
Molecular Dynamic Simulations to Probe Stereoselectivity of Tiagabine Binding with Human GAT1



Abstract
:
1. Introduction
2. Results and Discussion
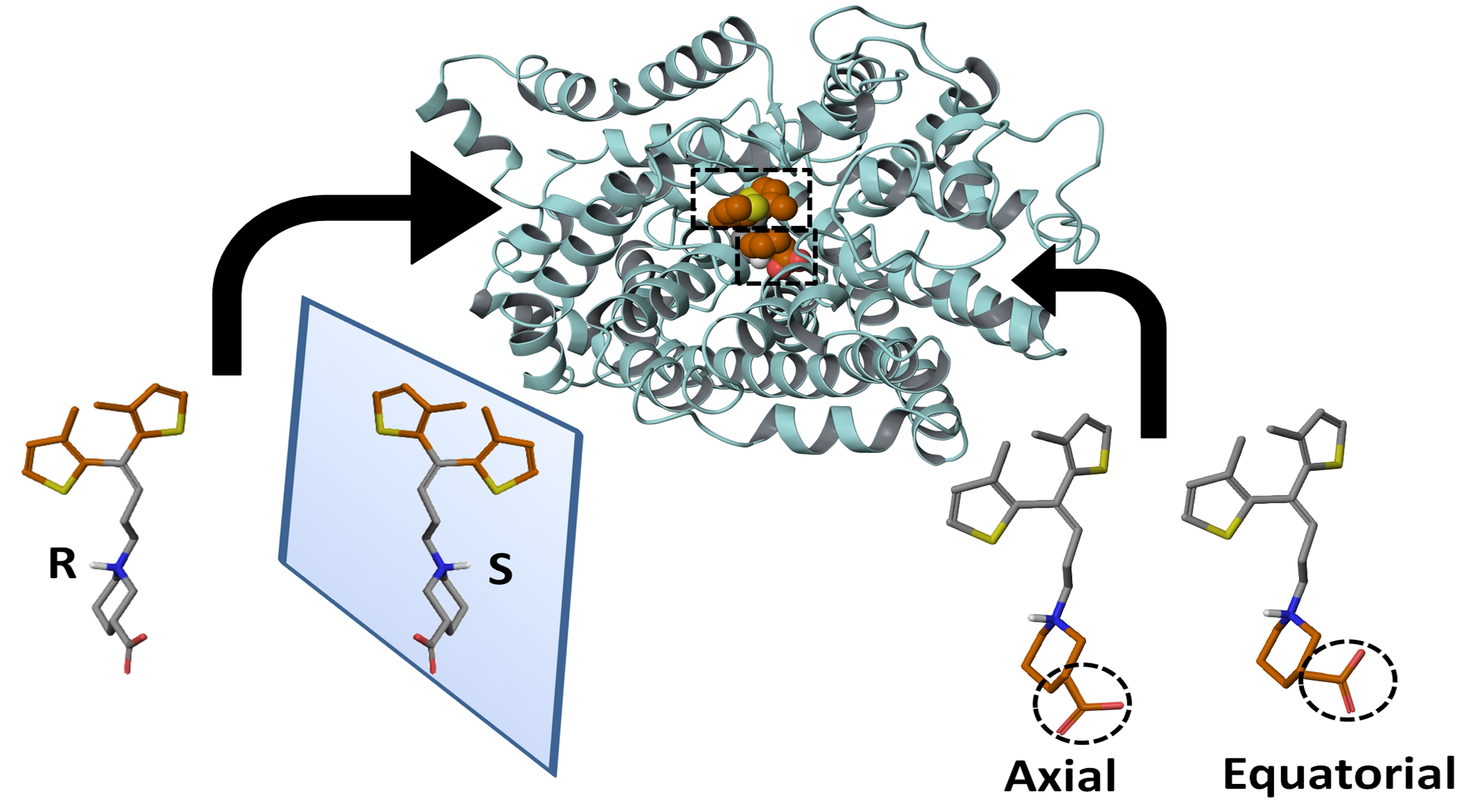
2.1. Docking and Clustering of R- and S-enantiomers of Tiagabine in hGAT1
2.2. Ligand–Protein Interaction Analysis of Unconstraint Docking Solutions
2.3. Ligand–Protein Interaction Analysis of Constraint Docking Solutions
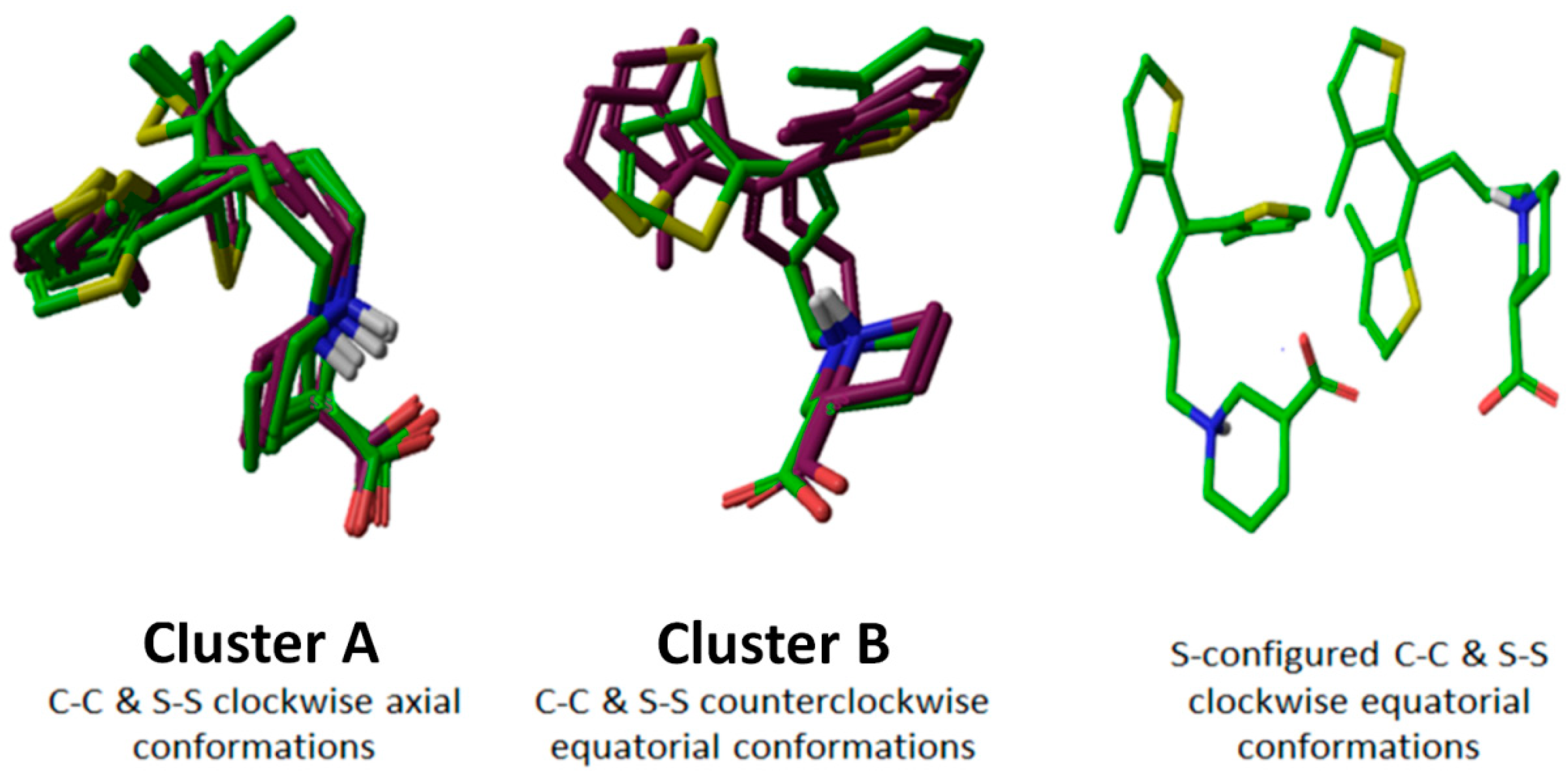
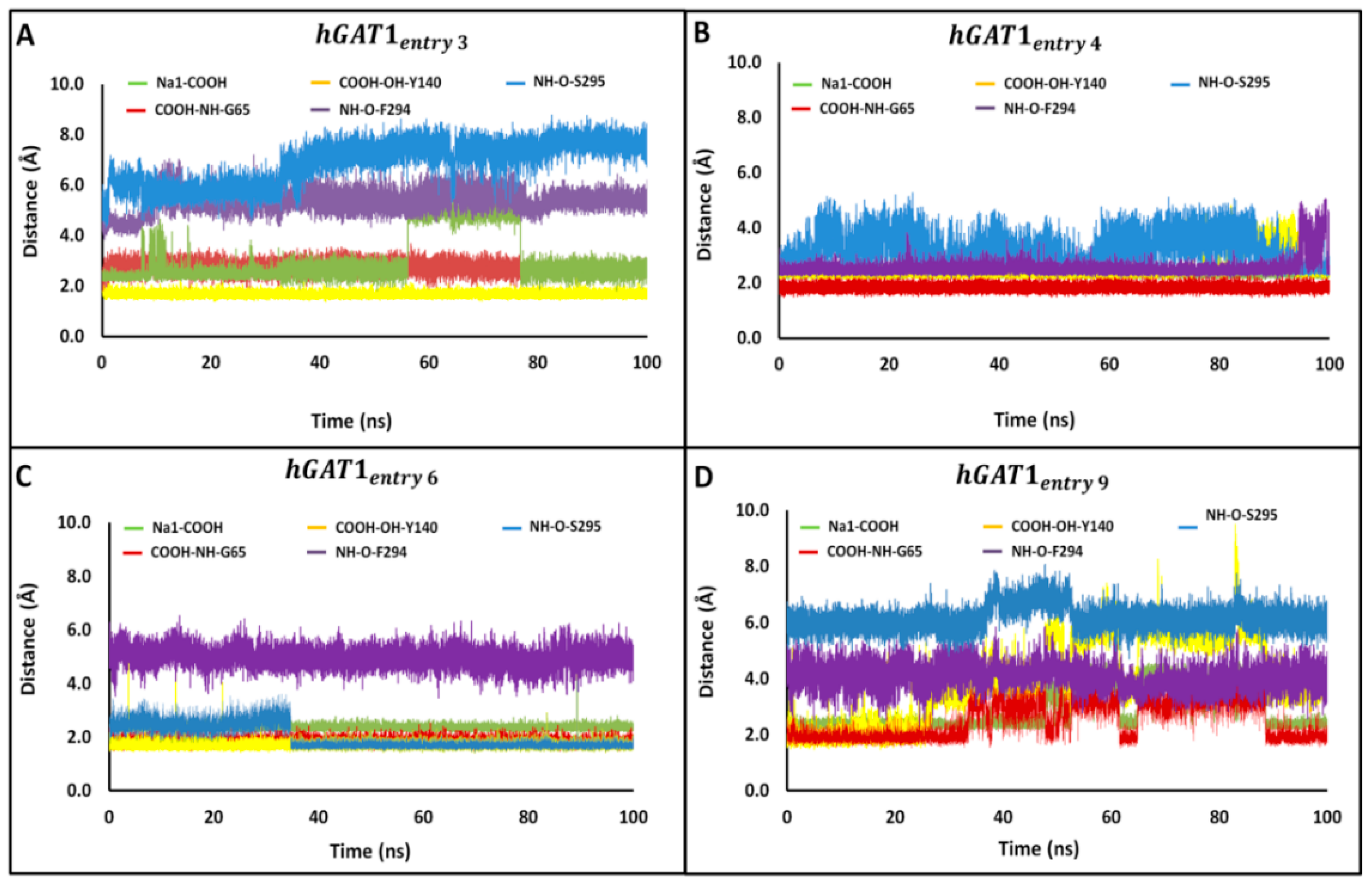
2.4. Molecular Dynamics of Selected Tiagabine Enantiomers in hGAT1
2.5. Cross Validation of hGAT1entry 4
3. Materials and Methods
3.1. Ligand Preparation
3.2. Protein Preparation and Molecular Docking Studies
3.2.1. Docking Protocol I (Unconstraint Docking Protocol)
3.2.2. Docking Protocol II (Constraint Docking Protocol)
3.3. Molecular Dynamics Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lovinger:, D.M. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 2010, 58, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckett, A.H. Chirality and Its Importance in Drug Development: What Are the Issues? Portland Press Limited: London, UK, 1991; pp. 443–446. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G. GABA transporter GAT1: A crucial determinant of GABAB receptor activation in cortical circuits. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 58, pp. 175–204. [Google Scholar]
- Zafar, S.; Jabeen, I. Structure, Function, and Modulation of γ-Aminobutyric Acid Transporter 1 (GAT1) in Neurological Disorders: A Pharmacoinformatic Prospective. Front. Chem. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Nägga, K.; Bogdanovic, N.; Marcusson, J. GABA transporters (GAT-1) in Alzheimer’s disease. J. Neural Transm. 