
Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases



Abstract
:1. Introduction
2. Structure of Adenosine Deaminase (ADA)
3. Inhibitors of ADA Activity
3.1. Transition-State Inhibitors
Coformycin and Deoxycoformycin Analogs
3.2. Ground-State Compounds
3.2.1. Deaza- and Dideazaadenosine Derivatives
3.2.2. EHNA-Like Compounds
3.3. Non-Nucleoside Inhibitors
3.4. Flavonoids and Sapogenins/Plant Extracts
3.5. Clinically Used Drugs Not Targeting ADA
3.6. The Challenges in Ecto-Adenosine Deaminase (eADA) Inhibition
4. Inhibitors of ADA Binding to Anchoring Proteins
5. Cardiovascular Pathologies Associated with Increased ADA Activity as Potential Targets for ADA Inhibition
5.1. Atherosclerosis
5.2. Thrombosis
5.3. Acute Myocardial Infarction and Myocardial Ischemia-Reperfusion Injury
5.4. Hypertension
5.5. Type 2 Diabetes Mellitus
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cortés, A.; Gracia, E.; Moreno, E.; Mallol, J.; Lluís, C.; Canela, E.I.; Casadó, V. Moonlighting Adenosine Deaminase: A Target Protein for Drug Development. Med. Res. Rev. 2015, 35, 85–125. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.Y.; Nygaard, P.; Chinault, A.C.; Kellems, R.E. Deduced amino acid sequence of Escherichia coli adenosine deaminase reveals evolutionarily conserved amino acid residues: Implications for catalytic function. Biochemistry 1991, 30, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Sander, C. An evolutionary treasure: Unification of a broad set of amidohydrolases related to urease. Proteins 1997, 28, 72–82. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Mierzejewska, P.; Sucajtys-Szulc, E.; Bulinska, A.; Zabielska, M.A.; Jablonska, P.; Serocki, M.; Koszalka, P.; Milczarek, R.; Jasztal, A.; et al. Inhibition of LPS-stimulated ecto-adenosine deaminase attenuates endothelial cell activation. J. Mol. Cell. Cardiol. 2019, 128. [Google Scholar] [CrossRef]
- Ebrahimi-Rad, M.; Khatami, S.; Ansari, S.; Jalylfar, S.; Valadbeigi, S.; Saghiri, R. Adenosine Deaminase 1 as a Biomarker for Diagnosis and Monitoring of Patients with Acute Lymphoblastic Leukemia. J. Med. Biochem. 2018, 37, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Canet, J.; Gracia, E.; Lluís, C.; Mallol, J.; Canela, E.I.; Cortés, A.; Casadó, V. Molecular Evidence of Adenosine Deaminase Linking Adenosine A2A Receptor and CD26 Proteins. Front. Pharmacol. 2018, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Kaljas, Y.; Liu, C.; Skaldin, M.; Wu, C.; Zhou, Q.; Lu, Y.; Aksentijevich, I.; Zavialov, A.V. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell. Mol. Life Sci. 2017, 74, 555–570. [Google Scholar] [CrossRef]
- Zavialov, A.V.; Engström, A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem. J. 2005, 391, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Correia-de-Sá, P.; Adães, S.; Timóteo, M.A.; Vieira, C.; Magalhães-Cardoso, T.; Nascimento, C.; Duarte-Araújo, M. Fine-tuning modulation of myenteric motoneurons by endogenous adenosine: On the role of secreted adenosine deaminase. Auton. Neurosci. 2006, 126–127, 211–224. [Google Scholar] [CrossRef]
- Gao, Z.-W.; Wang, X.; Lin, F.; Dong, K. Total adenosine deaminase highly correlated with adenosine deaminase 2 activity in serum. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Tardif, V.; Muir, R.; Cubas, R.; Chakhtoura, M.; Wilkinson, P.; Metcalf, T.; Herro, R.; Haddad, E.K. Adenosine deaminase-1 delineates human follicular helper T cell function and is altered with HIV. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Andreasyan, N.A.; Hairapetyan, H.L.; Sargisova, Y.G.; Mardanyan, S.S. ADA2 isoform of adenosine deaminase from pleural fluid. FEBS Lett. 2005, 579, 643–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavialov, A.V.; Yu, X.; Spillmann, D.; Lauvau, G.; Zavialov, A.V. Structural Basis for the Growth Factor Activity of Human Adenosine Deaminase ADA2. J. Biol. Chem. 2010, 285, 12367–12377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, K.L.; Moretti, F.A.; Carbonaro-Sarracino, D.A.; Gaspar, H.B.; Kohn, D.B. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID): Molecular Pathogenesis and Clinical Manifestations. J. Clin. Immunol. 2017, 37, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanetj. Rare Dis. 2018, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Köse, M.; Schiedel, A.C.; Bauer, A.A.; Poschenrieder, H.; Burbiel, J.C.; Akkinepally, R.R.; Stachel, H.-D.; Müller, C.E. Focused screening to identify new adenosine kinase inhibitors. Bioorg. Med. Chem. 2016, 24, 5127–5133. [Google Scholar] [CrossRef] [PubMed]
- Bin, A.; Caputi, V.; Bistoletti, M.; Montopoli, M.; Colucci, R.; Antonioli, L.; De Martin, S.; Castagliuolo, I.; Orso, G.; Giaroni, C.; et al. The ecto-enzymes CD73 and adenosine deaminase modulate 5′-AMP-derived adenosine in myofibroblasts of the rat small intestine. Purinergic Signal. 2018, 14, 409–421. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Pérez-Torras, S. Who Is Who in Adenosine Transport. Front. Pharmacol. 2018, 9, 627. [Google Scholar] [CrossRef] [Green Version]
- Camici, M.; Garcia-Gil, M.; Tozzi, M.G. The Inside Story of Adenosine. Int. J. Mol. Sci. 2018, 19, 784. [Google Scholar] [CrossRef] [Green Version]
- Soslau, G. Extracellular adenine compounds within the cardiovascular system: Their source, metabolism and function. Med. Drug Discov. 2019, 4, 100018. [Google Scholar] [CrossRef]
