
Dihomooxacalix[4]arene-Based Fluorescent Receptors for Anion and Organic Ion Pair Recognition

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Structural Analysis
2.2. Anion Complexation
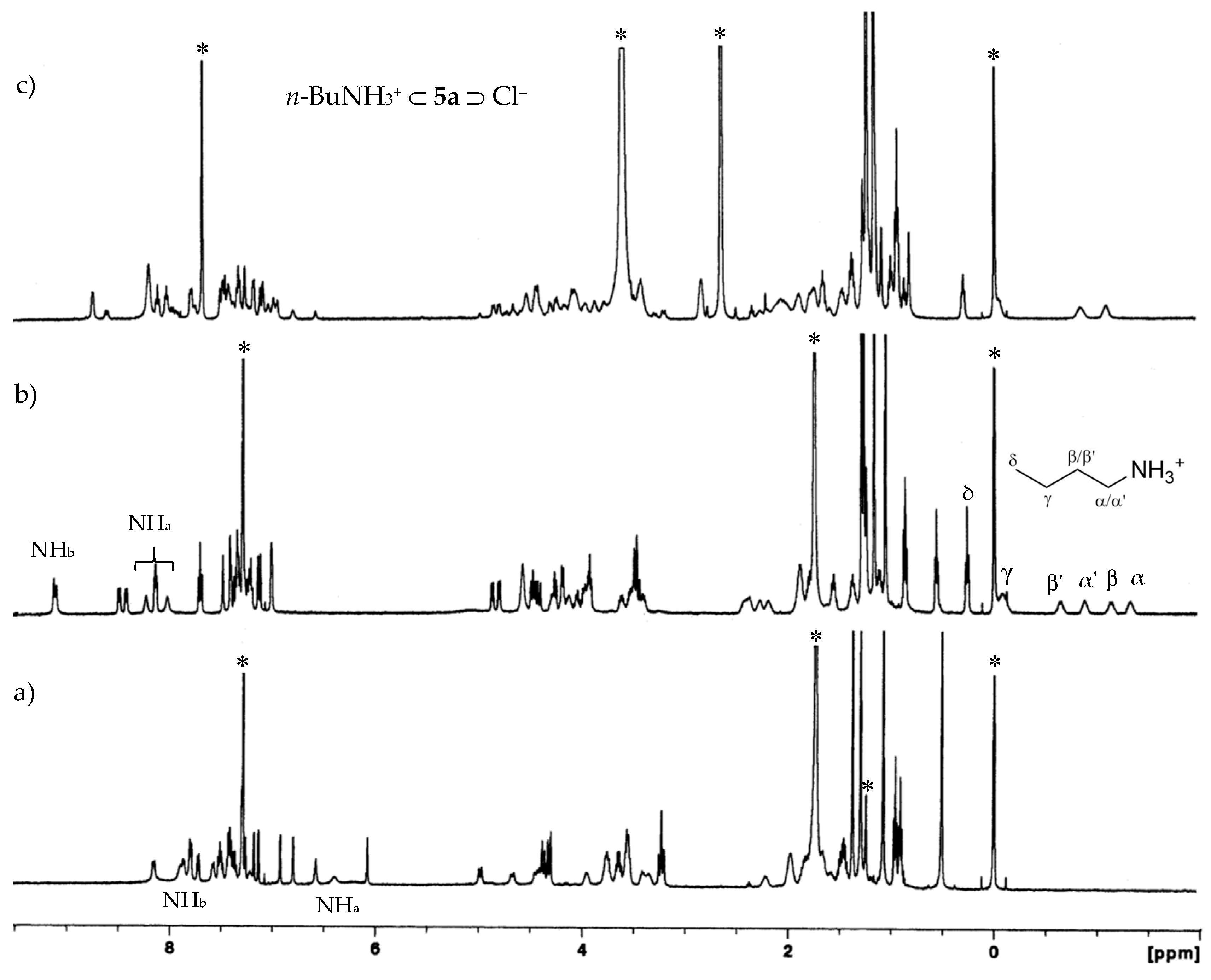
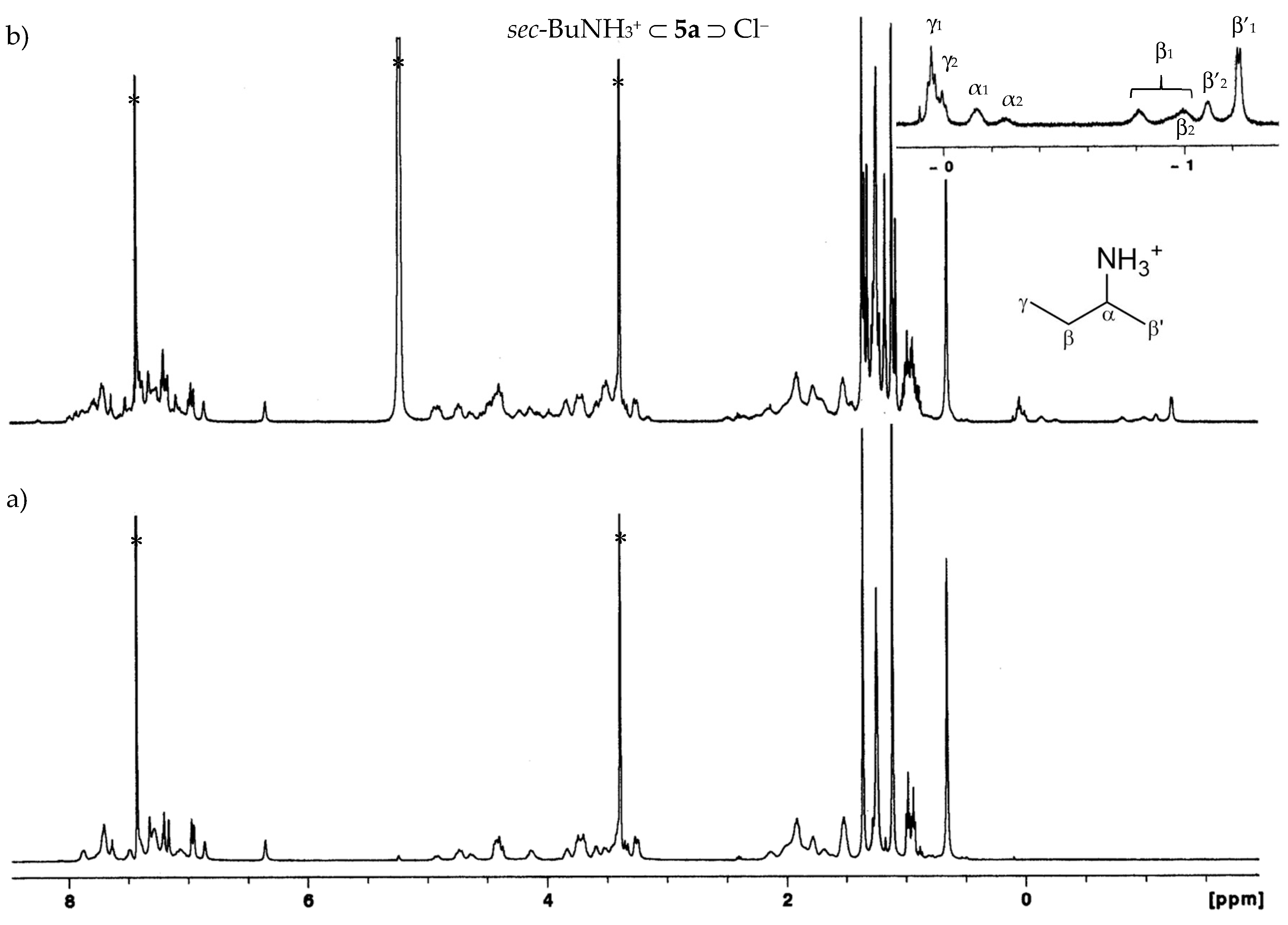
2.2.1. Proton NMR Studies
2.2.2. UV-Vis Absorption and Fluorescence Studies
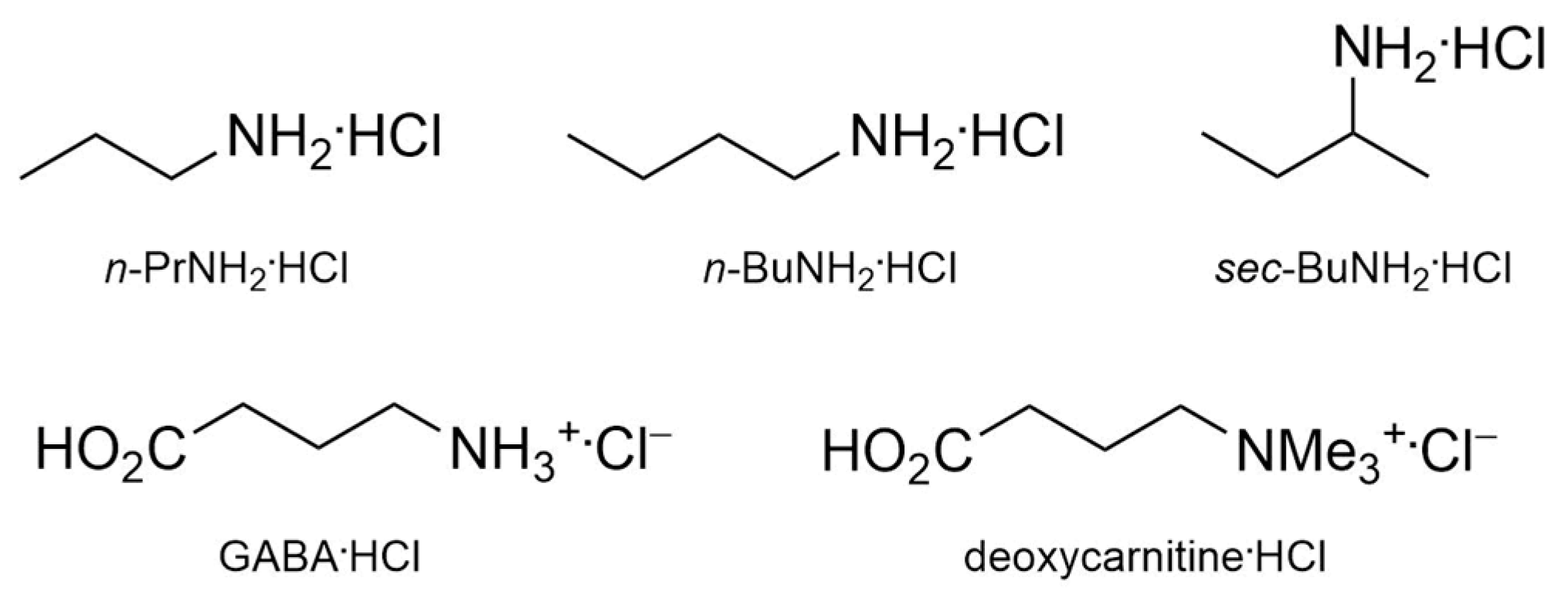
2.3. Organic Ion Pair Recognition
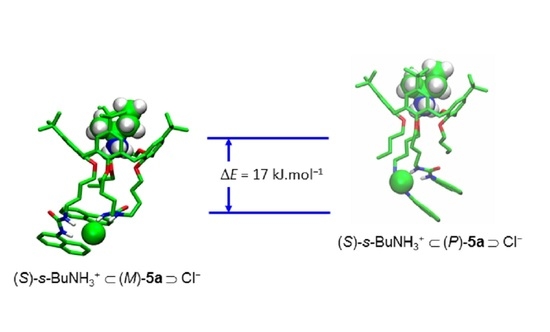
2.4. Theoretical Studies
3. Materials and Methods
3.1. Synthesis
3.1.1. Procedure for the Synthesis of (thio)ureas 5a and 5c
3.1.2. Procedure for the Synthesis of Symmetric Urea 5b. Precursor 2b has Already been Obtained and 3b and 4b were Synthesised as Described for 3a and 4a
3.2. Determination of the Crystallographic Structures of 5a and 5b
3.3. H-NMR Titrations
3.4. UV-Vis Absorption and Fluorescence Studies
3.5. Quantum Chemistry Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Gutsche, C.D. Calixarenes: An Introduction: Edition 2, 2nd ed.; The Royal Society of Chemistry: Cambridge, UK, 2008; Monographs in Supramolecular Chemistry. [Google Scholar]
- Neri, P.; Sessler, J.L.; Wang, M.-X. Calixarenes and Beyond; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Kumar, R.; Jung, Y.; Kim, J.S. Fluorescent calixarene hosts. In Calixarenes and Beyond; Neri, P., Sessler, J.L., Wang, M.-X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 743–760. [Google Scholar]
- Kumar, R.; Sharma, A.; Singh, H.; Suating, P.; Kim, H.S.; Sunwoo, K.; Shim, I.; Gibb, B.C.; Kim, J.S. Revisiting fluorescent calixarenes: From molecular sensors to smart materials. Chem. Rev. 2019, 119, 9657–9721. [Google Scholar] [CrossRef]