1999, 106, 1141–1149. [Google Scholar] [CrossRef]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, A.; Auta, J.; Davis, J.M.; Dong, E.; Grayson, D.R.; Veldic, M.; Zhang, X.; Costa, E. GABAergic dysfunction in schizophrenia: New treatment strategies on the horizon. Psychopharmacology 2005, 180, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Galvan, A.; Wichman, T.; Smith, Y. Localization and function of GABA transporters GAT-1 and GAT-3 in the basal ganglia. Front. Syst. Neurosci. 2011, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Richerson, G.B.; Wu, Y. Role of the GABA transporter in epilepsy. In Recent Advances in Epilepsy Research; Springer: Berlin/Heidelberg, Germany, 2004; Volume 548, pp. 76–91. [Google Scholar]
- Baglo, Y.; Gabrielsen, M.; Sylte, I.; Gederaas, O.A. Homology modeling of human γ-butyric acid transporters and the binding of pro-drugs 5-aminolevulinic acid and methyl aminolevulinic acid used in photodynamic therapy. PLoS ONE 2013, 8, e65200. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.; Kanner, B.I. The substrates of the γ-aminobutyric acid transporter GAT-1 induce structural rearrangements around the interface of transmembrane domains 1 and 6. J. Biol. Chem. 2008, 283, 14376–14383. [Google Scholar] [CrossRef] [Green Version]
- Savtchenko, L.; Megalogeni, M.; Rusakov, D.A.; Walker, M.C.; Pavlov, I. Synaptic GABA release prevents GABA transporter type-1 reversal during excessive network activity. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Deisz, R. PHARMACORESISTANCE. Cellular and Molecular Mechanisms Contributing to Pharmacoresistance in Human Neocortical Tissue. Encycl. Basic Epilepsy Res. 2009, 1133–1138. [Google Scholar] [CrossRef]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl-dependent neurotransmitter transporters. Nature 2005, 437, 215. