
Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2: An In Silico Study

 , ,
, ,
Abstract
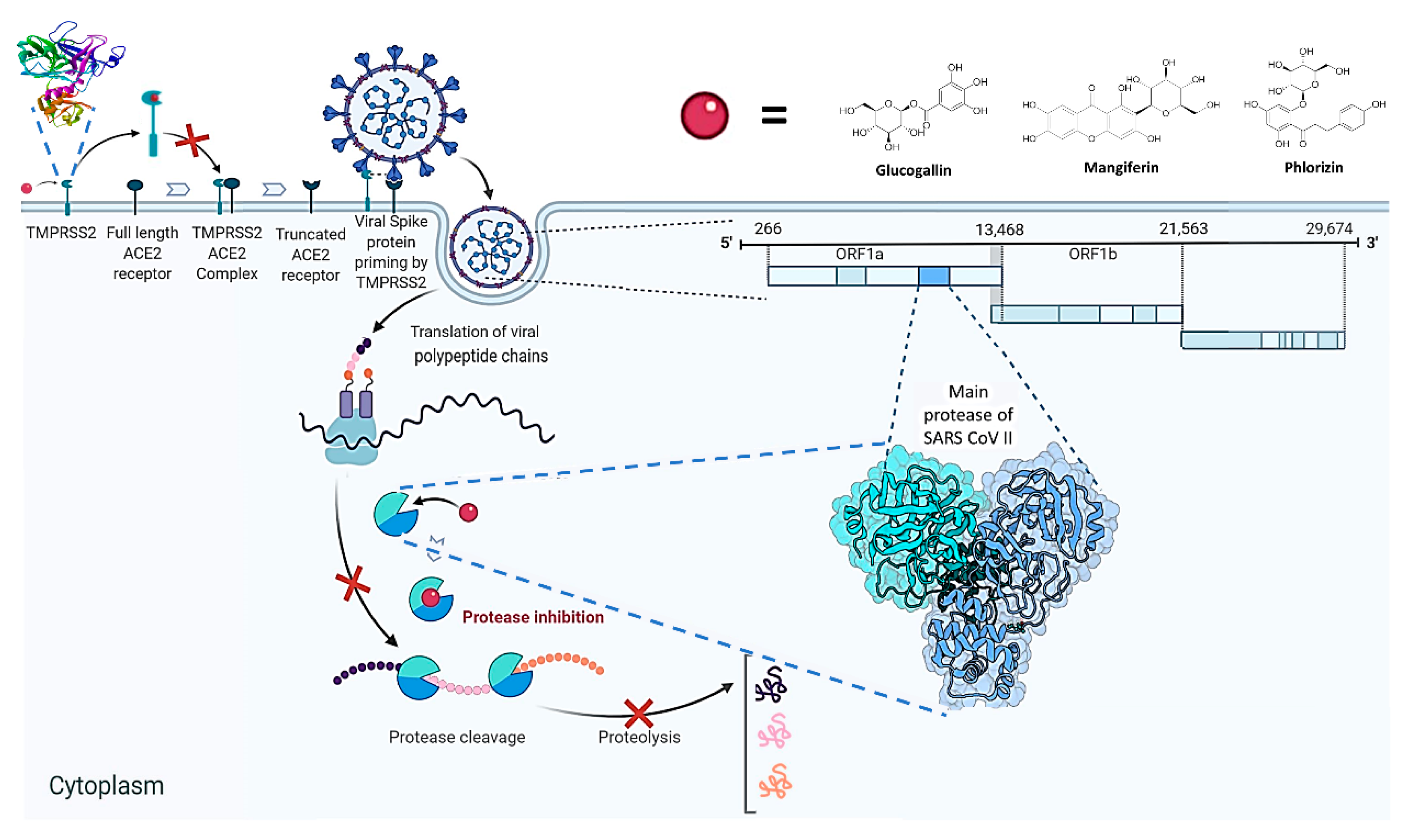
1. Introduction
2. Results
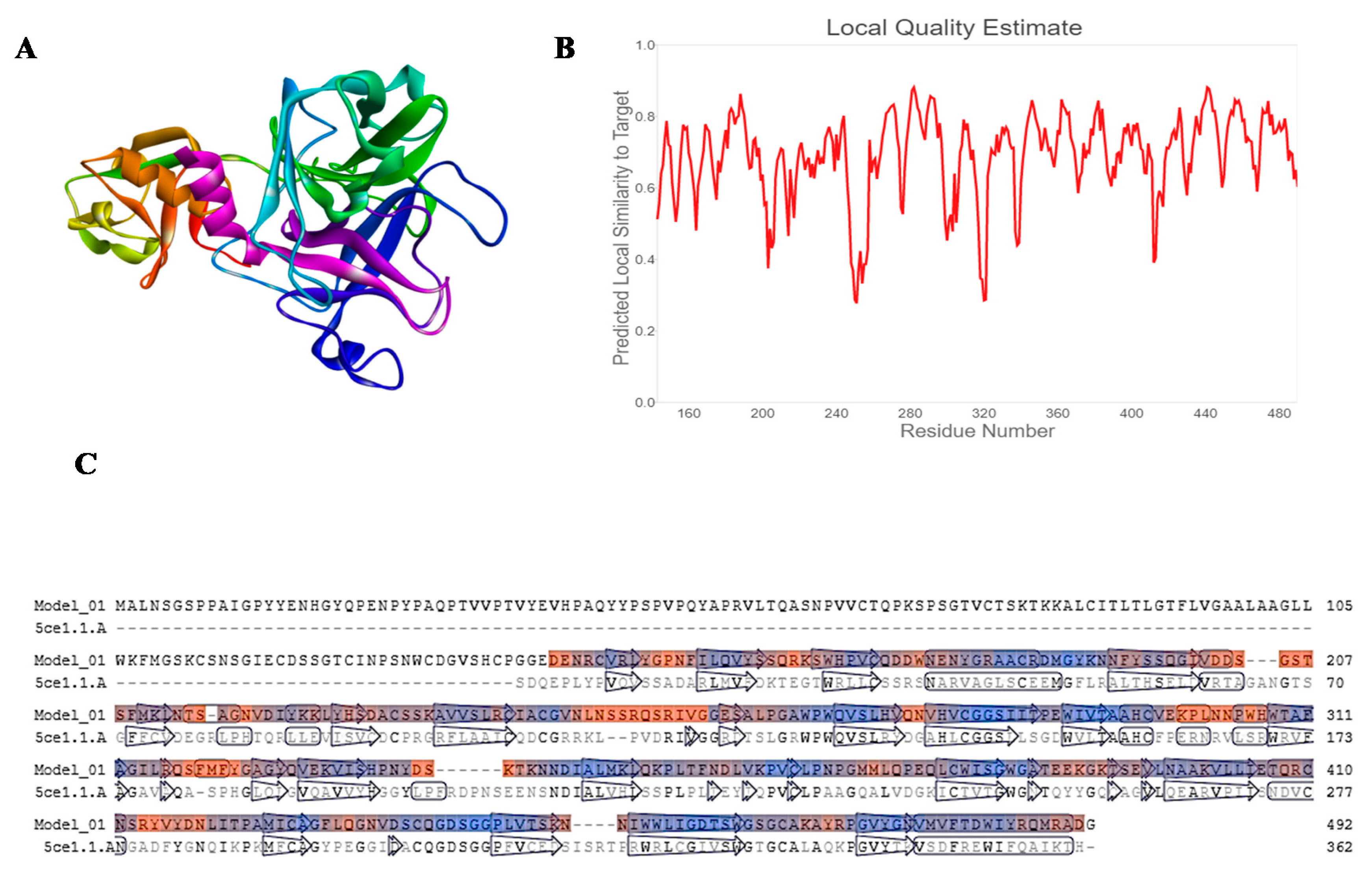
2.1. Homology Modelling
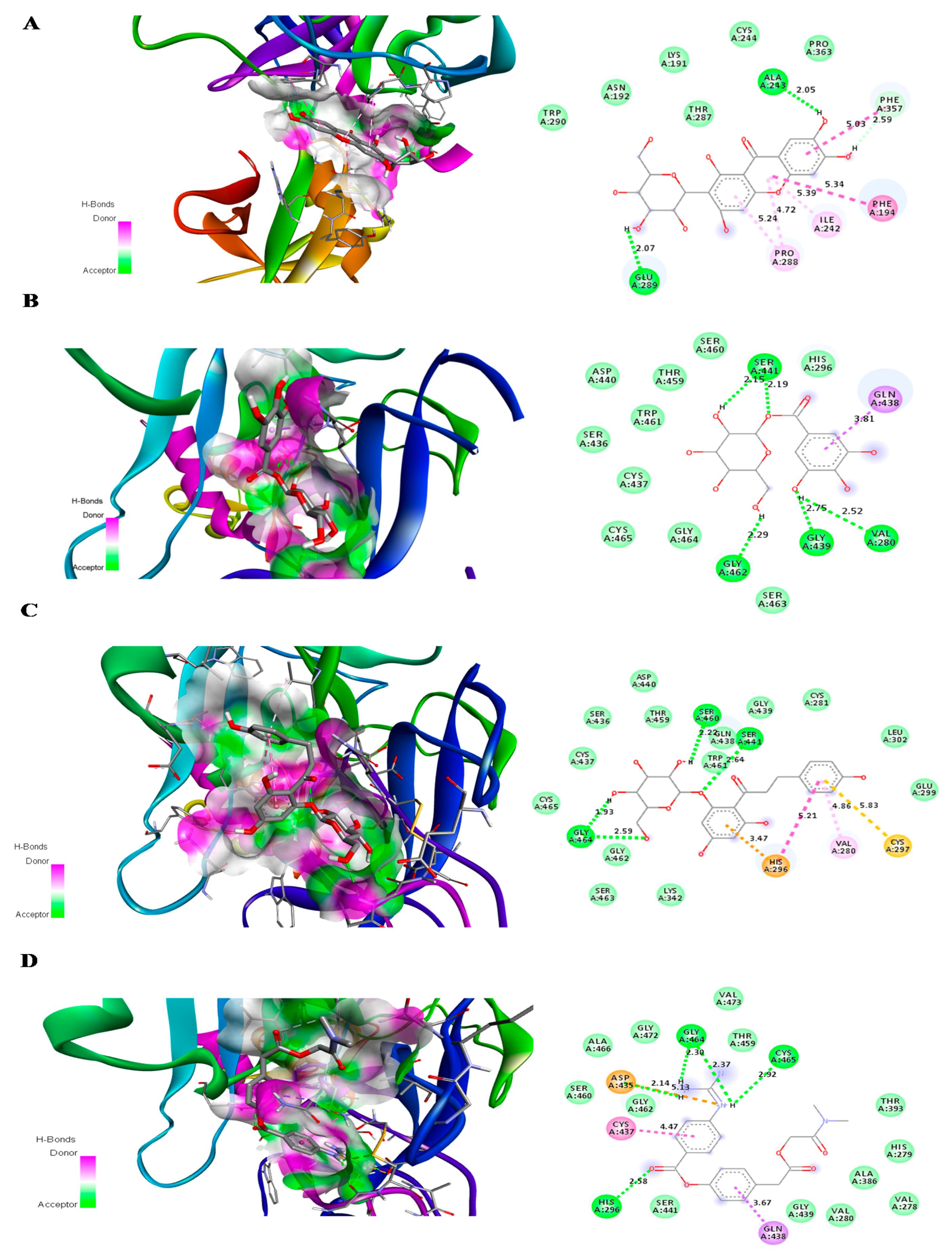
2.2. Molecular Docking
2.3. ADME Prediction
2.4. Prediction of Targets
2.5. Prediction of Toxicity
2.6. Molecular Dynamics Simulation and MM-PBSA Analysis
3. Discussion
4. Material and Methods
4.1. Preparation of Ligand and Protein
4.2. Homology Modelling
4.3. Procedure for Molecular Docking of Ligands and Protein
4.4. ADME Prediction Study
4.5. Prediction of Target
4.6. Prediction of Toxicity
4.7. Molecular Simulation and Energy Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CoV | Coronavirus |
| COVID-19 | Coronavirus disease-19 |
| GROMACS | Groningen Machine for Chemical Simulation |
| hERG | Human Ether-a-go-go-Related Gene |
| HSV-1 | Herpes Simplex Virus-1 |
| log P | Partition Co-efficient |
| MD | Molecular Dynamics |
| Mpro | main protease |
| RMSD | root mean square division |
| RMSF | root mean square fluctuation |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome-Coronavirus-2 |
References
- Zeng, W.; Gautam, A.; Huson, D.H. Enhanced COVID-19 data for improved prediction of survival. bioRxiv 2020. [Google Scholar] [CrossRef]
- Oliver, S.; Vittorio, O.; Cirillo, G.; Boyer, C. Enhancing the therapeutic effects of polyphenols with macromolecules. Polym. Chem. 2016, 7, 1529–1544. [Google Scholar] [CrossRef]
- Tangney, C.C.; Rasmussen, H.E. Polyphenols, inflammation, and cardiovascular disease. Curr. Atheroscler. Rep. 2013, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Aryaeian, N.; Sedehi, S.K.; Arablou, T. Polyphenols and their effects on diabetes management: A review. Med. J. Islam Repub. Iran. 2017, 31, 134. [Google Scholar] [CrossRef] [PubMed]