- Kim, S.K.; Bok, J.H.; Bartsch, R.A.; Lee, J.Y.; Kim, J.S. A fluoride-selective PCT chemosensor based on formation of a static pyrene excimer. Org. Lett. 2005, 7, 4839–4842. [Google Scholar] [CrossRef] [PubMed]
- Jeon, N.J.; Ryu, B.J.; Lee, B.H.; Nam, K.C. Fluorescent sensing of tetrahedral anions with a pyrene urea derivative of calix[4]arene chemosensor. Bull. Kor. Chem. Soc. 2009, 30, 1675–1677. [Google Scholar]
- Ryu, B.J.; Jeon, N.J.; Nam, K.C. 1,3-Alternate calix[4]arene bifuntional fluorescent receptor containing urea and crown ether moieties. Bull. Korean Chem. Soc. 2010, 31, 3445–3447. [Google Scholar] [CrossRef] [Green Version]
- Patra, S.; Gunupuru, R.; Lo, R.; Suresh, E.; Ganguly, B.; Paul, P. Cation-induced fluorescent excimer emission in calix[4]arene-chemosensors bearing quinoline as a fluorogenic unit: Experimental, molecular modeling and crystallographic studies. New J. Chem. 2012, 36, 988–1002. [Google Scholar] [CrossRef]
- Sutariya, P.G.; Pandya, A.; Lodha, A.; Menon, S.K. A pyrenyl linked calix[4]arene fluorescence probe for recognition of ferric and phosphate ions. RSC Advances 2014, 4, 34922–34926. [Google Scholar] [CrossRef]
- Uttam, B.; Kandi, R.; Hussain, M.A.; Rao, C.P. Fluorescent lower rim 1,3-dibenzooxadiazole conjugate of calix[4]arene in selective sensing of fluoride in solution and in biological cells using confocal microscopy. J. Org. Chem. 2018, 83, 11850–11859. [Google Scholar] [CrossRef]
- Capici, C.; De Zorzi, R.; Gargiulli, C.; Gattuso, G.; Geremia, S.; Notti, A.; Pappalardo, S.; Parisi, M.F.; Puntoriero, F. Calix[5]crown-3-based heteroditopic receptors for n-butylammonium halides. Tetrahedron 2010, 66, 4987–4993. [Google Scholar] [CrossRef]