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellenbrand, T.; Höfner, G.; Wein, T.; Wanner, K.T. Synthesis of 4-substituted nipecotic acid derivatives and their evaluation as potential GABA uptake inhibitors. Bioorg. Med. Chem. 2016, 24, 2072–2096. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.; Wein, T.; Höfner, G.; Wanner, K.T. Development of Highly Potent GAT1 Inhibitors: Synthesis of Nipecotic Acid Derivatives with N-Arylalkynyl Substituents. Chem. Med. Chem. 2017, 12, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, L.J.; Andersen, K.E.; Lau, J.; Lundt, B.F.; Henry, R.F.; Morton, H.E.; Nærum, L.; Petersen, H.; Stephensen, H.; Suzdak, P.D. Synthesis of novel GABA uptake inhibitors. 3. Diaryloxime and diarylvinyl ether derivatives of nipecotic acid and guvacine as anticonvulsant agents. J. Med. Chem. 1999, 42, 3447–3462. [Google Scholar] [CrossRef] [PubMed]
- Sałat, K.; Podkowa, A.; Mogilski, S.; Zaręba, P.; Kulig, K.; Sałat, R.; Malikowska, N.; Filipek, B. The effect of GABA transporter 1 (GAT1) inhibitor, tiagabine, on scopolamine-induced memory impairments in mice. Pharmacol. Rep. 2015, 67, 1155–1162. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, W.; Díez-Sampedro, A.; Richerson, G.B. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 2007, 56, 851–865. [Google Scholar] [CrossRef] [Green Version]
- Jacob, S.; Nair, A.B. An updated overview on therapeutic drug monitoring of recent antiepileptic drugs. Drugs RD 2016, 16, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Brady, M.L.; Pilli, J.; Lorenz-Guertin, J.M.; Das, S.; Moon, C.E.; Graff, N.; Jacob, T.C. Depolarizing, inhibitory GABA type A receptor activity regulates GABAergic synapse plasticity via ERK and BDNF signaling. Neuropharmacology 2018, 128, 324–339. [Google Scholar] [CrossRef]
