
Machine Learning Methods in Drug Discovery




Abstract
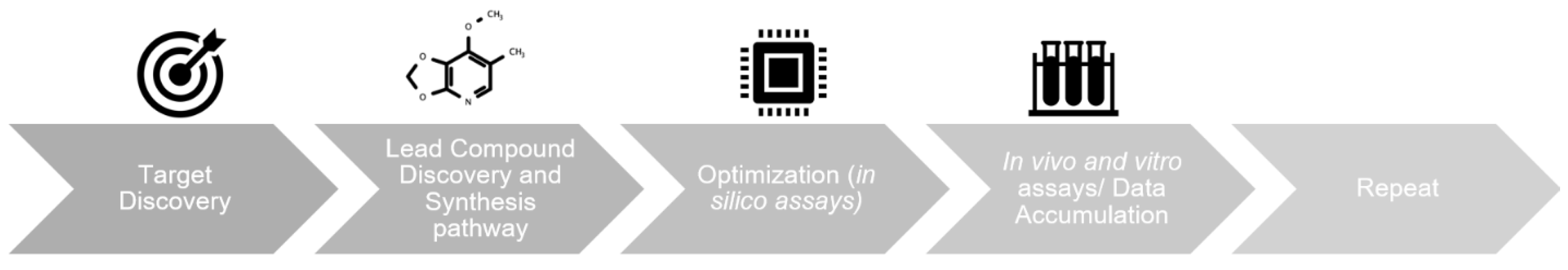
1. Introduction
2. ML Algorithms Used in Drug Discovery
3. Random Forest (RF)
4. Naive Bayesian (NB)
5. Support Vector Machine (SVM)
6. Limitations
7. Deep Learning (DL) Methods
8. Examples of Drug Discovery (Paper Summaries and Relevance to Topic)
9. Conclusions
Funding
Conflicts of Interest
References
- Fayyad, J.; Jaradat, M.A.; Gruyer, D.; Najjaran, H. Deep Learning Sensor Fusion for Autonomous Vehicle Perception and Localization: A Review. Sensors 2020, 20, 4220. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Li, X. Machine Learning Paradigms for Speech Recognition: An Overview. IEEE Trans. Audio Speech Lang. Process. 2013, 21, 1060–1089. [Google Scholar] [CrossRef]
- Afouras, T.; Chung, J.S.; Senior, A.; Vinyals, O.; Zisserman, A. Deep Audio-visual Speech Recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Joachims, T.; Radlinski, F. Search Engines that Learn from Implicit Feedback. Computer 2007, 40, 34–40. [Google Scholar] [CrossRef]
- Morgan, S.; Grootendorst, P.; Lexchin, J.; Cunningham, C.; Greyson, D. The cost of drug development: A systematic review. Health Policy 2011, 100, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Zhang, W.; Shu, M.; Luo, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Competitive molecular docking approach for predicting estrogen receptor subtype α agonists and antagonists. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Shu, M.; Luo, H.; Ye, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Estrogenic activity data extraction and in silico prediction show the endocrine disruption potential of bisphenol A replacement compounds. Chem. Res. Toxicol. 2015, 28, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Neamati, N.; Winslow, H.E.; Christensen, J.L.; Orr, A.; Pommier, Y.; Milne, G.W.A. Identification of HIV-1 integrase inhibitors based on a four-point pharmacophore. Antivir. Chem. Chemother. 1998, 9, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tong, W.; Xie, Q.; Fang, H.; Perkins, R. An in silico ensemble method for lead discovery: Decision forest. SAR QSAR Environ. Res. 2005, 16, 339–347. [Google Scholar] [CrossRef]
- Hong, H.; Fang, H.; Xie, Q.; Perkins, R.; Sheehan, D.M.; Tong, W. Comparative molecular field analysis (CoMFA) model using a large diverse set of natural, synthetic and environmental chemicals for binding to the androgen receptor. SAR QSAR Environ. Res. 2003, 14, 373–388. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A.; Morales, J.F.; Hather, G.; Podichetty, J.T.; Kim, S.; Bloomingdale, P.C.; Kim, S.; Burton, J.; Brown, J.D.; Winterstein, A.G.; et al. Machine Learning in Drug Discovery and Development Part 1: A Primer. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Gertrudes, J.C.; Maltarollo, V.G.; Silva, R.A.; Oliveira, P.R.; Honório, K.M.; da Silva, A.B. Machine learning techniques and drug design. Curr. Med. Chem. 2012, 19, 4289–4297. [Google Scholar] [CrossRef]
- Agarwal, S.; Dugar, D.; Sengupta, S. Ranking chemical structures for drug discovery: A new machine learning approach. J. Chem. Inf. Model. 2010, 50, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Bernardes, G.J.L. Machine learning for target discovery in drug development. Curr. Opin. Chem. Biol. 2020, 56, 16–22. [Google Scholar] [CrossRef]