- Jeon, N.J.; Ryu, B.J.; Park, K.D.; Lee, Y.J.; Nam, K.C. Tetrahedral anions selective fluorescent calix[6]arene receptor containing urea and pyrene moieties. Bull. Korean Chem. Soc. 2010, 31, 3809–3811. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, E.; Picron, J.-F.; Flidrova, K.; Bruylants, G.; Bartik, K.; Jabin, I. Fluorescent chemosensors for anions and contact ion pairs with a cavity-based selectivity. J. Org. Chem. 2014, 79, 6179–6188. [Google Scholar]
- Brunetti, E.; Moerkerke, S.; Wouters, J.; Bartik, K.; Jabin, I. A selective calix[6]arene-based fluorescent chemosensor for phosphatidylcholine type lipids. Org. Biomol. Chem. 2016, 14, 10201–10207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of supramolecular anion recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef] [PubMed]
- Gale, P.A.; Howe, E.N.W.; Wu, X. Anion receptor chemistry. Chem 2016, 1, 351–422. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Sessler, J.L. Ion pair receptors. Chem. Soc. Rev. 2010, 39, 3784–3809. [Google Scholar] [CrossRef] [PubMed]
- McConnell, A.J.; Beer, P.D. Heteroditopic receptors for ion-pair recognition. Angew. Chem. Int. Ed. 2012, 51, 5052–5061. [Google Scholar] [CrossRef] [PubMed]
- Marcos, P.M.; Teixeira, F.A.; Segurado, M.A.P.; Ascenso, J.R.; Bernardino, R.J.; Michel, S.; Hubscher-Bruder, V. Bidentate urea derivatives of p-tert-butyldihomooxacalix[4]arene: Neutral receptors for anion complexation. J. Org. Chem. 2014, 79, 742–751. [Google Scholar] [CrossRef]
- Gattuso, G.; Notti, A.; Parisi, M.F.; Pisagatti, I.; Marcos, P.M.; Ascenso, J.R.; Brancatelli, G.; Geremia, S. Selective recognition of biogenic amine hydrochlorides by heteroditopic dihomooxacalix[4]arenes. New J. Chem. 2015, 39, 817–821. [Google Scholar] [CrossRef]