- Zafar, S.; Jabeen, I. GRID-independent molecular descriptor analysis and molecular docking studies to mimic the binding hypothesis of γ-aminobutyric acid transporter 1 (GAT1) inhibitors. Peer J. 2019, 7, e6283. [Google Scholar] [CrossRef]
- Ben-Yona, A.; Bendahan, A.; Kanner, B.I. A glutamine residue conserved in the neurotransmitter: Sodium: Symporters is essential for the interaction of chloride with the GABA transporter GAT-1. J. Biol. Chem. 2011, 286, 2826–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovstrup, S.; Taboureau, O.; Bräuner-Osborne, H.; Jørgensen, F.S. Homology modelling of the GABA transporter and analysis of tiagabine binding. Chem. Med. Chem. 2010, 5, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Madsen, K.; White, H.; Clausen, R.P.; Larsson, O.M.; Krogsgaard-Larsen, P.; Schousboe, A. Functional and pharmacological aspects of GABA transporters. In Handbook of Nerochemistry and Molecular Neurobiology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 285–304. [Google Scholar]
- Jurik, A.; Zdrazil, B.; Holy, M.; Stockner, T.; Sitte, H.H.; Ecker, G.F. A binding mode hypothesis of tiagabine confirms liothyronine effect on γ-aminobutyric acid transporter 1 (GAT1). J. Med. Chem. 2015, 58, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.; Tropsha, A.; Winkler, D.A. Beware of R 2: Simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters, SC’06. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Singh, S.K.; Piscitelli, C.L.; Yamashita, A.; Gouaux, E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 322, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.D.; Caserio, M.C. Basic Principles of Organic Chemistry; Benjamin, WA Inc.: San Francisco, CA, USA, 1977; Volume 42. [Google Scholar]
