and unresolved mysteries. Curr. Opin. Immunol. 2019, 60, 37–45. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Griswold, A.R.; Bachovchin, D.A. The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 2020, 297, 13–25. [Google Scholar] [CrossRef]
- Harris, P.A.; Duraiswami, C.; Fisher, D.T.; Fornwald, J.; Hoffman, S.J.; Hofmann, G.; Jiang, M.; Lehr, R.; McCormick, P.M.; Nickels, L.; et al. High throughput screening identifies ATP-competitive inhibitors of the NLRP1 inflammasome. Bioorganic Med. Chem. Lett. 2015, 25, 2739–2743. [Google Scholar] [CrossRef]
- Liao, K.C.; Mogridge, J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect. Immun. 2013, 81, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Yamashita, A.; Matsuda, M.; Kawai, K.; Sawa, T.; Amaya, F. NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain 2019, 160, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Bruey-Sedano, N.; Newman, R.; Chandler, S.; Stehlik, C.; Reed, J.C. PAN1/ NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-κB and caspase-1 activation in macrophages. J. Biol. Chem. 2004, 279, 51897–51907. [Google Scholar] [CrossRef] [Green Version]
- Tilburgs, T.; Meissner, T.B.; Ferreira, L.M.R.; Mulder, A.; Musunuru, K.; Ye, J.; Strominger, J.L. NLRP2 is a suppressor of NF-κB signaling and HLA-C expression in human trophoblasts. Biol. Reprod. 2017, 96, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.N.; Pascarella, A.; Licursi, V.; Caiello, I.; Taranta, A.; Rega, L.R.; Levtchenko, E.; Emma, F.; De Benedetti, F.; Prencipe, G. NLRP2 Regulates Proinflammatory and Antiapoptotic Responses in Proximal Tubular Epithelial Cells. Front. Cell Dev. Biol. 2019, 7, 252. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Chang, B.; Lu, C.; Su, J.; Wu, Y.; Lv, P.; Wang, Y.; Liu, J.; Zhang, B.; Quan, F.; et al. Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PLoS ONE 2012, 7, 30344. [Google Scholar] [CrossRef] [Green Version]
- Kuchmiy, A.A.; D’Hont, J.; Hochepied, T.; Lamkanfi, M. NLRP2 controls age-associated maternal fertility. J. Exp. Med. 2016, 213, 2851–2860. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Liu, H.; Liu, F.; Gao, Y.; Chen, J.; Huo, J.; Han, J.; Xiao, T.; Zhang, W. NLRP2 and FAF1 deficiency blocks early embryogenesis in the mouse. Reproduction 2017, 154, 245–251. [Google Scholar] [CrossRef]
- Mahadevan, S.; Sathappan, V.; Utama, B.; Lorenzo, I.; Kaskar, K.; Van Den Veyver, I.B. Maternally expressed NLRP2 links the subcortical maternal complex (SCMC) to fertility, embryogenesis and epigenetic reprogramming. Sci. Rep. 2017, 7, 44667. [Google Scholar] [CrossRef]
- Mu, J.; Wang, W.; Chen, B.; Wu, L.; Li, B.; Mao, X.; Zhang, Z.; Fu, J.; Kuang, Y.; Sun, X.; et al. Mutations in NLRP2 and NLRP5 cause female infertility characterised by early embryonic arrest. J. Med. Genet. 2019, 56, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Fontalba, A.; Gutierrez, O.; Fernandez-Luna, J.L. NLRP2, an inhibitor of the NF-κB pathway, is transcriptionally activated by NF-κB and exhibits a nonfunctional allelic variant. J. Immunol. 2007, 179, 8519–8524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- van den Berg, D.F.; te Velde, A.A. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Deftereos, S.G.; Siasos, G.; Giannopoulos, G.; Vrachatis, D.A.; Angelidis, C.; Giotaki, S.G.; Gargalianos, P.; Giamarellou, H.; Gogos, C.; Daikos, G.; et al. The Greek study in the effects of colchicine in Covid-19 complications prevention (GRECCO-19 study): Rationale and study design. Hell. J. Cardiol. 2020, 61, 42–45. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Magliarelli, H.F.; Mailly, L.; Mihlan, M.; Liu, Y.; Gámez, M.P.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Hui Bu, C.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Tartey, S.; Kanneganti, T.D. Inflammasomes in the pathophysiology of autoinflammatory syndromes. J. Leukoc. Biol. 2020, 107, 379–391. [Google Scholar] [CrossRef]