- Vecchio, E.A.; White, P.J.; May, L.T. Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headrick, J.P.; Ashton, K.J.; Rose′meyer, R.B.; Peart, J.N. Cardiovascular adenosine receptors: Expression, actions and interactions. Pharmacol. Ther. 2013, 140, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Yáñez, I.; Castillo, C.A.; Merighi, S.; Gessi, S. The role of adenosine receptors in psychostimulant addiction. Front. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutryb-Zajac, B.; Jablonska, P.; Serocki, M.; Bulinska, A.; Mierzejewska, P.; Friebe, D.; Alter, C.; Jasztal, A.; Lango, R.; Rogowski, J.; et al. Nucleotide ecto-enzyme metabolic pattern and spatial distribution in calcific aortic valve disease; its relation to pathological changes and clinical presentation. Clin. Res. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, H.; Chellappan, D.K.; Sengupta, P.; Pandey, M.; Gorain, B. Adenosine Receptors in Modulation of Central Nervous System Disorders. Curr. Pharm. Des. 2019, 25, 2808–2827. [Google Scholar] [CrossRef]
- Grenz, A.; Homann, D.; Eltzschig, H.K. Extracellular adenosine: A safety signal that dampens hypoxia-induced inflammation during ischemia. Antioxid Redox Signal 2011, 15, 2221–2234. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 151–199. [Google Scholar] [CrossRef] [Green Version]
- Welihinda, A.A.; Kaur, M.; Greene, K.; Zhai, Y.; Amento, E.P. The adenosine metabolite inosine is a functional agonist of the adenosine A2A receptor with a unique signaling bias. Cell. Signal. 2016, 28, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Da Rocha Lapa, F.; de Oliveira, A.P.L.; Accetturi, B.G.; de Oliveira Martins, I.; Domingos, H.V.; de Almeida Cabrini, D.; de Lima, W.T.; Santos, A.R.S. Anti-inflammatory effects of inosine in allergic lung inflammation in mice: Evidence for the participation of adenosine A2A and A 3 receptors. Purinergic Signal. 2013, 9, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Herman-de-Sousa, C.; Pinheiro, A.R.; Paramos-de-Carvalho, D.; Costa, M.A.; Ferreirinha, F.; Magalhães-Cardoso, T.; Ribeiro, S.; Pelletier, J.; Sévigny, J.; Correia-de-Sá, P. Opposing Effects of Adenosine and Inosine in Human Subcutaneous Fibroblasts May Be Regulated by Third Party ADA Cell Providers. Cells 2020, 9, 651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia, E.; Farré, D.; Cortés, A.; Ferrer-Costa, C.; Orozco, M.; Mallol, J.; Lluís, C.; Canela, E.I.; McCormick, P.J.; Franco, R.; et al. The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. Faseb J. 2013, 27, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, R.S.; Hong, M. How important is the N-3 sugar moiety in the tight-binding interaction of coformycin with adenosine deaminase? Biochem. Biophys. Res. Commun. 1997, 236, 88–93. [Google Scholar] [CrossRef]
- Wang, Z.; Quiocho, F.A. Complexes of Adenosine Deaminase with Two Potent Inhibitors: X-ray Structures in Four Independent Molecules at pH of Maximum Activity†,‡. Biochemistry 1998, 37, 8314–8324. [Google Scholar] [CrossRef]
- Sideraki, V.; David, K.W.; Linda, C.K.; Florante, A.Q.; Frederick, B.R. Site-Directed Mutagenesis of Histidine 238 in Mouse Adenosine Deaminase: Substitution of Histidine 238 Does Not Impede Hydroxylate Formation†,‡. Biochemistry 1996, 35, 15028–15091. [Google Scholar] [CrossRef]
- Nagano, N.; Orengo, C.A.; Thornton, J.M. One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 2002, 321, 741–765. [Google Scholar] [CrossRef]
- Niu, W.; Shu, Q.; Chen, Z.; Mathews, S.; Di Cera, E.; Frieden, C. The role of Zn2+ on the structure and stability of murine adenosine deaminase. J. Phys. Chem. B 2010, 114, 16156–16165. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Z.; Walker, H.; Lau, J.Y.N.; Hong, Z. Activation and Deactivation of a Broad-Spectrum Antiviral Drug by a Single Enzyme: Adenosine Deaminase Catalyzes Two Consecutive Deamination Reactions. Antimicrob. Agents Chemother. 2003, 47, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Mallol, J.; Casadó, V.; Lluis, C.; Canela, E.I.; Saura, C.; Blanco, J.; Ciruela, F. Ecto-adenosine deaminase: An ecto-enzyme and a costimulatory protein acting on a variety of cell surface receptors. Drug Dev. Res. 1998, 45, 261–268. [Google Scholar] [CrossRef]
- Cristalli, G.; Costanzi, S.; Lambertucci, C.; Lupidi, G.; Vittori, S.; Volpini, R.; Camaioni, E. Adenosine deaminase: Functional implications and different classes of inhibitors. Med. Res. Rev. 2001, 21, 105–128. [Google Scholar] [CrossRef]
- Lupidi, G.; Colao, C.; Marmocchi, F.; Cristalli, G. Photoinactivation studies on adenosine deaminase. Iubmb Life 1998, 44, 1031–1043. [Google Scholar] [CrossRef]
- Mardanyan, S.; Sharoyan, S.; Antonyan, A.; Armenyan, A.; Cristalli, G.; Lupidi, G. Tryptophan environment in adenosine deaminase: I. Enzyme modification with N-bromosuccinimide in the presence of adenosine and EHNA analogues. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2001, 1546, 185–195. [Google Scholar] [CrossRef]
- Muraoka, T.; Katsuramaki, T.; Shiraishi, H.; Yokoyama, M.M. Automated enzymatic measurement of adenosine deaminase isoenzyme activities in serum. Anal. Biochem. 1990, 187, 268–272. [Google Scholar] [CrossRef]
- Van der Weyden, M.B.; Kelley, W.N. Human adenosine deaminase. Distribution and properties. J. Biol. Chem. 1976, 251, 5448–5456. [Google Scholar] [PubMed]