- Gao, D.; Chen, Q.; Zeng, Y.; Jiang, M.; Zhang, Y. Application of Machine Learning on Drug Target Discovery. Curr. Drug Metab. 2020. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Zoffmann, S.; Vercruysse, M.; Benmansour, F.; Maunz, A.; Wolf, L.; Marti, R.B.; Heckel, T.; Ding, H.; Truong, H.H.; Prummer, M.; et al. Machine learning-powered antibiotics phenotypic drug discovery. Sci. Rep. 2019, 9, 5013. [Google Scholar] [CrossRef]
- Ekins, S.; Puhl, A.C.; Zorn, K.M.; Lane, T.R.; Russo, D.P.; Klein, J.J.; Hickey, A.J.; Clark, A.M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019, 18, 435–441. [Google Scholar] [CrossRef]
- Khamis, M.A.; Gomaa, W.; Ahmed, W.F. Machine learning in computational docking. Artif. Intell. Med. 2015, 63, 135–152. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Talambedu, U.; Shanmugarajan, D.; Goyal, A.K.; Kumar, C.S.; Middha, S.K. Recent Updates on Computer-aided Drug Discovery: Time for a Paradigm Shift. Curr. Top. Med. Chem. 2017, 17, 3296–3307. [Google Scholar] [CrossRef]
- Réda, C.; Kaufmann, E.; Delahaye-Duriez, A. Machine learning applications in drug development. Comput. Struct. Biotechnol. J. 2020, 18, 241–252. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Webb, G.I. Naïve Bayes. In Encyclopedia of Machine Learning; Sammut, C., Webb, G.I., Eds.; Springer: Boston, MA, USA, 2010. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; Jamuar, S.S.; Moody, A.R.; Karnes, J.H.; Varga, O.; Hedensted, S.; Spreafico, R.; Hafler, D.A.; McKinney, E.F. From Big Data to Precision Medicine. Front. Med. 2019, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; He, H.; Luo, H.; Zhang, T.; Jiang, J. Artificial intelligence and big data facilitated targeted drug discovery. Stroke Vasc. Neurol. 2019, 4, 206. [Google Scholar] [CrossRef]
- Cirillo, D.; Valencia, A. Big data analytics for personalized medicine. Curr. Opin. Biotechnol. 2019, 58, 161–167. [Google Scholar] [CrossRef]
- Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine Learning for Drug-Target Interaction Prediction. Molecules 2018, 23, 2208. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Adelstein, S.J.; Kassis, A.I. Target discovery from data mining approaches. Drug Discov. Today 2009, 14, 147–154. [Google Scholar] [CrossRef]
- Yella, J.K.; Yaddanapudi, S.; Wang, Y.; Jegga, A.G. Changing Trends in Computational Drug Repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef]
- Sarica, A.; Cerasa, A.; Quattrone, A. Random Forest Algorithm for the Classification of Neuroimaging Data in Alzheimer’s Disease: A Systematic Review. Front. Aging Neurosci. 2017, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Cano, G.; Garcia-Rodriguez, J.; Garcia-Garcia, A.; Perez-Sanchez, H.; Benediktsson, J.; Thapa, A.; Barr, A. Automatic selection of molecular descriptors using random forest: Application to drug discovery. Expert Syst. Appl. 2017, 72, 151–159. [Google Scholar] [CrossRef]
- Rahman, R.; Otridge, J.; Pal, R. IntegratedMRF: Random forest-based framework for integrating prediction from different data types. Bioinformatics 2017, 33, 1407–1410. [Google Scholar] [CrossRef]