- Touitou, I.; Lesage, S.; McDermott, M.; Cuisset, L.; Hoffman, H.; Dode, C.; Shoham, N.; Aganna, E.; Hugot, J.P.; Wise, C.; et al. Infevers: An evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 2004, 24, 194–198. [Google Scholar] [CrossRef]
- Meng, G.; Strober, W. New insights into the nature of autoinflammatory diseases from mice with Nlrp3 mutations. Eur. J. Immunol. 2010, 40, 649–653. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.K.; Kanneganti, T.D. NLRP6 in infection and inflammation. Microbes Infect. 2013, 15, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef]
- Ghimire, L.; Paudel, S.; Jin, L.; Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020, 13, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhu, S.; Yang, L.; Cui, S.; Pan, W.; Jackson, R.; Zheng, Y.; Rongvaux, A.; Sun, Q.; Yang, G.; et al. Nlrp6 regulates intestinal antiviral innate immunity. Science. 2015, 350, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seregin, S.S.; Golovchenko, N.; Schaf, B.; Chen, J.; Eaton, K.A.; Chen, G.Y. NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury. Mucosal Immunol. 2017, 10, 434–445. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.K.; Subbarao Malireddi, R.K.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Lu, A.; Xie, W.J.; Ruan, J.; Negro, R.; Egelman, E.H.; Fu, T.M.; Wu, H. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2052–2057. [Google Scholar] [CrossRef] [Green Version]
- Leng, F.; Yin, H.; Qin, S.; Zhang, K.; Guan, Y.; Fang, R.; Wang, H.; Li, G.; Jiang, Z.; Sun, F.; et al. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci. Rep. 2020, 10, 198. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-Containing Inflammasome Mediates Recognition of Microbial Lipopeptides in Human Macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shah, S.Z.A.; Yang, L.; Zhang, Z.; Zhou, X.; Zhao, D. Virulent Mycobacterium bovis Beijing strain activates the NLRP7 inflammasome in THP-1 macrophages. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, A.S.; Proell, M.; Eibl, C.; Page, R.; Schwarzenbacher, R.; Peti, W. Three-dimensional structure of the NLRP7 pyrin domain insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J. Biol. Chem. 2010, 285, 27402–27410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, H.; Biswas, A.; Zimmer, N.; Messaed, C.; Oldenburg, J.; Slim, R.; El-Maarri, O. NLRP7 inter-domain interactions: The NACHT-associated domain is the physical mediator for oligomeric assembly. Mol. Hum. Reprod. 2014, 20, 990–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.J.; Park, H.H. Crystal structure of the human NLRP9 pyrin domain reveals a bent N-terminal loop that may regulate inflammasome assembly. FEBS Lett. 2020, 594, 2396–2405. [Google Scholar] [CrossRef] [PubMed]
- Marleaux, M.; Anand, K.; Latz, E.; Geyer, M. Crystal structure of the human NLRP9 pyrin domain suggests a distinct mode of inflammasome assembly. FEBS Lett. 2020, 594, 2383–2395. [Google Scholar] [CrossRef]
- Wang, L.; Manji, G.A.; Grenier, J.M.; Al-Garawi, A.; Merriam, S.; Lora, J.M.; Geddes, B.J.; Briskin, M.; DiStefano, P.S.; Bertin, J. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J. Biol. Chem. 2002, 277, 29874–29880. [Google Scholar] [CrossRef] [Green Version]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.M.; Vanaja, S.K.; Rathinam, V.A.K.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 Inflammasome Recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Tuncer, S.; Fiorillo, M.T.; Sorrentino, R. The multifaceted nature of NLRP12. J. Leukoc. Biol. 2014, 96, 991–1000. [Google Scholar] [CrossRef]