- Gakis, C. Adenosine deaminase (ADA) isoenzymes ADA1 and ADA2: Diagnostic and biological role. Eur. Respir. J. 1996, 9, 632–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.A.; Ahluwalia, G.; Connelly, M.C.; Cooney, D.A.; Broder, S.; Johns, D.G.; Fridland, A. Metabolic pathways for the activation of the antiretroviral agent 2′,3′-dideoxyadenosine in human lymphoid cells. J. Biol. Chem. 1988, 263, 15354–15357. [Google Scholar]
- Bagheri, S.; Saboury, A.A.; Haertlé, T. Adenosine deaminase inhibition. Int. J. Biol. Macromol. 2019, 141, 1246–1257. [Google Scholar] [CrossRef]
- Terasaka, T. Non-nucleoside adenosine deaminase inhibitors: 2000—2004. Expert Opin. Ther. Pat. 2005, 15, 817–828. [Google Scholar] [CrossRef]
- Sawa, T.; Fukagawa, Y.; Homma, I.; Takeuchi, T.; Umezawa, H. Mode of inhibition of coformycin on adenosine deaminase. J. Antibiot. (Tokyo) 1967, 20, 227–231. [Google Scholar]
- Woo, P.W.K.; Dion, H.W.; Lange, S.M.; Dahl, L.F.; Durham, L.J. A novel adenosine and ara-a deaminase inhibitor, (R)-3-(2-deoxy-β-d-erythro -pentofuranosyl)-3,6,7,8-tetrahydroimidazo [4,5-d] [1,3]diazepin-8-ol. J. Heterocycl. Chem. 1974, 11, 641–643. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Thomas, H.J.; Zell, A.L.; Einsphar, H.M.; Bugg, C.E. Study on the inhibition of adenosine deaminase. J. Med. Chem. 1985, 28, 1751–1753. [Google Scholar] [CrossRef] [PubMed]
- Warzocha, K.; Fabianowska-Majewska, K.; Bloński, J.; Krykowski, E.; Robak, T. 2-Chlorodeoxyadenosine inhibits activity of adenosine deaminase and S-adenosylhomocysteine hydrolase in patients with chronic lymphocytic leukaemia. Eur. J. Cancer 1997, 33, 170–173. [Google Scholar] [CrossRef]
- Ma, L.; Zhong, J.; Zhao, Z.; Luo, Z.; Ma, S.; Sun, J.; He, H.; Zhu, T.; Liu, D.; Zhu, Z.; et al. Activation of TRPV1 reduces vascular lipid accumulation and attenuates atherosclerosis. Cardiovasc. Res. 2011, 92, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, S.; Imamura, N.; Kuga, H.; Ishikawa, H.; Yamazaki, Y.; Okano, K.; Kimura, K.; Takahashi, Y.; Tanaka, H. Adechlorin, a new adenosine deaminase inhibitor containing chlorine production, isolation and properties. J. Antibiot. (Tokyo) 1985, 38, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, S.; Ishikawa, H.; Kuga, H.; Imamura, N.; Taga, S.; Takahashi, Y.; Tanaka, H. Adecypenol, a unique adenosine deaminase inhibitor containing homopurine and cyclopentene rings. Taxonomy, production and enzyme inhibition. J. Antibiot. (Tokyo) 1986, 39, 1219–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristalli, G.; Eleuteri, A.; Vittori, S.; Volpini, R.; Camaioni, E.; Lupidi, G. Adenosine deaminase inhibitors: Structure-activity relationships in 1-deazaadenosine and erythro-9-(2-hydroxy-3-nonyl)adenine analogues. Drug Dev. Res. 1993, 28, 253–258. [Google Scholar] [CrossRef]
- Cristalli, G.; Vittori, S.; Eleuteri, A.; Grifantini, M.; Volpini, R.; Lupidi, G.; Capolongo, L.; Pesenti, E. Purine and 1-deazapurine ribonucleosides and deoxyribonucleosides: synthesis and biological activity. J. Med. Chem. 1991, 34, 2226–2230. [Google Scholar] [CrossRef]
- Cristalli, G.; Vittori, S.; Eleuteri, A.; Volpini, R.; Camaioni, E.; Lupidi, G.; Mahmood, N.; Bevilacqua, F.; Palu, G. Synthesis and biological evaluation of N6-cycloalkyl derivatives of 1-deazaadenine nucleosides: A new class of anti-human immunodeciency virus agents. J Med Chem 1995, 38, 4019–4025. [Google Scholar] [CrossRef]
- Volpini, R.; Costanzi, S.; Vittori, S.; Cristalli, G.; Lupidi, G. Synthesis and Adenosine Deaminase Inhibitory Activity of 3′-Deoxy-1-deazaadenosines. Helv. Chim. Acta 1999, 82, 2112–2118. [Google Scholar] [CrossRef]
- Porter, D.J.; Abushanab, E. Kinetics of inhibition of calf intestinal adenosine deaminase by (+)- and (–)-erythro-9-(2-hydroxy-3-nonyl)adenine. Biochemistry 1992, 31, 8216–8220. [Google Scholar] [CrossRef] [PubMed]
- Barankiewicz, J.; Danks, A.M.; Abushanab, E.; Makings, L.; Wiemann, T.; Wallis, R.A.; Pragnacharyulu, P.V.; Fox, A.; Marangos, P.J. Regulation of adenosine concentration and cytoprotective effects of novel reversible adenosine deaminase inhibitors. J. Pharmacol. Exp. Ther. 1997, 283, 1230–1238. [Google Scholar] [PubMed]
- Caiolfa, V.R.; Gill, D.; Parola, A.H. Probing the active site of adenosine deaminase by a pH responsive fluorescent competitive inhibitor. Biophys. Chem. 1998, 70, 41–56. [Google Scholar] [CrossRef]
- Cristalli, G.; Franchetti, P.; Grifantini, M.; Vittori, S.; Lupidi, G.; Riva, F.; Bordoni, T.; Geroni, C.; Verini, M.A. Adenosine deaminase inhibitors. Synthesis and biological activity of deaza analogues of erythro-9-(2-hydroxy-3-nonyl)adenine. J. Med. Chem. 1988, 31, 390–393. [Google Scholar] [CrossRef]
- Cristalli, G.; Eleuteri, A.; Franchetti, P.; Grifantini, M.; Vittori, S.; Lupidi, G. Adenosine deaminase inhibitors: synthesis and structure-activity relationships of imidazole analogues of erythro-9-(2-hydroxy-3-nonyl)adenine. J. Med. Chem. 1991, 34, 1187–1192. [Google Scholar] [CrossRef]
- Cristalli, G.; Eleuteri, A.; Volpini, R.; Vittori, S.; Camaioni, E.; Lupidi, G. Adenosine deaminase inhibitors: Synthesis and structure-activity relationships of 2-hydroxy-3-nonyl derivatives of azoles. J. Med. Chem. 1994, 37, 201–205. [Google Scholar] [CrossRef]
- Terasaka, T.; Nakanishi, I.; Nakamura, K.; Eikyu, Y.; Kinoshita, T.; Nishio, N.; Sato, A.; Kuno, M.; Seki, N.; Sakane, K. Structure-based de novo design of non-nucleoside adenosine deaminase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1115–1118. [Google Scholar] [CrossRef]