- Carocho, M.; Ferreira, I.C. The role of phenolic compounds in the fight against cancer—A review. Anticancer Agents Med. Chem. 2013, 13, 1236–1258. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Napolitano, M.; Tedesco, I.; Moccia, S.; Milito, A.; Russo, G.L. Neuroprotectiverole of natural polyphenols. Curr. Top. Med. Chem. 2016, 16, 1943–1950. [Google Scholar] [CrossRef]
- Bouarab-Chibane, L.; Forquet, V.; Lantéri, P.; Clément, Y.; Léonard-Akkari, L.; Oulahal, N.; Degraeve, P.; Bordes, C. Antibacterial properties of polyphenols: Characterization and QSAR (quantitative structure-activity relationship) models. Front. Microbiol. 2019, 10, 829. [Google Scholar] [CrossRef]
- King, A.; Young, G. Characteristics and occurrence of phenolic phytochemicals. J. Am. Diet. Assoc. 1999, 99, 213–218. [Google Scholar] [CrossRef]
- Havranek, B.; Islam, S.M. An in silico approach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J. Biomol. Struct. Dyn. 2020, 1–2. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Tsegaye, T.S.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 2009, 83, 3200–3211. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.S.; Lu, Z.Y. Antiviral effect of mangiferin and isomangiferin on herpes simplex virus. Chin. Med. J. 1990, 103, 160. [Google Scholar]
- Zhu, X.M.; Song, J.X.; Huang, Z.Z.; Wu, Y.M.; Yu, M.J. Antiviral activity of mangiferin against herpes simplex virus type 2 in vitro. Zhongguoyao Li Xue Bao Acta Pharmacol. Sin. 1993, 14, 452–454. [Google Scholar]
- Muruganandan, S.; Lal, J.; Gupta, P.K. Immunotherapeutic effects of mangiferin mediated by the inhibition of oxidative stress to activated lymphocytes, neutrophils and macrophages. Toxicology 2005, 215, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.B.; Coelho, L.P.; Insuela, D.B.; Carvalho, V.F.; dos Santos, M.H.; Silva, P.M.; Martins, M.A. Mangiferin prevents guinea pig tracheal contraction via activation of the nitric oxide-cyclic GMP pathway. PLoS ONE 2013, 8, e71759. [Google Scholar] [CrossRef]