- Jéru, I.; Le Borgne, G.; Cochet, E.; Hayrapetyan, H.; Duquesnoy, P.; Grateau, G.; Morali, A.; Sarkisian, T.; Amselem, S. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum. 2011, 63, 1459–1464. [Google Scholar] [CrossRef]
- Jéru, I.; Duquesnoy, P.; Fernandes-Alnemri, T.; Cochet, E.; Yu, J.W.; Lackmy-Port-Lis, M.; Grimprel, E.; Landman-Parker, J.; Hentgen, V.; Marlin, S.; et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 2008, 105, 1614–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Whiteheart, S.W.; Wilkinson, A.J. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 2004, 146, 106–112. [Google Scholar] [CrossRef]
- Yang, X.; Yang, F.; Wang, W.; Lin, G.; Hu, Z.; Han, Z.; Qi, Y.; Zhang, L.; Wang, J.; Sui, S.F.; et al. Structural basis for specific flagellin recognition by the NLR protein NAIP5. Cell Res. 2018, 28, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Davoodi, J.; Lin, L.; Kelly, J.; Liston, P.; MacKenzie, A.E. Neuronal apoptosis-inhibitory protein does not interact with Smac and requires ATP to bind caspase-9. J. Biol. Chem. 2004, 279, 40622–40628. [Google Scholar] [CrossRef] [Green Version]
- Karimpour, S.; Davoodi, J.; Ghahremani, M.H. Integrity of ATP binding site is essential for effective inhibition of the intrinsic apoptosis pathway by NAIP. Biochem. Biophys. Res. Commun. 2011, 407, 158–162. [Google Scholar] [CrossRef]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [Green Version]
- Sahillioglu, A.C.; Sumbul, F.; Ozoren, N.; Haliloglu, T. Structural and dynamics aspects of ASC speck assembly. Structure 2014, 22, 1722–1734. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Li, Y.; Yin, Q.; Ruan, J.; Yu, X.; Egelman, E.; Wu, H. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 2015, 1, 15013. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Li, Y.; Schmidt, F.I.; Yin, Q.; Chen, S.; Fu, T.M.; Tong, A.B.; Ploegh, H.L.; Mao, Y.; Wu, H. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 2016, 23, 416–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharana, J.; Dehury, B.; Sahoo, J.R.; Jena, I.; Bej, A.; Panda, D.; Sahoo, B.R.; Patra, M.C.; Pradhan, S.K. Structural and functional insights into CARDs of zebrafish (Danio rerio) NOD1 and NOD2, and their interaction with adaptor protein RIP2. Mol. Biosyst. 2015, 11, 2324–2336. [Google Scholar] [CrossRef] [PubMed]
- Maharana, J.; Pradhan, S.K.; De, S. NOD1CARD might be using multiple interfaces for RIP2-mediated CARD-CARD interaction: Insights from molecular dynamics simulation. PLoS ONE 2017, 12, e0170232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharana, J.; Patra, M.C.; De, B.C.; Sahoo, B.R.; Behera, B.K.; De, S.; Pradhan, S.K. Structural insights into the MDP binding and CARD-CARD interaction in zebrafish (Danio rerio) NOD2: A molecular dynamics approach. J. Mol. Recognit. 2014, 27, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Maharana, J.; Vats, A.; Gautam, S.; Nayak, B.P.; Kumar, S.; Sendha, J.; De, S. POP1 might be recruiting its type-Ia interface for NLRP3-mediated PYD-PYD interaction: Insights from MD simulation. J. Mol. Recognit. 2017, 30, e2632. [Google Scholar] [CrossRef]
- Maharana, J. Elucidating the interfaces involved in CARD-CARD interactions mediated by NLRP1 and Caspase-1 using molecular dynamics simulation. J. Mol. Graph. Model. 2018, 80, 7–14. [Google Scholar] [CrossRef]
- Huber, R.G.; Eibl, C.; Fuchs, J.E. Intrinsic flexibility of NLRP pyrin domains is a key factor in their conformational dynamics, fold stability, and dimerization. Protein Sci. 2015, 24, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Reubold, T.F.; Wohlgemuth, S.; Eschenburg, S. Crystal structure of full-length Apaf-1: How the death signal is relayed in the mitochondrial pathway of apoptosis. Structure 2011, 19, 1074–1083. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Topf, M.; Reubold, T.F.; Eschenburg, S.; Akey, C.W. Changes in Apaf-1 conformation that drive apoptosome assembly. Biochemistry 2013, 52, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Halff, E.F.; Diebolder, C.A.; Versteeg, M.; Schouten, A.; Brondijk, T.H.C.; Huizinga, E.G. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 2012, 287, 38460–38472. [Google Scholar] [CrossRef] [Green Version]