- Kuno, M.; Seki, N.; Tsujimoto, S.; Nakanishi, I.; Kinoshita, T.; Nakamura, K.; Terasaka, T.; Nishio, N.; Sato, A.; Fujii, T. Anti-inflammatory activity of non-nucleoside adenosine deaminase inhibitor FR234938. Eur. J. Pharmacol. 2006, 534, 241–249. [Google Scholar] [CrossRef]
- Melzig, M. Inhibition of Adenosine Deaminase Activity of Aortic Endothelial Cells by Selected Flavonoids. Planta Med. 1996, 62, 20–21. [Google Scholar] [CrossRef]
- Arun, K.G.; Sharanya, C.S.; Sandeep, P.M.; Sadasivan, C. Inhibitory activity of hibifolin on adenosine deaminase- experimental and molecular modeling study. Comput. Biol. Chem. 2016, 64, 353–358. [Google Scholar] [CrossRef]
- Li, G.; Nakagome, I.; Hirono, S.; Itoh, T.; Fujiwara, R. Inhibition of adenosine deaminase (ADA)-mediated metabolism of cordycepin by natural substances. Pharmacol. Res. Perspect. 2015, 3, e121. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, H.; Safipour, A. Curcumin inhibits the expression of ornithine decarboxylase and adenosine deaminase genes in MCF-7 human breast cancer cells. Arch. Biol. Sci. 2018, 70, 639–645. [Google Scholar] [CrossRef]
- Ni, H.; Li, Y.-H.; Hao, R.-L.; Li, H.; Hu, S.-Q.; Li, H.-H. Identification of adenosine deaminase inhibitors from Tofu wastewater and litchi peel and their synergistic anticancer and antibacterial activities with cordycepin. Int. J. Food Sci. Technol. 2016, 51, 1168–1176. [Google Scholar] [CrossRef]
- Koch, H.P.; Aichinger, A.; Bohne, B.; Plank, G. In vitro inhibition of adenosine deaminase by a group of steroid and Triterpenoid compounds. Phyther. Res. 1994, 8, 109–111. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Tang, P.; Liu, Z.; Guo, G.-J.; Sun, Q.-Y.; Yin, J. Identification of a New Uncompetitive Inhibitor of Adenosine Deaminase from Endophyte Aspergillus niger sp. Curr. Microbiol. 2018, 75, 565–573. [Google Scholar] [CrossRef]
- Ataie, G.; Safaranian, S.; Divsalar, A.; Saboury, A.A.; Moosavi-Movahedi, A.A.; Ranjbar, B.; Cristalli, G.; Mardanian, S. Kinetic and Structural Analysis of the Inhibition of Adenosine Deaminase by Acetaminophen. J. Enzyme Inhib. Med. Chem. 2004, 19, 71–78. [Google Scholar] [CrossRef]
- Ajloo, D.; Saboury, A.A.; Haghi-Asli, N.; Ataei-Jafarai, G.; Moosavi-Movahedi, A.A.; Ahmadi, M.; Mahnam, K.; Namaki, S. Kinetic, thermodynamic and statistical studies on the inhibition of adenosine deaminase by aspirin and diclofenac. J. Enzyme Inhib. Med. Chem. 2007, 22, 395–406. [Google Scholar] [CrossRef]
- Centelles, J.J.; Franco, R.; Bozal, J. Purification and partial characterization of brain adenosine deaminase: Inhibition by purine compounds and by drugs. J. Neurosci. Res. 1988, 19, 258–267. [Google Scholar] [CrossRef]
- Sheid, B. Trazodone, a nontricyclic antidepressant, is an inhibitor of adenosine deaminase. Res. Commun. Chem. Pathol. Pharmacol. 1985, 47, 149–152. [Google Scholar]
- Agarwal, R.P.; Spector, T.; Parks, R.E. Tight-binding inhibitors--IV. Inhibition of adenosine deaminases by various inhibitors. Biochem. Pharmacol. 1977, 26, 359–367. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Ferella, M.; Lundén-Miguel, H.; Betha, E.; van Reet, N.; Amin, D.N.; Oberg, B.; Andersson, B.; Kristensson, K.; Wigzell, H.; et al. Preclinical assessment of the treatment of second-stage African trypanosomiasis with cordycepin and deoxycoformycin. PLoS Negl. Trop. Dis. 2009, 3, e495. [Google Scholar] [CrossRef] [PubMed]
- Lupidi, G.; Cristalli, G.; Marmocchi, F.; Riva, F.; Grifantini, M. Inhibition of adenosine deaminase from several sources by deaza derivatives of adenosine and EHNA. J. Enzyme Inhib. 1985, 1, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.K.; Rudolph, F.B.; Quiocho, F.A. Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science 1991, 252, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, M.; Maguire, M.H. Studies on adenosine deaminase. I. Purification and properties of ox heart adenosine deaminase. Mol. Pharmacol. 1966, 2, 574–584. [Google Scholar] [PubMed]
- Jeong, S.Y.; Ahn, S.G.; Lee, J.H.; Kim, H.S.; Kim, J.W.; Rhim, H.; Jeong, S.W.; Kim, I.K. 3-deazaadenosine, a S-adenosylhomocysteine hydrolase inhibitor, has dual effects on NF-kappaB regulation. Inhibition of NF-kappaB transcriptional activity and promotion of IkappaBalpha degradation. J. Biol. Chem. 1999, 274, 18981–18988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnaluri, V.K.C.; Estève, P.-O.; Ruse, C.I.; Pradhan, S. S-adenosylhomocysteine Hydrolase Participates in DNA Methylation Inheritance. J. Mol. Biol. 2018, 430, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Jurgensen, C.H.; Huber, B.E.; Zimmerman, T.P.; Wolberg, G. 3-deazaadenosine inhibits leukocyte adhesion and ICAM-1 biosynthesis in tumor necrosis factor-stimulated human endothelial cells. J. Immunol. 1990, 144, 653–661. [Google Scholar]
- Kandalkar, S.R.; Ramaiah, P.A.; Joshi, M.; Wavhal, A.; Waman, Y.; Raje, A.A.; Tambe, A.; Ansari, S.; De, S.; Palle, V.P.; et al. Modifications of flexible nonyl chain and nucleobase head group of (+)-erythro-9-(2’s-hydroxy-3’s-nonyl)adenine [(+)-EHNA] as adenosine deaminase inhibitors. Bioorg. Med. Chem. 2017, 25, 5799–5819. [Google Scholar] [CrossRef]
- Curtis, M.A.; Varkhedkar, V.; Pragnacharyulu, P.V.; Abushanab, E. Adenosine deaminase inhibitors. Synthesis and biological evaluation of aralkyladenines (ARADS). Bioorg. Med. Chem. Lett. 1998, 8, 1639–1642. [Google Scholar] [CrossRef]
- Bhansali, S.G.; Kulkarni, V.M. Combined 2D and 3D-QSAR, molecular modelling and docking studies of pyrazolodiazepinones as novel phosphodiesterase 2 inhibitors. SAR QSAR Environ. Res. 2014, 25, 905–937. [Google Scholar] [CrossRef]