- Teixeira, F.A.; Marcos, P.M.; Ascenso, J.R.; Brancatelli, G.; Hickey, N.; Geremia, S. Selective binding of spherical and linear anions by tetraphenyl(thio)urea-based dihomooxacalix[4]arene receptors. J. Org. Chem. 2017, 82, 11383–11390. [Google Scholar] [CrossRef]
- Augusto, A.S.; Miranda, A.S.; Ascenso, J.R.; Miranda, M.Q.; Félix, V.; Brancatelli, G.; Hickey, N.; Geremia, S.; Marcos, P.M. Anion recognition by partial cone dihomooxacalix[4]arene-based receptors bearing urea groups: Remarkable affinity for benzoate ion. Eur. J. Org. Chem. 2018, 2018, 5657–5667. [Google Scholar] [CrossRef]
- Miranda, A.S.; Serbetci, D.; Marcos, P.M.; Ascenso, J.R.; Berberan-Santos, M.N.; Hickey, N.; Geremia, S. Ditopic receptors based on dihomooxacalix[4]arenes bearing phenylurea moieties with electron-withdrawing groups for anions and organic ion pairs. Front. Chem. 2019, 7, 758. [Google Scholar] [CrossRef]
- Miranda, A.S.; Martelo, L.M.; Fedorov, A.A.; Berberan-Santos, M.N.; Marcos, P.M. Fluorescence properties of p-tert-butyldihomooxacalix[4]arene derivatives and the effect of anion complexation. New J. Chem. 2017, 41, 5967–5973. [Google Scholar] [CrossRef]
- Jaime, C.; de Mendoza, J.; Prados, P.; Nieto, P.; Sanchez, C. Carbon-13 NMR chemical shifts. A single rule to determine the conformation of calix[4]arenes. J. Org. Chem. 1991, 56, 3372–3376. [Google Scholar] [CrossRef]
- Hynes, M.J. EQNMR: A computer program for the calculation of stability constants from nuclear magnetic resonance chemical shift data. J.Chem. Soc., Dalton Trans. 1993, 311–312. [Google Scholar] [CrossRef]
- Stibor, I.; Budka, J.; Michlova, V.; Tkadlecova, M.; Pojarova, M.; Curinova, P.; Lhotak, P. Systematic approach to new ligands for anion recognition based on ureido-calix[4]arenes. New J. Chem. 2008, 32, 1597–1607. [Google Scholar] [CrossRef]