- Gupta, P.; Kanwal, A.; Putcha, U.K.; Bulani, Y.; Sojitra, B.; Khatua, T.N.; Kuncha, M.; Banerjee, S.K. Cardioprotective effect of ritonavir, an antiviral drug, in isoproterenol induced myocardial necrosis: A new therapeutic implication. J. Transl. Med. 2013, 11, 1–6. [Google Scholar] [CrossRef]
- Jourdes, M.; Pouységu, L.; Deffieux, D.; Teissedre, P.L.; Quideau, S. Hydrolyzable tannins: Gallotannins and ellagitannins. In Natural Products; Ramawat, K., Mérillon, J.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the local structural quality of transmembrane protein models using statistical potentials (QMEANBrane). Bioinformatics 2014, 30, i505-11. [Google Scholar] [CrossRef]
- Hall, D.C., Jr.; Ji, H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med Infect Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]
- Arun, K.G.; Sharanya, C.S.; Abhithaj, J.; Francis, D.; Sadasivan, C. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Majumder, R.; Mandal, M. Screening of plant-based natural compounds as a potential COVID-19 main protease inhibitor: An in silico docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Zhang, L.; Duan, Y.; Yu, J.; Wang, L.; Yang, K.; Liu, F.; You, T.; Liu, X.; Yang, X.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293.40. [Google Scholar]
- Mesecar, A.D. A taxonomically-driven approach to development of potent, broad-spectrum inhibitors of coronavirus main protease including SARS-CoV-2 (COVID-19). Be Publ. 2020. [Google Scholar] [CrossRef]
- Santos, K.B.; Guedes, I.A.; Karl, A.L.M.; Dardenne, L.E. Highly flexible ligand docking: Benchmarking of the DockThorprogram on the LEADS-PEP protein-peptide data set. J. Chem. Inf. Model. 2020, 60, 667–683. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 1–3. [Google Scholar] [CrossRef]
- Da Silva Antonio, A.; Wiedemann, L.S.; Veiga-Junior, V.F. Natural products’ role against COVID-19. RSC Adv. 2020, 10, 23379–23393. [Google Scholar] [CrossRef]