- Reubold, T.F.; Wohlgemuth, S.; Eschenburg, S. A new model for the transition of APAF-1 from inactive monomer to caspase-activating apoptosome. J. Biol. Chem. 2009, 284, 32717–32724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanasyeva, A.; Hirtreiter, A.; Schreiber, A.; Grohmann, D.; Pobegalov, G.; McKay, A.R.; Tsaneva, I.; Petukhov, M.; Käs, E.; Grigoriev, M.; et al. Lytic water dynamics reveal evolutionarily conserved mechanisms of ATP hydrolysis by TIP49 AAA+ ATPases. Structure 2014, 22, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, S.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Crystal structure of NOD2 and its implications in human disease. Nat. Commun. 2016, 7, 11813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenthorey, J.L.; Haloupek, N.; López-Blanco, J.R.; Grob, P.; Adamson, E.; Hartenian, E.; Lind, N.A.; Bourgeois, N.M.; Chacón, P.; Nogales, E.; et al. The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 2017, 358, 888–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner-Bratkovič, I.; Sušjan, P.; Lainšček, D.; Tapia-Abellán, A.; Cerović, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrίn, P.; Jerala, R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018, 9, 5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowds, T.A.; Masumoto, J.; Zhu, L.; Inohara, N.; Núñez, G. Cryopyrin-induced interleukin 1β secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 2004, 279, 21924–21928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Agostini, L.; Meylan, E.; Tschopp, J. Identification of bacterial muramyl dipeptide as activator of the NALP3/Cryopyrin inflammasome. Curr. Biol. 2004, 14, 1929–1934. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Cuddy, M.; Shu, V.C.W.; Yip, K.W.; Madiraju, C.; Diaz, P.; Matsuyama, T.; Kaibara, M.; Taniyama, K.; Vasile, S.; et al. Versatile assays for high throughput screening for activatorsor inhibitors of intracellular proteases and their cellular regulators. PLoS ONE 2009, 4, e7655. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comp. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E. CHARMM General Force Field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsletter on Protein Crystallography 2002, 40, 82–92. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulte, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological inhibitors of the NLRP3 inflammasome. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Wang, W.; Liu, F.; Li, H. Recent progress on the discovery of NLRP3 inhibitors and their therapeutic potential. Curr. Med. Chem. 2020, 27, 1–14. [Google Scholar] [CrossRef]
- Perregaux, D.G.; McNiff, P.; Laliberte, R.; Hawryluk, N.; Peurano, H.; Stam, E.; Eggler, J.; Griffiths, R.; Dombroski, M.A.; Gabel, C.A. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 2001, 299, 187–197. [Google Scholar]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; de Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Groß, C.J.; Mishra, R.; Schneider, K.S.; Médard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.A.; Fromm, S.; et al. K+ Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human monocytes engage an alternative inflammasome pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quang, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Rhee, M.H.; Kim, E.; Cho, J.Y. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat. Inflamm. 2012, 2012, 416036. [Google Scholar] [CrossRef]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent modifiers: A chemical perspective on the reactivity of α, β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef]