- Marcos, P.M.; Teixeira, F.A.; Segurado, M.A.P.; Ascenso, J.R.; Bernardino, R.J.; Brancatelli, G.; Geremia, S. Synthesis and anion binding properties of new dihomooxacalix[4]arene diurea and dithiourea receptors. Tetrahedron 2014, 70, 6497–6505. [Google Scholar] [CrossRef]
- Teixeira, F.A.; Ascenso, J.R.; Cragg, P.J.; Hickey, N.; Geremia, S.; Marcos, P.M. Recognition of anions, monoamine neurotransmitter and trace amine hydrochlorides by ureido-hexahomotrioxacalix[3]arene ditopic receptors. Eur. J. Org. Chem. 2020, 1930–1940. [Google Scholar] [CrossRef]
- Scheerder, J.; Fochi, M.; Engbersen, F.J.; Reinhoudt, D.N. Urea-derivatized p-tert-butylcalix[4]arenes: Neutral ligands for selective anion complexation. J. Org. Chem. 1994, 59, 7815–7820. [Google Scholar] [CrossRef] [Green Version]
- Hamon, M.; Ménand, M.; Le Gac, S.; Luhmer, M.; Dalla, V.; Jabin, I. Calix[6]tris(thio)ureas: Heteroditopic receptors for the cooperative binding of organic ion pairs. J. Org. Chem. 2008, 73, 7067–7071.
- Bryantsev, V.S.; Hay, B.P. Conformational preferences and internal rotation in alkyl- and phenyl-substituted thiourea derivatives. J. Phys. Chem. A 2006, 110, 4678–4688. [Google Scholar] [CrossRef]
- Mei, M.; Wu, S. Fluorescent sensor for α,ω-dicarboxylate anions. New J Chem. 2001, 25, 471–475. [Google Scholar] [CrossRef]
- Gaeta, C.; Talotta, C.; Farina, F.; Teixeira, F.A.; Marcos, P.M.; Ascenso, J.R.; Neri, P. Alkylammonium cation complexation into the narrow cavity of dihomooxacalix[4]arene macrocycle. J. Org. Chem. 2012, 77, 10285–10293. [Google Scholar] [CrossRef] [PubMed]