- Ngwa, W.; Kumar, R.; Thompson, D.; Lyerly, W.; Moore, R.; Reid, T.-E.; Lowe, H.; Toyang, N. Potential of flavonoid-inspired phytomedicines against COVID-19. Molecules 2020, 25, 2707. [Google Scholar] [CrossRef]
- Leegwater, E.; Strik, A.; Wilms, E.B.; Bosma, L.B.; Burger, D.M.; Ottens, T.H.; van Nieuwkoop, C. Drug-induced liver injury in a COVID-19 patient: Potential interaction of remdesivir with P-glycoprotein inhibitors. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug disCoVery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. Swiss target prediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lee, S.; Tran, A.; Allsopp, M.; Lim, J.B.; Hénin, J.; Klauda, J.B. CHARMM36 united atom chain model for lipids and surfactants. J. Phys. Chem. B 2014, 118, 547–556. [Google Scholar] [CrossRef]
- Boonstra, S.; Onck, P.R.; van der Giessen, E. CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. Open source drug discovery consortium g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the corresponding authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Number of Conventional H Bonding | Residue Receptor | Bond Length(Å) | Docking Score (kcal/mol) | Actual Residue by Experimental Crystal Structure | Ref. |
|---|---|---|---|---|---|---|
| Remdesivir | 5 | THR199(A), ASP289(A), LYS137(A), ARG 131(A), LEU287(A) | 2.51, 2.66, 2.91 2.75, 3.10 | −7.9 | - | - |
| N3 | 8 | HIS41(A), THR190(A), GLU166(A), GLN189(A), THR26(A), GLY143(A), CYS145(A) (2H bonding) | 2.65, 2.42, 2.65, 1.81, 3.10, 1.98, 3.40, 3.45 | −7.5 | GLN 189 (A),THR190(A),GLU166(A),PHE140(A),HIS164(A) and GLY 143(A) | [24] |
| X77 | 5 | HIS164(A), GLY143(A), ASN142(A), GLU166(A), PHE140(A) | 2.35, 3.09, 2.05, 2.19, 2.73 | −8.6 | GLY143(A), GLU166(A), ASN142(A) and CYS145(A) | [25] |
| Glucogallin | 6 | CYS145(A), SER144(A), GLY143(A) (2H bonding), HIS163(A), MET165(A) | 3.3, 2.37, 2.40, 1.97, 2.40, 3.05 | −7.0 | - | - |