- Rahman, R.; Dhruba, S.R.; Ghosh, S.; Pal, R. Functional random forest with applications in dose-response predictions. Sci. Rep. 2019, 9, 1628. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Lee, K.; Lee, M.; Kim, D. Utilizing random Forest QSAR models with optimized parameters for target identification and its application to target-fishing server. BMC Bioinform. 2017, 18, 567. [Google Scholar] [CrossRef]
- Bielza, C.; Larrañaga, P. Discrete Bayesian Network Classifiers: A Survey. ACM Comput. Surv. 2014, 47, 43. [Google Scholar] [CrossRef]
- Gilboa, E.; Saatçi, Y.; Cunningham, J.P. Scaling Multidimensional Inference for Structured Gaussian Processes. IEEE Trans. Pattern Anal. Mach. Intell. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sun, H. A naive bayes classifier for prediction of multidrug resistance reversal activity on the basis of atom typing. J. Med. Chem. 2005, 48, 4031–4039. [Google Scholar] [CrossRef] [PubMed]
- Ratanamahatana, C.A.; Gunopulos, D. Feature selection for the naive bayesian classifier using decision trees. Appl. Artif. Intell. 2010. [Google Scholar] [CrossRef]
- Kim, S.-B.; Han, K.-S.; Rim, H.-C.; Myaeng, S.-H. Some Effective Techniques for Naive Bayes Text Classification. IEEE Trans. Knowl. Data Eng. 2006, 18, 1457–1466. [Google Scholar] [CrossRef]
- Anagaw, A.; Chang, Y.-L. A new complement naïve Bayesian approach for biomedical data classification. J. Ambient Intell. Hum. Comput. 2019, 10, 3889–3897. [Google Scholar] [CrossRef]
- Nigsch, F.; Bender, A.; Jenkins, J.L.; Mitchell, J.B.O. Ligand-Target Prediction Using Winnow and Naive Bayesian Algorithms and the Implications of Overall Performance Statistics. J. Chem. Inf. Model. 2008, 48, 2313–2325. [Google Scholar] [CrossRef]
- Pang, X.; Fu, W.; Wang, J.; Kang, D.; Xu, L.; Zhao, Y.; Liu, A.L.; Du, G.H. Identification of Estrogen Receptor α Antagonists from Natural Products via In Vitro and In Silico Approaches. Oxid. Med. Cell. Longev. 2018, 2018, 6040149. [Google Scholar] [CrossRef]
- Wei, Y.; Li, W.; Du, T.; Hong, Z.; Lin, J. Targeting HIV/HCV Coinfection Using a Machine Learning-Based Multiple Quantitative Structure-Activity Relationships (Multiple QSAR) Method. Int. J. Mol. Sci. 2019, 20, 3572. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. Support vector machines for drug discovery. Expert Opin. Drug Discov. 2014, 9, 93–104. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Kronenberger, T.; Espinoza, G.Z.; Oliveira, P.R.; Honorio, K.M. Advances with support vector machines for novel drug discovery. Expert Opin. Drug Discov. 2019, 14, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.N.; Philot, E.A.; Trossini, G.H.; Scott, L.P.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, S.; Zararsiz, G.; Goksuluk, D. Drug/nondrug classification using Support Vector Machines with various feature selection strategies. Comput. Methods Programs Biomed. 2014, 117, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, A.M.; Geppert, H.; Bajorath, J. Application of support vector machine-based ranking strategies to search for target-selective compounds. Methods Mol. Biol. 2011, 672, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Hinselmann, G.; Rosenbaum, L.; Jahn, A.; Fechner, N.; Ostermann, C.; Zell, A. Large-scale learning of structure-activity relationships using a linear support vector machine and problem-specific metrics. J. Chem. Inf. Model. 2011, 51, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Zhang, C.H.; Deng, N.Y.; Wang, Y. Kernel-based data fusion improves the drug-protein interaction prediction. Comput. Biol. Chem. 2011, 35, 353–362. [Google Scholar] [CrossRef]