- Borden, L.A.; Dhar, T.M.; Smith, K.E.; Weinshank, R.L.; Branchek, T.A.; Gluchowski, C. Tiagabine, SK&F 89976-A, CI-966, and NNC-711 are selective for the cloned GABA transporter GAT-1. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 269, 219–224. [Google Scholar] [CrossRef]
- Suzdak, P.D.; Frederiksen, K.; Andersen, K.E.; Sørensen, P.O.; Knutsen, L.J.; Nielsen, E.B. NNC-711, a novel potent and selective γ-aminobutyric acid uptake inhibitor: Pharmacological characterization. Eur. J. Pharmacol. 1992, 224, 189–198. [Google Scholar] [CrossRef]
- LigPrep; Schrödinger, LLC: New York, NY, USA. Available online: https://www.schrodinger.com/ligprep (accessed on 15 October 2020).
- Zafar, S.; Nguyen, M.E.; Muthyala, R.; Jabeen, I.; Sham, Y.Y. Modeling and simulation of hGAT1: A Mechanistic Investigation of the GABA Transport Process. Comput. Struct. Biotechnol. J. 2018. [Google Scholar] [CrossRef]
- Quick, M.; Winther, A.M.L.; Shi, L.; Nissen, P.; Weinstein, H.; Javitch, J.A. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc. Natl. Acad. Sci. USA 2009, 106, 5563–5568. [Google Scholar] [CrossRef] [Green Version]
- Krogsgaard-Larsen, P.; Strømgaard, K.; Madsen, U. Textbook of Drug Design and Discovery, 2010, 5th ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- The Advantages of Computational Docking. Available online: https://www.schrodinger.com/glide (accessed on 15 October 2020).
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Bismuth, Y.; Kavanaugh, M.P.; Kanner, B.I. Tyrosine 140 of the γ-aminobutyric acid transporter GAT-1 plays a critical role in neurotransmitter recognition. J. Biol. Chem. 1997, 272, 16096–16102. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
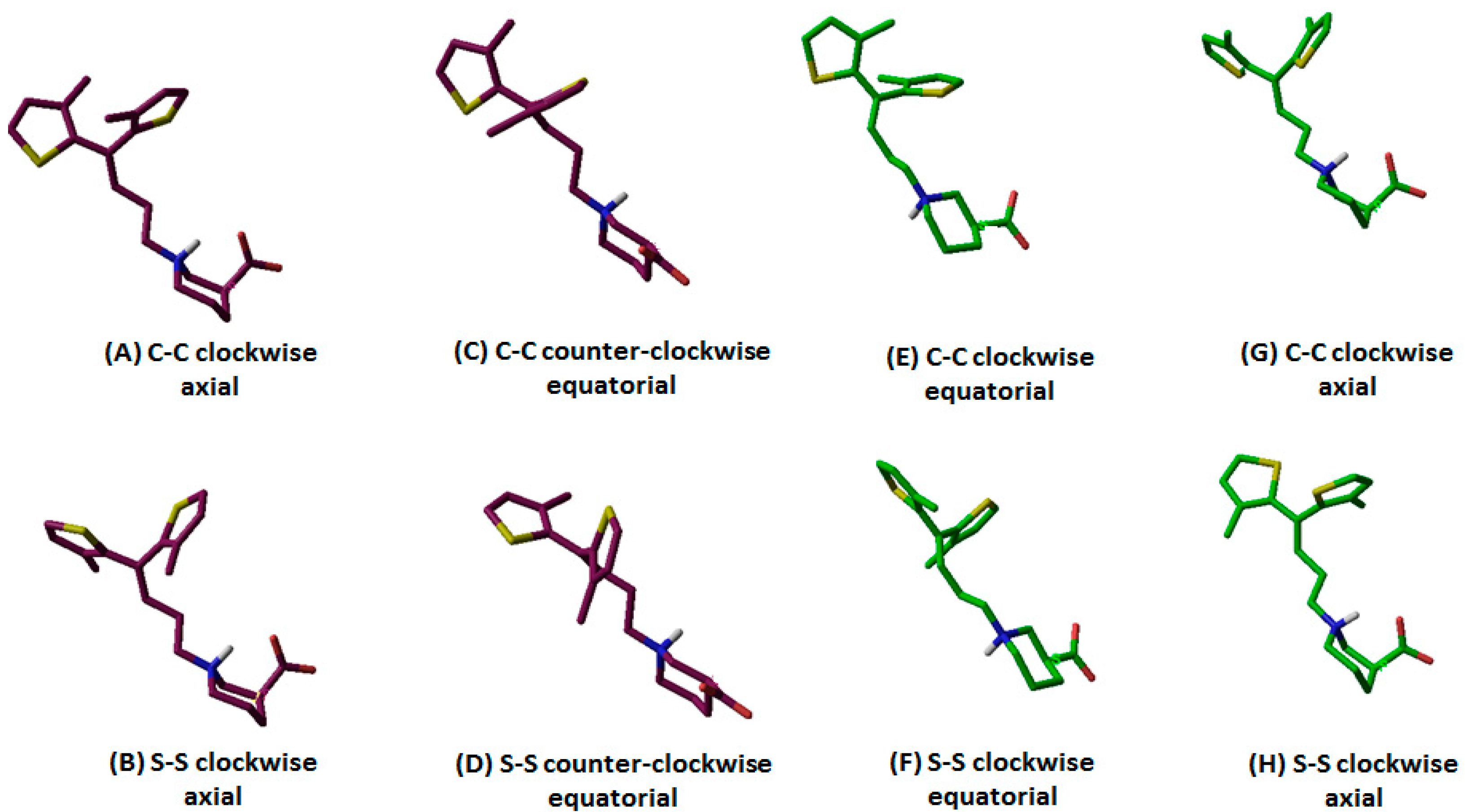