| Mangiferin | 6 | LEU141(A), SER144(A), ASN142(A) (2H bonding),THR 190(A) (2H bonding) | 2.26,2.80, 2.72 2.12, 2.46,1.91 | −8.5 | - | - |
| Phlorizin | 6 | GLN189(A), MET49(A), CYS145(A), MET165(A), GLY143(A), SER144(A) | 2.25, 2.98,3.75,2.88,2.59, 2.73 | −7.9 | - | - |
| Compound | Number of H Bonding | Residue Receptor | Bond Length (Å) | Docking Score (kcal/mol) |
|---|---|---|---|---|
| Camostat mesylate | 4 | GLY464(A) (2H bonding), CYS465(A), HIS296(A), ASP 435(A) | 2.30, 2.37, 2.92, 2.58,2.14 | −7.1 |
| Glucogallin | 4 | SER441(A) (2H bonding), VAL280(A), GLY439(A), GLY 462(A) | 2.15, 2.19, 2.52, 2.75, 2.29 | −6.9 |
| Mangiferin | 2 | ALA 243(A), GLU 289(A) | 2.05, 2.07 | −6.9 |
| Phlorizin | 4 | SER460(A), SER441(A), GLY 464(A) (2H bonding) | 2.20, 2.64, 1.97, 2.59 | −7.7 |
| Compound | Maximum Tolerated Dose(Human) (log mg/kg/day) | Oral Rat Acute Toxicity (LD50) (mol/kg) | Oral Rat Chronic Toxicity (LOAEL) (log mg/kg bw/day) | T.PyriformisToxicity (log ug/L) | Minnow Toxicity (log mM) | Ames Toxicity | Hepato-Toxicity | Skin Sensitivity | hERGI/II |
|---|---|---|---|---|---|---|---|---|---|
| Remdesivir | 0.15 | 2.043 | 1.639 | 0.285 | 0.291 | NO | YES | NO | NO/YES |
| X77 | 0.601 | 2.396 | 1.528 | 0.285 | 2.563 | YES | YES | NO | NO/YES |
| N3 | 0.305 | 2.344 | 3.084 | 0.287 | −2.205 | YES | YES | NO | NO/YES |
| Camostat mesylate | 0.133 | 2.319 | 2.81 | 0.285 | 0.524 | NO | NO | NO | NO |
| Mangiferin | 0.58 | 2.396 | 4.277 | 0.285 | 5.898 | NO | NO | NO | NO |
| Glucogallin | 0.238 | 2.314 | 3.112 | 0.285 | 6.151 | NO | NO | NO | NO |
| Phlorizin | 0.555 | 2.494 | 4.667 | 0.285 | 6.334 | NO | NO | NO | NO |
| Target-Ligand Complex | Binding Energy (kcal/mol) | Solvation Energy (kcal/mol) | Electrostatic Energy (kcal/mol) | Van der WaalsEnergy (kcal/mol) | SASA Energy (kcal/mol) |
|---|---|---|---|---|---|
| Mpro-Remdesivir | −5.4127 ± 4.24 | 36.394 ± 8.92 | −12.87 ± 4.90 | −25.58 ± 5.40 | −3.342 ± 0.53 |