- Kawai, K.; Fujishima, S.; Takahashi, Y. Predictive activity profiling of drugs by topological-fragment-spectra-based support vector machines. J. Chem. Inf. Model. 2008, 48, 1152–1160. [Google Scholar] [CrossRef]
- Kawai, K.; Takahashi, Y. Identification of the Dual Action Antihypertensive Drugs Using TFS-Based Support Vector Machines. Chem. Bio Inform. J. 2009, 9, 41–51. [Google Scholar] [CrossRef][Green Version]
- Rossi, G.P.; Dual, A.C.E. NEP inhibitors: A review of the pharmacological properties of MDL 100240. Cardiovasc. Drug Rev. 2003, 21, 51–66. [Google Scholar] [CrossRef]
- Kaiser, T.M.; Burger, P.B. Error Tolerance of Machine Learning Algorithms across Contemporary Biological Targets. Molecules 2019, 24, 2115. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Model selection and overfitting. Nat. Methods 2016, 13, 703–704. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Awale, M.; Probst, D.; Reymond, J.L. Exploring Chemical Space with Machine Learning. Chimia 2019, 73, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Von Lilienfeld, O.A.; Müller, K.; Tkatchenko, A. Exploring chemical compound space with quantum-based machine learning. Nat. Rev. Chem. 2020, 4, 347–358. [Google Scholar] [CrossRef]
- Dobson, C. Chemical space and biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Henson, A.B.; Granda, J.M.; Cronin, L. How to explore chemical space using algorithms and automation. Nat. Rev. Chem. 2019, 3, 119–128. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Dana, D.; Gadhiya, S.V.; St Surin, L.G.; Li, D.; Naaz, F.; Ali, Q.; Paka, L.; Yamin, M.A.; Narayan, M.; Goldberg, I.D. Deep Learning in Drug Discovery and Medicine; Scratching the Surface. Molecules 2018, 23, 2384. [Google Scholar] [CrossRef]
- Korotcov, A.; Tkachenko, V.; Russo, D.P.; Ekins, S. Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol. Pharm. 2017, 14, 4462–4475. [Google Scholar] [CrossRef]
- Ekins, S. The Next Era: Deep Learning in Pharmaceutical Research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar] [CrossRef]
- D’Souza, S.; Prema, K.V.; Balaji, S. Machine learning models for drug-target interactions: Current knowledge and future directions. Drug Discov. Today 2020, 25, 748–756. [Google Scholar] [CrossRef]
- Baskin, I.I.; Winkler, D.; Tetko, I.V. A renaissance of neural networks in drug discovery. Expert Opin. Drug Discov. 2016, 11, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design—A review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Mehridehnavi, A.; Pérez-Garrido, A.; Pérez-Sánchez, H. Neural network and deep-learning algorithms used in QSAR studies: Merits and drawbacks. Drug Discov. Today 2018, 23, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Jiang, D.; Nambiar, D.K.; Liew, L.P.; Hay, M.P.; Bloomstein, J.; Lu, P.; Turner, B.; Le, Q.T.; Tibshirani, R.; et al. Chemical Space Mimicry for Drug Discovery. J. Chem. Inf. Model. 2017, 57, 875–882. [Google Scholar] [CrossRef]