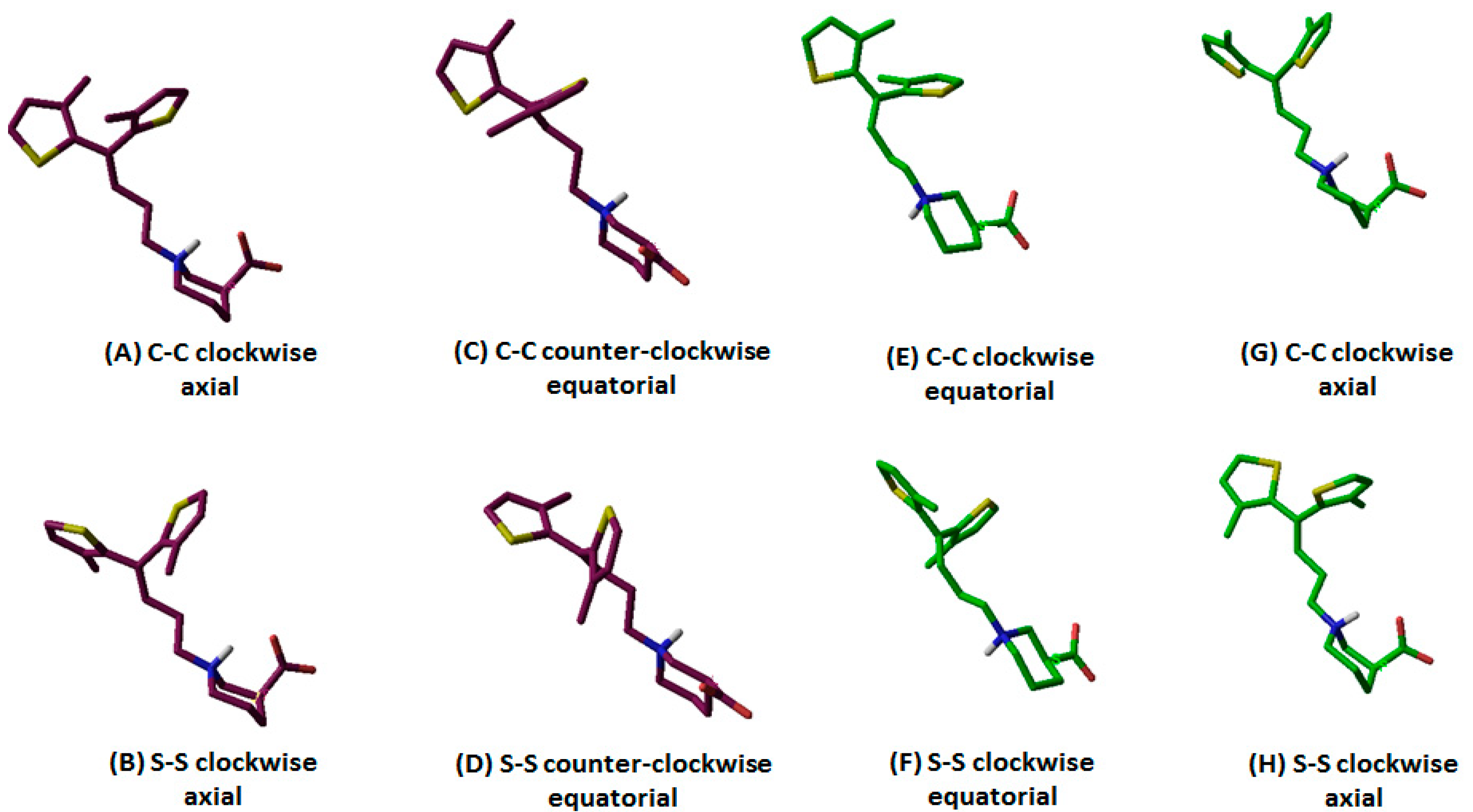
| Configuration of Protonated N Atom | Direction of Thiophene Rings with Respect to the Observer’s Eye | Orientation of Thiophene Rings | –COOH Configuration | Terminology | Entry Code |
|---|---|---|---|---|---|
| R (maroon purple) | C–C | Clockwise | Axial | R-configured C–C clockwise axial | 1 |
| S–S | R-configured S–S clockwise axial | 2 | |||
| C–C | Counterclockwise | Equatorial | R-configured C–C counterclockwise equatorial | 3 | |
| S–S | R-configured S–S counterclockwise equatorial | 4 | |||
| S (green) | C–C | Clockwise | Axial | S-configured C–C clockwise axial | 5 |
| S–S | S-configured S–S clockwise axial | 6 | |||
| C–C | Counterclockwise | Equatorial | S-configured C–C counterclockwise equatorial | 7 | |
| S–S | S-configured S–S counterclockwise equatorial | 8 | |||
| S (green) | C–C | Clockwise | Equatorial | S-configured C–C clockwise equatorial | 9 |
| S–S | S-configured S–S clockwise equatorial | 10 |
| Code | Entries Taken from Table 1 | Gscore (Kcal/mol) | |||
|---|---|---|---|---|---|
| Unconstraint | Hydrophobic Region Constraint | Hydrogen Bonding Constraint | Hydrophobic and Hydrogen Bonding Constraints | ||
| 1 | R-configured C–C clockwise axial | −7.13 | −7.00 | −7.37 | −7.37 |
| 2 | R-configured S–S clockwise axial | −7.09 | −5.25 | −7.38 | −5.25 |
| 3 | R-configured C–C counterclockwise equatorial | −7.73 | −9.36 | −6.86 | −6.46 |
| 4 | R-configured S–S counterclockwise equatorial | −7.98 | −6.21 | −6.43 | −6.83 |
| 5 | S-configured C–C clockwise axial | −6.97 | −7.7 | −7.07 | −6.35 |
| 6 | S-configured S–S clockwise axial | −8.08 | −8.14 | −5.53 | −6.39 |
| 7 | S-configured C–C counterclockwise equatorial | −7.00 | −7.16 | −6.86 | −6.20 |
| 8 | S-configured S–S counterclockwise equatorial | −6.15 | −5.83 | −6.22 | −5.37 |