- Iffland, A.; Kohls, D.; Low, S.; Luan, J.; Zhang, Y.; Kothe, M.; Cao, Q.; Kamath, A.V.; Ding, Y.-H.; Ellenberger, T. Structural Determinants for Inhibitor Specificity and Selectivity in PDE2A Using the Wheat Germ in Vitro Translation System †. Biochemistry 2005, 44, 8312–8325. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, N.U.; O’Donnell, J.M. Hydrolysis of N-methyl-D-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus. J. Pharmacol. Exp. Ther. 2002, 302, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feneck, R. Phosphodiesterase inhibitors and the cardiovascular system. Contin. Educ. Anaesth. Crit. Care Pain 2007, 7, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, T.; Tsuji, K.; Kato, T.; Nakanishi, I.; Kinoshita, T.; Kato, Y.; Kuno, M.; Inoue, T.; Tanaka, K.; Nakamura, K. Rational Design of Non-Nucleoside, Potent, and Orally Bioavailable Adenosine Deaminase Inhibitors: Predicting Enzyme Conformational Change and Metabolism. J. Med. Chem. 2005, 48, 4750–4753. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, T.; Okumura, H.; Tsuji, K.; Kato, T.; Nakanishi, I.; Kinoshita, T.; Kato, Y.; Kuno, M.; Seki, N.; Naoe, Y.; et al. Structure-Based Design and Synthesis of Non-Nucleoside, Potent, and Orally Bioavailable Adenosine Deaminase Inhibitors. J. Med. Chem. 2004, 47, 2728–2731. [Google Scholar] [CrossRef]
- Koch, H.P.; Jager, W.; Hysek, J.; Korpert, B. Garlic and onion extracts. In vitro inhibition of adenosine deaminase. Phyther. Res. 1992, 6, 50–52. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Koszalka, P.; Slominska, E.M.; Smolenski, R.T. The effects of pro- and anti-atherosclerotic factors on intracellular nucleotide concentration in murine endothelial cells. Nucleosides Nucleotides Nucleic Acids 2018, 37, 645–652. [Google Scholar] [CrossRef]
- Kowalczyk, E.; Kopff, M.; Kowalski, J.; Kopff, A.; Mikhailidis, D.P.; Barylski, M.; Banach, M. Effect of Cardiovascular Drugs on Adenosine Deaminase Activity. Angiology 2009, 59, 740–744. [Google Scholar] [CrossRef]
- Kong, W.; Engel, K.; Wang, J. Mammalian nucleoside transporters. Curr. Drug Metab. 2004, 5, 63–84. [Google Scholar] [CrossRef]
- Noji, T.; Karasawa, A.; Kusaka, H. Adenosine uptake inhibitors. Eur. J. Pharmacol. 2004, 495, 1–16. [Google Scholar] [CrossRef]
- Alsharif, K.F.; Thomas, M.R.; Judge, H.M.; Khan, H.; Prince, L.R.; Sabroe, I.; Ridger, V.C.; Storey, R.F. Ticagrelor potentiates adenosine-induced stimulation of neutrophil chemotaxis and phagocytosis. Vascul. Pharmacol. 2015, 71, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harker, L.A.; Kadatz, R.A. Mechanism of action of dipyridamole. Thromb. Res. Suppl. 1983, 4, 39–46. [Google Scholar] [CrossRef]
- Jackson, E.K.; Cheng, D.; Jackson, T.C.; Verrier, J.D.; Gillespie, D.G. Extracellular guanosine regulates extracellular adenosine levels. Am. J. Physiol. Physiol. 2013, 304, C406–C421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd-Elfattah, A.S.; Tuchy, G.E.; Jessen, M.E.; Salter, D.R.; Goldstein, J.P.; Brunsting, L.A.; Wechsler, A.S. Hot shot induction and reperfusion with a specific blocker of the es-ENT1 nucleoside transporter before and after hypothermic cardioplegia abolishes myocardial stunning in acutely ischemic hearts despite metabolic derangement: Hot shot drug delivery before hypothermic cardioplegia. J. Thorac. Cardiovasc. Surg. 2013, 146, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, J.; Valenzuela, A.; Herrera, C.; Lluís, C.; Hovanessian, A.G.; Franco, R. The HIV-1 gp120 inhibits the binding of adenosine deaminase to CD26 by a mechanism modulated by CD4 and CXCR4 expression. FEBS Lett. 2000, 477, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Martín, M.; Huguet, J.; Centelles, J.J.; Franco, R. Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine deaminase as costimulatory molecule. J. Immunol. 1995, 155, 4630–4643. [Google Scholar] [PubMed]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Haskó, G. Adenosine Regulation of the Immune System. In The Adenosine Receptors; Springer International Publishing: New York, NY, USA, 2018; pp. 499–514. [Google Scholar]
- Dubey, R.K.; Baruscotti, I.; Stiller, R.; Fingerle, J.; Gillespie, D.G.; Mi, Z.; Leeners, B.; Imthurn, B.; Rosselli, M.; Jackson, E.K. Adenosine, Via A 2B Receptors, Inhibits Human (P-SMC) Progenitor Smooth Muscle Cell Growth. Hypertension 2020, 75, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Leiva, A.; Guzman-Gutierrez, E.; Contreras-Duarte, S.; Fuenzalida, B.; Cantin, C.; Carvajal, L.; Salsoso, R.; Gutierrez, J.; Pardo, F.; Sobrevia, L. Adenosine Receptors: Modulators of Lipid Availability That Are Controlled by Lipid Levels. Mol. Aspects Med. 2017, 55, 26–44. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine signaling and the immune system: When a lot could be too much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef]
- Görlach, A. Control of adenosine transport by hypoxia. Circ. Res. 2005, 97, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Sajjan, N.B.; Makandar, A. Evaluation of serum adenosine deaminase levels with components of metabolic syndrome. Orig. Res. Artic. Int. J. Clin. Biochem. Res. 2016, 3, 285–291. [Google Scholar] [CrossRef]