- Due to the inherent chirality of dihomooxacalix[4]arenes, (P) and (M) descriptors were used as an alternative to the usual (R) and (S) notation. For this subject see: (a) Szumna, A. Inherently chiral concave molecules – from synthesis to applications. Chem. Soc. Rev. 2010, 39, 4274–4285, (b) ref. 22. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallog. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallog. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallog. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Rev. D.01; Gaussian Inc.: Wallingford, CT, UAS, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A (°) | B (°) | C (°) | D (°) | |
|---|---|---|---|---|
| 5a | 124 a | 69 | 143 a | 99 |
| 5b | 125 | 83 | 140 a | 88 a |
| p-CF3-Phurea b | 125 a | 66 | 131 a | 101 |
| Phurea c (I) | 123 a | 64 | 133 a | 100 |
| Phure ac (II) | 121 a | 74 | 137 a | 97 |
| Spherical | Trigonal Planar | Tetrahedral | |||||||
|---|---|---|---|---|---|---|---|---|---|
| F− | Cl− | Br− | I− | NO3− | AcO− | BzO− | HSO4− | H2PO4− | |
| I. Radius/Å b | 1.33 | 1.81 | 1.96 | 2.20 | 1.79 | 2.32 | — | 1.90 | 2.00 |
| 5a | 2.80 | 2.60 | 2.12 | 1.78 | 2.12 | 2.51 | 2.76 | 2.54 | 2.25 |
| 5b | 3.12 | 2.91 | 2.46 | 1.94 | 2.38 | 3.17 | 3.07 | 2.52 | 2.67 |
| 5c | 2.67 | 1.75 | 1.06 | 1.09 | 1.06 | 2.17 | 2.01 | 1.89 | 2.02 |
| λmax,abs (nm) | ε (M−1 cm−1) | λmax,em (nm) | Stokes Shift a (nm) | τf (ns) | ΦF | kr (ns−1) | knr (ns−1) | |
|---|---|---|---|---|---|---|---|---|
| 5a | 283 | 382 | 99 | 8.93 | 0.31 b | 0.035 | 0.077 | |
| 5a + Cl− | 303 | 382 | 79 | 6.26 | 0.085 b | — | — | |
| 5a + BzO− | 303 | 382 | 79 | 1.98 | 0.048 b | 0.024 | 0.48 | |
| 5b | 282 | 379 | 97 | 7.57 | 0.26 b | 0.034 | 0.098 | |
| 5b + Cl− | 302 | 379 | 77 | 6.63 | 0.16 b | — | — | |
| 5b + BzO− | 302 | 379 | 77 | 3.27 | 0.078 b | 0.024 | 0.28 |
| ∆E (kJ.mol−1) | ||
|---|---|---|
| Host | 5a | 5b |
| F− | −498.9 | −557.1 |
| Cl− | −202.5 | −219.0 |