| 9 | S-configured C–C clockwise equatorial | −7.70 | −6.58 | −5.91 | −5.91 |
| 10 | S-configured S–S clockwise equatorial | −6.91 | −7.00 | −5.53 | −5.68 |
| Cluster A | Cluster B | S-Configured C–C/S–S Clockwise Equatorial Tiagabine Enantiomers | ||||||
|---|---|---|---|---|---|---|---|---|
| GAT1 Residues | Tiagabine’s –COOH Group (Å) | Tiagabine’s –NH Group (Å) | Tiagabine’s –COOH Group (Å) | Tiagabine’s –NH Group (Å) | Tiagabine’s –COOH Group (Å) | Tiagabine’s –NH Group (Å) | ||
| Unconstraint | G65 (–NH) | 1.8 | - | 1.8 | - | 1.8–3.9 | - | |
| Y140 (–OH) | 3.7–4.0 | - | 1.8–3.5 | - | 1.8–5.7 | - | ||
| F294 (O) | - | - | - | 1.8–4.0 | - | 1.8–4.2 | ||
| S295 (O) | - | 1.8–3.6 | - | - | - | - | ||
| Na1 | 2.3–2.4 | - | 2.5–2.6 | - | 2.6–4.05 | - | ||
| Constraint | Hydrophobic region constraint | G65 (–NH) | 1.8 | - | 1.8 | - | 1.8 | - |
| Y140 (–OH) | 4.0–4.4 | - | 2.4–3.0 | - | 4.2–4.5 | - | ||
| F294 (O) | - | - | - | 3.8–3.9 | - | 3.6–3.8 | ||
| S295 (O) | - | 3.2–3.5 | - | - | - | - | ||
| Na1 | 2.3–2.6 | - | 2.3–2.6 | - | 2.5–3.2 | - | ||
| Hydrogen bonding constraint | G65 (–NH) | 1.8 | - | 1.8 | - | 2.2–2.4 | - | |
| Y140 (–OH) | 1.8 | - | 1.8 | - | 1.8 | - | ||
| F294 (O) | - | - | - | 4.8–6.3 | - | 4.6–5.2 | ||
| S295 (O) | - | 4.8–5.1 | - | - | - | - | ||
| Na1 | 3.2–6.0 | - | 3.2–5.9 | - | 3.2–4.8 | - | ||
| Hydrophobic region and hydrogen bonding constraints | G65 (–NH) | 1.8 | - | 1.8 | - | 3.8–4.2 | - | |
| Y140 (–OH) | 1.8 | - | 1.8 | - | 3.2 | - | ||
| F294 (O) | - | 4.0–5.1 | - | 4.0–5.0 | 4.2–4.6 | - | ||
| S295 (O) | - | - | - | - | - | - | ||
| Na1 | 3.2–4.2 | - | 3.5–4.8 | - | 3.2–4.6 | - | ||
| Before MD | After MD | ||||
|---|---|---|---|---|---|