- Simard, T.; Jung, R.; Labinaz, A.; Faraz, M.A.; Ramirez, F.D.; Di Santo, P.; Perry-Nguyen, D.; Pitcher, I.; Motazedian, P.; Gaudet, C.; et al. Evaluation of Plasma Adenosine as a Marker of Cardiovascular Risk: Analytical and Biological Considerations. J. Am. Heart Assoc. 2019, 8, e12228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutryb-Zajac, B.; Bulinska, A.; Zabielska, M.A.; Mierzejewska, P.; Slominska, E.M.; Smolenski, R.T. Vascular extracellular adenosine metabolism in mice correlates with susceptibility to atherosclerosis. Nucleosides Nucleotides Nucleic Acids 2018, 37, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Kutryb-Zajac, B.; Mateuszuk, L.; Zukowska, P.; Jasztal, A.; Zabielska, M.; Toczek, M.; Zakrzewska, A.; Sitek, B.; Rogowski, J.; Lango, R.; et al. Increased activity of vascular adenosine deaminase in atherosclerosis and therapeutic potential of its inhibition. Cardiovasc. Res. 2016, 112, 590–605. [Google Scholar] [CrossRef]
- Samra, Y.A.; Saleh, H.M.; Hussein, K.A.; Elsherbiny, N.M.; Ibrahim, A.S.; Elmasry, K.; Fulzele, S.; El-Shishtawy, M.M.; Eissa, L.A.; Al-Shabrawey, M.; et al. Adenosine Deaminase-2–Induced Hyperpermeability in Human Retinal Vascular Endothelial Cells Is Suppressed by MicroRNA-146b-3p. Investig. Opthalmol. Vis. Sci. 2017, 58, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Ginés, S.; Mariño, M.; Mallol, J.; Canela, E.I.; Morimoto, C.; Callebaut, C.; Hovanessian, A.; Casadó, V.; Lluis, C.; Franco, R. Regulation of epithelial and lymphocyte cell adhesion by adenosine deaminase-CD26 interaction. Biochem. J. 2002, 361, 203–209. [Google Scholar]
- Bouma, M.G.; van den Wildenberg, F.A.; Buurman, W.A. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am J Physiol 1996, 270, C522–C529. [Google Scholar] [CrossRef]
- Franco, R.; Pacheco, R.; Gatell, J.M.; Gallart, T.; Lluis, C. Enzymatic and extraenzymatic role of adenosine deaminase 1 in T-cell-dendritic cell contacts and in alterations of the immune function. Crit. Rev. Immunol. 2007, 27, 495–509. [Google Scholar] [CrossRef]
- Pacheco, R.; Martinez-Navio, J.M.; Lejeune, M.; Climent, N.; Oliva, H.; Gatell, J.M.; Gallart, T.; Mallol, J.; Lluis, C.; Franco, R. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 9583–9588. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zou, H.; Zhao, P.; Sun, B.; Wang, J.; Kong, Q.; Mu, L.; Zhao, S.; Wang, G.; Wang, D.; et al. Activation of the adenosine A2A receptor attenuates experimental autoimmune encephalomyelitis and is associated with increased intracellular calcium levels. Neuroscience 2016, 330, 150–161. [Google Scholar] [CrossRef]
- Zavialov, A.V.; Gracia, E.; Glaichenhaus, N.; Franco, R.; Lauvau, G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J. Leukoc. Biol. 2010, 88, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Pakala, R.; Benedict, C.R. Endothelial cells regulate the proliferation of monocytes in vitro. Atherosclerosis 1999, 147, 25–32. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-Onset Stroke and Vasculopathy Associated with Mutations in ADA2. N. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-G.; Oh, J.; Bong, S.-K.; Kim, J.-S.; Park, S.; Kim, S.; Park, S.; Lee, S.-H.; Jang, Y. Macrophage polarization and acceleration of atherosclerotic plaques in a swine model. PLoS ONE 2018, 13, e193005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tits, L.J.H.; Stienstra, R.; van Lent, P.L.; Netea, M.G.; Joosten, L.A.B.; Stalenhoef, A.F.H. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: A crucial role for Krüppel-like factor 2. Atherosclerosis 2011, 214, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, A.B.; Rahman, M.M.; Chan, E.S.L.; Montesinos, M.C.; Awadallah, N.W.; Cronstein, B.N. Adenosine A2A receptor occupancy stimulates expression of proteins involved in reverse cholesterol transport and inhibits foam cell formation in macrophages. J. Leukoc. Biol. 2004, 76, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Gawaz, M. Platelets in Atherosclerosis. In Platelets in Thrombotic and Non-Thrombotic Disorders; Springer International Publishing: New York, NY, USA, 2017; pp. 993–1013. [Google Scholar]
- Li, J.; Wu, H.; Hao, Y.; Yao, X. Unstable Carotid Plaque is Associated With Coagulation Function and Platelet Activity Evaluated by Thrombelastography. J. Stroke Cerebrovasc. Dis. 2019, 28, 104336. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [Green Version]
- Souza, V.; Do, C.G.; Schlemmer, K.B.; Noal, C.B.; Jaques, J.A.S.; Bagatini, M.D.; Pimentel, V.C.; Carli, L.F.D.; Leal, C.A.M.; Fleck, J.; et al. Purinergic system ecto-enzymes participate in the thromboregulation of patients with indeterminate form of Chagas disease. Purinergic Signal. 2012, 8, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Leal, C.A.M.; Leal, D.B.R.; Adefegha, S.A.; Morsch, V.M.; da Silva, J.E.P.; Rezer, J.F.P.; Schrekker, C.M.L.; Abdalla, F.H.; Schetinger, M.R.C. Platelet aggregation and serum adenosine deaminase (ADA) activity in pregnancy associated with diabetes, hypertension and HIV. Cell Biochem. Funct. 2016, 34, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Stafford, N.P.; Pink, A.E.; White, A.E.; Glenn, J.R.; Heptinstall, S. Mechanisms Involved in Adenosine Triphosphate–Induced Platelet Aggregation in Whole Blood. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1928–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, E.; Pereira, J.; Mezzano, D.; Alarcón, M.; Caballero, J.; Palomo, I. Inhibition of platelet activation and thrombus formation by adenosine and inosine: studies on their relative contribution and molecular modeling. PLoS ONE 2014, 9, e112741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, F.W.; Vijayan, K.V.; Rumbaut, R.E. Platelets and Their Interactions with Other Immune Cells. Compr. Physiol. 2015, 5, 1265–1280. [Google Scholar] [CrossRef] [Green Version]