| AcO− | −261.5 | −291.4 |
| BzO− | −232.1 | −283.3 |
| HSO4− | −192.3 | −206.2 |
| H2PO4− | −220.6 | −236.6 |
| n-PrNH3+·Cl− | −736.3 | −811.6 |
| n-BuNH3+·Cl− | −656.5 | −709.5 |
| (R)-sec-BuNH3+·Cl−/(M)-5a | −704.1 | — |
| (S)-sec-BuNH3+·Cl−/(M)-5a | −712.4 | — |
| (R)-sec-BuNH3+·Cl−/(P)-5a | −691.1 | — |
| (S)-sec-BuNH3+·Cl−/(P)-5a | −695.4 | — |
| Spherical | Trigonal Planar | Tetrahedral | |||||||
|---|---|---|---|---|---|---|---|---|---|
| F− | Cl− | Br− | NO3− | AcO− | BzO− | HSO4− | H2PO4− | ||
| 5a | Abs | 4.21 | 3.59 | 3.20 | 3.19 | 3.79 | 3.94 | 3.13 | 3.06 |
| Emi | 4.05 | 3.48 | 3.14 | 3.12 | 3.66 | 4.00 | 3.01 | 2.90 | |
| 5b | Abs | 4.36 | 3.69 | 3.37 | 3.31 | 4.16 | 4.08 | 2.90 | 3.16 |
| Emi | 4.34 | 3.67 | 3.23 | 3.23 | 4.21 | 3.84 | 2.97 | 3.14 | |
| 5c | Abs | 4.18 | 3.01 | 2.71 | 3.07 | 3.66 | 3.71 | 2.87 | 3.14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, A.S.; Marcos, P.M.; Ascenso, J.R.; Berberan-Santos, M.N.; Schurhammer, R.; Hickey, N.; Geremia, S. Dihomooxacalix[4]arene-Based Fluorescent Receptors for Anion and Organic Ion Pair Recognition. Molecules 2020, 25, 4708. https://doi.org/10.3390/molecules25204708
Miranda AS, Marcos PM, Ascenso JR, Berberan-Santos MN, Schurhammer R, Hickey N, Geremia S. Dihomooxacalix[4]arene-Based Fluorescent Receptors for Anion and Organic Ion Pair Recognition. Molecules. 2020; 25(20):4708. https://doi.org/10.3390/molecules25204708
Chicago/Turabian StyleMiranda, Alexandre S., Paula M. Marcos, José R. Ascenso, Mário N. Berberan-Santos, Rachel Schurhammer, Neal Hickey, and Silvano Geremia. 2020. "Dihomooxacalix[4]arene-Based Fluorescent Receptors for Anion and Organic Ion Pair Recognition" Molecules 25, no. 20: 4708. https://doi.org/10.3390/molecules25204708