| hGAT1 Residues | Tiagabine’s –COOH Group | Tiagabine’s –NH Group | Tiagabine’s –COOH Group | Tiagabine’s –NH Group | |
| hGAT1entry 3 | G65(–NH) | 1.8 Å | - | 1.8 Å | - |
| Y140 (–OH) | 1.8 Å | - | 1.8 Å | - | |
| F294 (O) | - | 1.8 Å | - | 3.2 Å | |
| S295 (O) | - | - | - | - | |
| Na1 | 2.4 Å | - | 2.3 Å | - | |
| hGAT1entry 4 | G65(–NH) | 1.8 Å | - | 1.8 Å | - |
| Y140 (–OH) | 1.8 Å | - | 1.8 Å | - | |
| F294 (O) | - | 1.8 Å | - | 1.8 Å | |
| S295 (O) | - | - | - | - | |
| Na1 | 2.3 Å | - | 2.4 Å | - | |
| hGAT1entry 6 | G65(–NH) | 1.8 Å | - | 1.8 Å | - |
| Y140 (–OH) | 1.8 Å | - | 1.8 Å | - | |
| F294 (O) | - | - | - | 1.8 Å | |
| S295 (O) | - | 1.8 Å | - | - | |
| Na1 | 2.3 Å | - | 2.2 Å | - | |
| hGAT1entry 9 | G65(–NH) | 1.8 Å | - | 1.8 Å | - |
| Y140 (–OH) | 1.8 Å | - | 5.2 Å | - | |
| F294 (O) | - | 1.8 Å | - | 4.6 Å | |
| S295 (O) | - | - | - | - | |
| Na1 | 2.4 Å | - | 2.3 Å | - | |
Sample Availability: Samples of the compounds 1–10 of Table 1 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafar, S.; Jabeen, I. Molecular Dynamic Simulations to Probe Stereoselectivity of Tiagabine Binding with Human GAT1. Molecules 2020, 25, 4745. https://doi.org/10.3390/molecules25204745
Zafar S, Jabeen I. Molecular Dynamic Simulations to Probe Stereoselectivity of Tiagabine Binding with Human GAT1. Molecules. 2020; 25(20):4745. https://doi.org/10.3390/molecules25204745
Chicago/Turabian StyleZafar, Sadia, and Ishrat Jabeen. 2020. "Molecular Dynamic Simulations to Probe Stereoselectivity of Tiagabine Binding with Human GAT1" Molecules 25, no. 20: 4745. https://doi.org/10.3390/molecules25204745
APA StyleZafar, S., & Jabeen, I. (2020). Molecular Dynamic Simulations to Probe Stereoselectivity of Tiagabine Binding with Human GAT1. Molecules, 25(20), 4745. https://doi.org/10.3390/molecules25204745