- Jyothy, A.; Rani, H.S.; Rao, V.D.; Reddy, P.P. Serum Adenosine Deaminase Activity in Myocardial Infarction. Int. J. Hum. Genet. 2003, 3, 65–67. [Google Scholar] [CrossRef]
- Patil, N.; Chavan, V.; Karnik, N.D. Antioxidant status in patients with acute myocardial infarction. Indian J. Clin. Biochem. 2007, 22, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Jordan, J.E.; Zhao, Z.Q.; Vinten-Johansen, J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc. Res. 1999, 43, 860–878. [Google Scholar] [CrossRef] [Green Version]
- Barletta, K.E.; Ley, K.; Mehrad, B. Regulation of neutrophil function by adenosine. Arter. Thromb. Vasc. Biol. 2012, 32, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Piras, B.A.; Kron, I.L.; French, B.A.; Yang, Z. Adenosine 2B Receptor Activation Reduces Myocardial Reperfusion Injury by Promoting Anti-Inflammatory Macrophages Differentiation via PI3K/Akt Pathway. Oxid. Med. Cell. Longev. 2015, 2015, 585297. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Hansen, P.R. Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995, 91, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.C. Free radicals generated by xanthine oxidase-hypoxanthine damage adenylate cyclase and ATPase in gerbil cerebral cortex. Metab. Brain Dis. 1987, 2, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Torrellas, Y.; Pérez Aguilar, M.C.; Ramos, B.; Franco Useche, A.; Ibarra, A.; Rodríguez Bonfante, C.; Bonfante Cabarcas, R. Increased adenosine deaminase serum activity in patients with acute myocardial infarction. Latinoam. Hipertens. 2012, 5, 38–42. [Google Scholar]
- Merrill, G.F.; Downey, H.F.; Jones, C.E. Adenosine deaminase attenuates canine coronary vasodilation during systemic hypoxia. Am. J. Physiol. Circ. Physiol. 1986, 250, H579–H583. [Google Scholar] [CrossRef] [PubMed]
- Bernauer, W. Effect of exogenous adenosine deaminase on arrhythmias and the release of adenine nucleotide catabolites in isolated rat hearts with coronary occlusion and reperfusion. Naunyn. Schmiedebergs. Arch. Pharmacol. 1991, 344, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Smolenski, R.T.; Raisky, O.; Slominska, E.M.; Abunasra, H.; Kalsi, K.K.; Jayakumar, J.; Suzuki, K.; Yacoub, M.H. Protection from reperfusion injury after cardiac transplantation by inhibition of adenosine metabolism and nucleotide precursor supply. Circulation 2001, 104, I246–I252. [Google Scholar] [CrossRef] [Green Version]
- Smolenski, R.T.; Kalsi, K.K.; Zych, M.; Kochan, Z.; Yacoub, M.H. Adenine/ribose supply increases adenosine production and protects ATP pool in adenosine kinase-inhibited cardiac cells. J. Mol. Cell. Cardiol. 1998, 30, 673–683. [Google Scholar] [CrossRef]
- McClanahan, T.B.; Ignasiak, D.P.; Martin, B.J.; Mertz, T.E.; Gallagher, K.P. Effect of adenosine deaminase inhibition with pentostatin on myocardial stunning in dogs. Basic Res. Cardiol. 1995, 90, 176–183. [Google Scholar] [CrossRef]
- Oladipo, O.O.; Afolabi, B.B.; Okorodudu, A.O. Adenosine Deaminase Activity in Subjects With Normal Pregnancy, Pregnancy Induced Hypertension and Pre-Eclampsia. West Afr. J. Med. 2009, 28. [Google Scholar] [CrossRef] [Green Version]
- Abadier, M.; Pramod, A.B.; McArdle, S.; Marki, A.; Fan, Z.; Gutierrez, E.; Groisman, A.; Ley, K. Effector and Regulatory T Cells Roll at High Shear Stress by Inducible Tether and Sling Formation. Cell Rep. 2017, 21, 3885–3899. [Google Scholar] [CrossRef] [Green Version]
- Kutryb-Zajac, B.; Koszalka, P.; Mierzejewska, P.; Bulinska, A.; Zabielska, M.A.; Brodzik, K.; Skrzypkowska, A.; Zelazek, L.; Pelikant-Malecka, I.; Slominska, E.M.; et al. Adenosine deaminase inhibition suppresses progression of 4T1 murine breast cancer by adenosine receptor-dependent mechanisms. J. Cell. Mol. Med. 2018, 22, 5939–5954. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Capisano, M.C.; Atchison, D.K.; Harding, P.; Lasley, R.D.; Beierwaltes, W.H. Adenosine inhibits renin release from juxtaglomerular cells via an A1 receptor-TRPC-mediated pathway. Am. J. Physiol. Renal Physiol. 2013, 305, F1209–F1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, C.-J.; Wells, J.N.; Jackson, E.K. Endogenous Adenosine Restrains Renin Release in Conscious Rats. Circ. Res. 1990, 66, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofovic, S.P.; Kusaka, H.; Li, P.; Jackson, E.K. Effects of adenosine deaminase inhibition on blood pressure in old spontaneously hypertensive rats. Clin Exp Hypertens 1998, 20, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Rao, X.; Zhong, J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. J. Diabetes Res. 2017, 2017, 6494795. [Google Scholar] [CrossRef]
- Niraula, A.; Thapa, S.; Kunwar, S.; Lamsal, M.; Baral, N.; Maskey, R. Adenosine deaminase activity in type 2 diabetes mellitus: Does it have any role? BMC Endocr. Disord. 2018, 18, 58. [Google Scholar] [CrossRef]
- Sapkota, L.B.; Thapa, S.; Subedi, N. Correlation study of adenosine deaminase and its isoenzymes in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2017, 5, e000357. [Google Scholar] [CrossRef] [Green Version]
- Larijani, B.; Heshmat, R.; Ebrahimi-Rad, M.; Khatami, S.; Valadbeigi, S.; Saghiri, R. Diagnostic Value of Adenosine Deaminase and Its Isoforms in Type II Diabetes Mellitus. Enzyme Res. 2016, 2016, 9526593. [Google Scholar] [CrossRef]