- Kerr, I.D.; Lee, J.J.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Bertinaria, M.; Gastaldi, S.; Marini, E.; Giorgis, M. Development of covalent NLRP3 inflammasome inhibitors: Chemistry and biological activity. Arch. Biochem. Biophys. 2019, 670, 116–139. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.J.V.; Verkman, A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin–induced intestinal fluid secretion. J. Clin. Invest. 2002, 110, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Varadarajan, S.; Mūnoz-Planillo Burberry, A.; Nakamura, Y.; Núñez, G. 3,4-Methylenedioxy-β’-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 2014, 289, 1142–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Y.; Wu, Y.C.; Wu, C.C. Prevention of platelet glycoprotein IIb/IIIa activation by 3,4-methylenedioxy-β-nitrostyrene, a novel tyrosine kinase inhibitor. Mol. Pharmacol. 2006, 70, 1380–1389. [Google Scholar] [CrossRef]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Koenders, M.I.; Azam, T.; Tengesdal, I.W.; Powers, N.; de Graff, D.M.; Dinarellow, C.A.; Joosten, L.A.B. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res. Ther. 2018, 20, 169. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [Green Version]
- Shim, D.-W.; Shin, W.-Y.; Yu, S.-H.; Kim, B.-H.; Ye, S.-K.; Koppula, S.; Won, H.-S.; Kang, T.-B.; Lee, K.-H. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci. Rep. 2017, 7, 15020. [Google Scholar] [CrossRef]
- Lee, H.G.; Cho, N.-C.; Jeong, A.J.; Li, Y.-C.; Rhie, S.-J.; Choi, J.S.; Lee, K.-H.; Kim, Y.; Kim, Y.-N.; Kim, M.-H.; et al. Immunomodulatory activities of the benzoxathiole derivative BOT-4-one ameliorate pathogenic skin inflammation in mice. J. Invest. Dermatol. 2016, 136, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-H.; Yoon, B.R.; Kim, E.K.; Noh, K.H.; Kwon, S.-H.; Yi, E.H.; Lee, H.G.; Choi, J.S.; Kang, S.W.; Park, I.-C.; et al. Alleviation of collagen-induced arthritis by the benzoxathiole derivative BOT-4-one in mice: Implication of the Th1-and Th17-cell-mediated immune responses. Biochem. Pharmacol. 2016, 110, 47–57. [Google Scholar] [CrossRef]
- Cocco, M.; Garella, D.; Di Stilo, A.; Borretto, E.; Stevanato, L.; Giorgis, M.; Marini, E.; Fantozzi, R.; Miglio, G.; Bertinaria, M. Electrophilic Warhead-Based Design of Compounds Preventing NLRP3 Inflammasome-Dependent Pyroptosis. J. Med. Chem. 2014, 57, 10366–10382. [Google Scholar] [CrossRef] [PubMed]
- Cocco, M.; Pellegrini, C.; Martínez-Banaclocha, H.; Giorgis, M.; Marini, E.; Costale, A.; Miglio, G.; Fornai, M.; Antonioli, L.; López-Castejón, G.; et al. Development of an Acrylate Derivative Targeting the NLRP3 Inflammasome for the Treatment of Inflammatory Bowel Disease. J. Med. Chem. 2017, 60, 3656–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, M.; Miglio, G.; Giorgis, M.; Garella, D.; Marini, E.; Costale, A.; Regazzoni, L.; Vistoli, G.; Orioli, M.; Massulaha-Ahmed, R.; et al. Design, Synthesis, and Evaluation of Acrylamide Derivatives as Direct NLRP3 Inflammasome Inhibitors. ChemMedChem 2016, 11, 1790–1803. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandall, C.F.; Ziehr, B.K.; MacDonald, J.A. ATP-Binding and Hydrolysis in Inflammasome Activation. Molecules 2020, 25, 4572. https://doi.org/10.3390/molecules25194572
Sandall CF, Ziehr BK, MacDonald JA. ATP-Binding and Hydrolysis in Inflammasome Activation. Molecules. 2020; 25(19):4572. https://doi.org/10.3390/molecules25194572
Chicago/Turabian StyleSandall, Christina F., Bjoern K. Ziehr, and Justin A. MacDonald. 2020. "ATP-Binding and Hydrolysis in Inflammasome Activation" Molecules 25, no. 19: 4572. https://doi.org/10.3390/molecules25194572