| Mpro-X77 | −14.16 ± 11.478 | 35.77 ± 20.88 | −9.35 ± 5.597 | −36.71 ± 19.14 | −3.879 ± 2.05 |
| Mpro-N3 | −16.58 ± 8.263 | 57.78 ± 26.110 | −18.49 ± 8.145 | −50.04 ± 20.54 | −5.834 ± 2.39 |
| CamostatMesylate-TMPRSS2 | −34.6558 ± 4.34 | −76.79 ± 16.5 | −53.95 ± 7.26 | −34.70 ± 4.34 | −4.349 ± 0.35 |
| Mpro-phlorizin | −9.65 ± 3.33 | 36.80 ± 6.52 | −10.1 ± 3.40 | −32.29 ± 5.15 | −4.02 ± 0.45 |
| Mpro-Glucogallin | −8.22 ± 2.52 | 34.190 ± 2.78 | −10.34 ± 6.28 | −29.56 ± 2.43 | −3.55 ± 0.25 |
| Mpro-Mangiferin | −4.50 ± 8.37 | 33.33 ± 23.71 | −6.419 ± 5.76 | −24.33 ± 12.7 | −3.15 ± 1.64 |
| TMPRSS2-Phlorizin | −1.57 ± 4.83 | 41.08 ± 9.90 | −11.67 ± 5.84 | −27.39 ± 5.29 | −3.59 ± 0.45 |
| TMPRSS2-Glucogallin | −1.10 ± 4.67 | 40.68 ± 7.25 | −15.63 ± 4.89 | −22.96 ± 3.52 | −3.18 ± 0.26 |
| TMPRSS2-Mangiferin | −3.04 ± 3.98 | 46.366 ± 10.41 | −19.34 ± 8.453 | −26.40 ± 3.01 | −3.65 ± 0.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Gautam, A.; Chandel, S.; Ghosh, A.; Dey, D.; Roy, S.; Ravichandiran, V.; Ghosh, D. Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2: An In Silico Study. Molecules 2020, 25, 4604. https://doi.org/10.3390/molecules25204604
Singh R, Gautam A, Chandel S, Ghosh A, Dey D, Roy S, Ravichandiran V, Ghosh D. Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2: An In Silico Study. Molecules. 2020; 25(20):4604. https://doi.org/10.3390/molecules25204604
Chicago/Turabian StyleSingh, Rajveer, Anupam Gautam, Shivani Chandel, Arijit Ghosh, Dhritiman Dey, Syamal Roy, Velayutham Ravichandiran, and Dipanjan Ghosh. 2020. "Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2: An In Silico Study" Molecules 25, no. 20: 4604. https://doi.org/10.3390/molecules25204604
APA StyleSingh, R., Gautam, A., Chandel, S., Ghosh, A., Dey, D., Roy, S., Ravichandiran, V., & Ghosh, D. (2020). Protease Inhibitory Effect of Natural Polyphenolic Compounds on SARS-CoV-2: An In Silico Study. Molecules, 25(20), 4604. https://doi.org/10.3390/molecules25204604