- Takhshid, M.A.; Zahediannejad, Z.; Aboualizadeh, F.; Moezzi, L.; Ranjbaran, R. G22A Polymorphism of Adenosine Deaminase and its Association with Biochemical Characteristics of Gestational Diabetes Mellitus in an Iranian Population. Iran. J. Med. Sci. 2015, 40, 170–174. [Google Scholar]
- Kather, H. Pathways of purine metabolism in human adipocytes. Further evidence against a role of adenosine as an endogenous regulator of human fat cell function. J. Biol. Chem. 1990, 265, 96–102. [Google Scholar]
- Koupenova, M.; Ravid, K. Adenosine, adenosine receptors and their role in glucose homeostasis and lipid metabolism. J. Cell. Physiol. 2013, 228, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Thliveris, J.A.; Begleiter, A.; Kobrinsky, N.L.; Verburg, L.; Dean, H.J.; Johnston, J.B. Prevention of insulin-dependent diabetes mellitus by 2′-deoxycoformycin in the BB Wistar rat. Biochem. Pharmacol. 1993, 46, 1071–1075. [Google Scholar] [CrossRef]
- Zhong, J.; Gong, Q.; Goud, A.; Srinivasamaharaj, S.; Rajagopalan, S. Recent Advances in Dipeptidyl-Peptidase-4 Inhibition Therapy: Lessons from the Bench and Clinical Trials. J. Diabetes Res. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufinatscha, K.; Radlinger, B.; Dobner, J.; Folie, S.; Bon, C.; Profanter, E.; Ress, C.; Salzmann, K.; Staudacher, G.; Tilg, H.; et al. Dipeptidyl peptidase-4 impairs insulin signaling and promotes lipid accumulation in hepatocytes. Biochem. Biophys. Res. Commun. 2017, 485, 366–371. [Google Scholar] [CrossRef]
- Lee, J.G.; Kang, D.G.; Yu, J.R.; Kim, Y.; Kim, J.; Koh, G.; Lee, D. Changes in Adenosine Deaminase Activity in Patients with Type 2 Diabetes Mellitus and Effect of DPP-4 Inhibitor Treatment on ADA Activity. Diabetes Metab. J. 2011, 35, 149–158. [Google Scholar] [CrossRef] [Green Version]
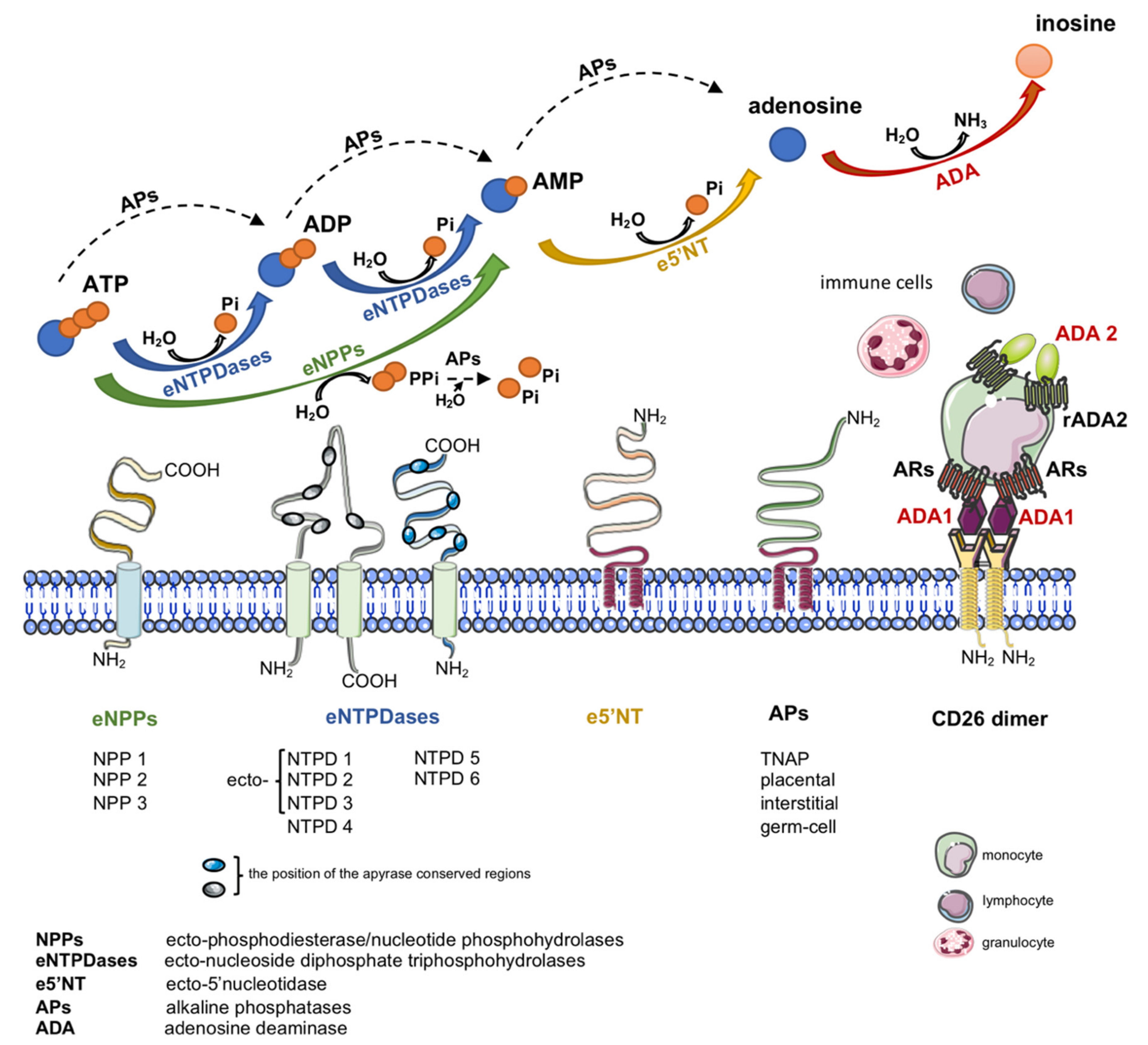

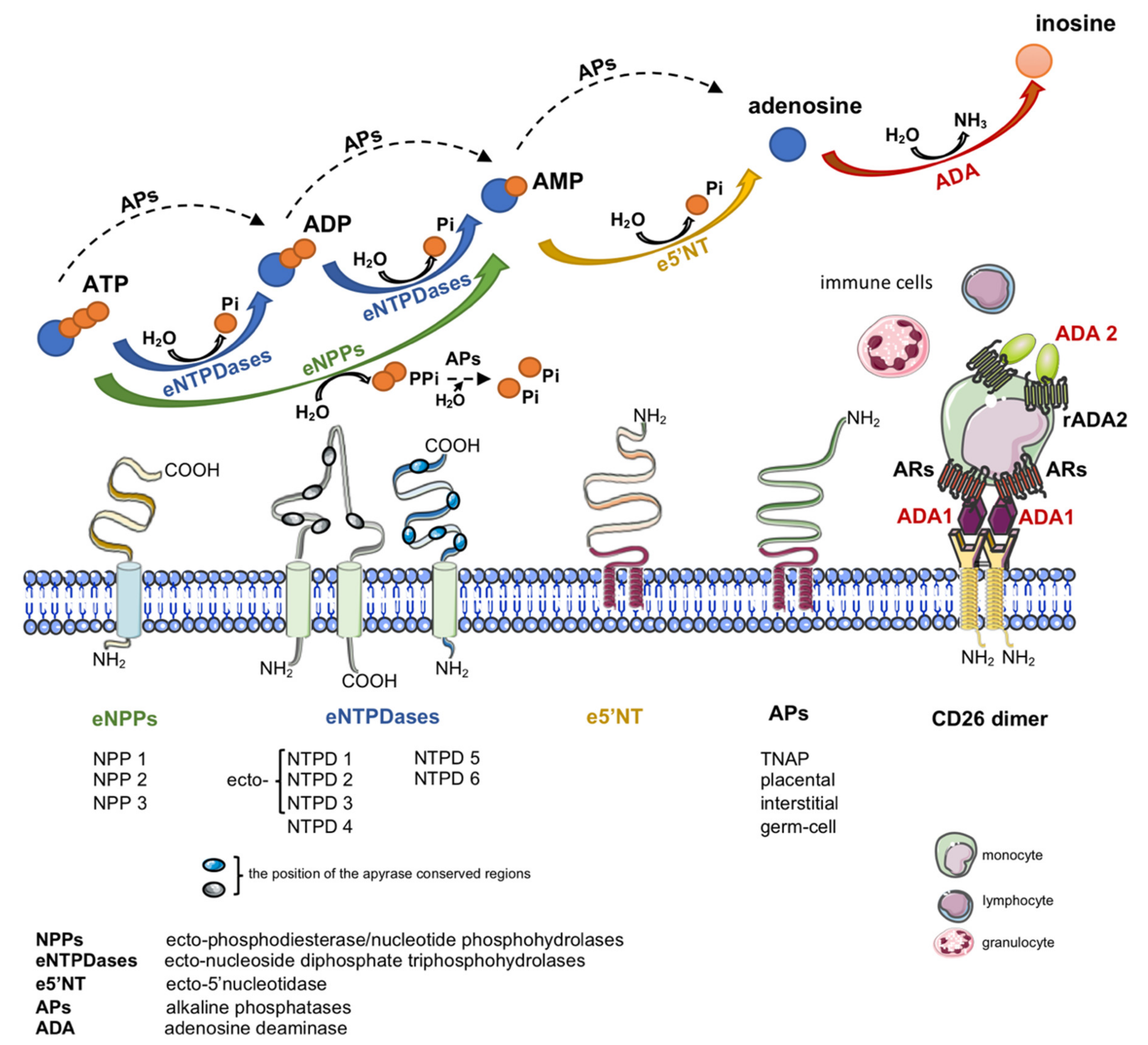{kind=link}
| Adenosine Deaminase Inhibitor | Ki Value (or IC50) | Ref. |
|---|---|---|
| Transition-state inhibitors | ||
| Coformycin | 10 pM | [50] |
| 2′-Deoxycoformycin | 2.5 pM | [51] |
| 4R-(1-Hydroxyethyl)-5-methyl-1-β-D-ribofuranosylimidazole | 61 µM | [52] |
| 2′-Chloro-2′deoxyadenosine (cladribine) | (0.2 µM) | [53,54] |
| 2′-Chloro-2′deoxycoformycin | 0.53 nM | [55] |
| Adecypenol | 47 nM | [56] |
| Ground-state inhibitors | ||
| 1-Deazaadenosine | 0.66 µM | [57] |
| 2′-Deoxy-1-deazaadenozine | 0.19 µM | [58] |
| N6-Hydroxy-2′deoxy-1-deazaadenozine | 0.25 µM | [59] |
| N6-Methyl-2′deoxy-1-deazaadenozine | 1.2 µM | [59] |
| N6-Cyclopropyl-2′deoxy-1-deazaadenozine | 5.9 µM | [59] |
| 3′-Deoxy-1-deazaadenosine | 2.6 µM | [60] |
| 2′3′-Dideoxy-1-deazaadenosine | 2.2 µM | [60] |
| Erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA) | 1.6 nM | [61] |
| 9′-Chloro-EHNA | 2.7 nM | [62] |
| 9′-Phthalimido-EHNA | 0.95 nM | [62] |
| Fluorescent derivatives of epsilon-EHNA | 2.8 µM | [63] |
| 1-DeazaEHNA | 0.16 µM | [64] |
| 3-DeazaEHNA | 0.01 µM | [64] |
| Erythro-1-(2-hydroxy-3-nonyl)imidazole | 0.90 µM | [65] |
| Erythro-9-(2-hydroxy-3-nonyl)imidazole-4-carboxamide | 0.035 µM | [65] |
| Erythro-9-(2-hydroxy-3-nonyl)1,2,4-triazole | 0.3 µM | [66] |
| Non-nucleoside inhibitors | ||
| 1-(1-Hydroxy-4-phenylbutan-2-yl)-1H-imidazole-4-carboxamide | 5.9 µM | [67] |
| 1-((1R,2S)-2-Hydroxy-1-(2-(1-naphthyl)ethyl)propyl)-1H-imidazole-4-carboxamide (FR234938) | 3.6 nM | [68] |
| Flavonoids and sapogenins/plant extracts | ||
| Kaempherol | (30 µM) | [69] |
| Quercetin | (30 µM) | [69] |
| Hibifolin | 50 µM | [70] |
| Naringrin | 200 µM | [71] |
| Curcumin | (13.6 µM) | [72] |
| Genistein | (1.5 mM) | [73] |
| Cyanidin-3-rutinoside | (0.95 mM) | [73] |
| Acidic sapogenins 3-(4-Nitrophenyl)-5-phenyl isoxazole | 1 µM (0.380 mM) | [74] [75] |
| Drugs | ||
| Acetaminophen | 214 µM | [76] |
| Diclofenac | 30 µM | [77] |
| Aspirin | 43 µM | [77] |
| Lidoflazine | 30 µM | [78] |
| Phenylbutazone | 54 µM | [78] |
| Chlordiazepoxide | 83 µM | [78] |
| Tradozon | 60 µM | [79] |
| Cardiovascular Pathology | ADA Activity | ADA Inhibitor | Therapeutic Effect of ADA Inhibition |
|---|---|---|---|
| Atherosclerosis | ↑ tADA (plasma) [112] ↑ ADA1 (vessel wall) [115] | dCF [115] | + |
| Thrombosis | ↑ tADA (plasma) [134] | n.d. | n.d. |
| AMI/IRI | ↑ tADA (plasma) [138,139] | dCF [151] | + |
| Hypertension | ↑ tADA (plasma) [112] | EHNA [157] | + |
| T2DM | ↑ tADA (plasma) [159] ↑ ADA1 (plasma) [160,161] ↑ ADA2 (plasma) [160,161] | dCF [165] | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutryb-Zajac, B.; Mierzejewska, P.; Slominska, E.M.; Smolenski, R.T. Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases. Molecules 2020, 25, 4652. https://doi.org/10.3390/molecules25204652
Kutryb-Zajac B, Mierzejewska P, Slominska EM, Smolenski RT. Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases. Molecules. 2020; 25(20):4652. https://doi.org/10.3390/molecules25204652
Chicago/Turabian StyleKutryb-Zajac, Barbara, Paulina Mierzejewska, Ewa M. Slominska, and Ryszard T. Smolenski. 2020. "Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases" Molecules 25, no. 20: 4652. https://doi.org/10.3390/molecules25204652
APA StyleKutryb-Zajac, B., Mierzejewska, P., Slominska, E. M., & Smolenski, R. T. (2020). Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases. Molecules, 25(20), 4652. https://doi.org/10.3390/molecules25204652