- Gupta, A.; Müller, A.T.; Huisman, B.J.H.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative Recurrent Networks for De Novo Drug Design. Mol. Inf. 2018, 37, 1700111. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- De Almeida, A.F.; Moreira, R.; Rodrigues, T. Synthetic organic chemistry driven by artificial intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, X.; Zare, R.N. Optimizing Chemical Reactions with Deep Reinforcement Learning. ACS Cent. Sci. 2017, 3, 1337–1344. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef]
- Lee, A.A.; Yang, Q.; Sresht, V.; Bolgar, P.; Hou, X.; Klug-McLeod, J.L.; Butler, C.R. Molecular Transformer unifies reaction prediction and retrosynthesis across pharma chemical space. Chem. Commun. 2019, 55, 12152–12155. [Google Scholar] [CrossRef]
- Reher, R.; Kim, H.W.; Zhang, C.; Mao, H.H.; Wang, M.; Nothias, L.F.; Caraballo-Rodriguez, A.M.; Glukhov, E.; Teke, B.; Leao, T.; et al. A Convolutional Neural Network-Based Approach for the Rapid Annotation of Molecularly Diverse Natural Products. J. Am. Chem. Soc. 2020, 142, 4114–4120. [Google Scholar] [CrossRef]
- Rathi, P.C.; Ludlow, R.F.; Verdonk, M.L. Practical High-Quality Electrostatic Potential Surfaces for Drug Discovery Using a Graph-Convolutional Deep Neural Network. J. Med. Chem. 2020, 63, 8778–8790. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef]
- Chang, P.; Grinband, J.; Weinberg, B.D.; Bardis, M.; Khy, M.; Cadena, G.; Su, M.Y.; Cha, S.; Filippi, C.G.; Bota, D.; et al. Deep-Learning Convolutional Neural Networks Accurately Classify Genetic Mutations in Gliomas. Am. J. Neuroradiol. 2018, 39, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Mencattini, A.; di Giuseppe, D.; Comes, M.C.; Casti, P.; Corsi, F.; Bertani, F.R.; Ghibelli, L.; Businaro, L.; di Natale, C.; Parrini, M.C.; et al. Discovering the hidden messages within cell trajectories using a deep learning approach for in vitro evaluation of cancer drug treatments. Sci. Rep. 2020, 10, 7653. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-H.; Zhang, S.-W.; Shi, J.-Y. DPDDI: A deep predictor for drug-drug interactions. BMC Bioinform. 2020, 21, 419. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackerman, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Fields, F.R.; Freed, S.D.; Carothers, K.E.; Hamid, M.N.; Hammers, D.E.; Ross, J.N.; Kalwajtys, V.R.; Gonzalez, A.J.; Hildreth, A.D.; Friedberg, I.; et al. Novel antimicrobial peptide discovery using machine learning and biophysical selection of minimal bacteriocin domains. Drug Dev. Res. 2020, 81, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Reker, D.; Bernardes, G.J.L.; Rodrigues, T. Computational advances in combating colloidal aggregation in drug discovery. Nat. Chem. 2019, 11, 402–418. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.; Kim, D.S.; DeLuca, A.M.; Alani, R.M. Comprehensive Expression Profiling of Tumor Cell Lines Identifies Molecular Signatures of Melanoma Progression. PLoS ONE 2007, 2, e594. [Google Scholar] [CrossRef]
- Sakellaropoulos, T.; Vougas, K.; Narang, S.; Koinis, F.; Kotsinas, A.; Polyzos, A.; Moss, T.J.; Piha-Paul, S.; Zhou, H.; Kardala, E.; et al. A Deep Learning Framework for Predicting Response to Therapy in Cancer. Cell Rep. 2019, 29, 3367–3373.e4. [Google Scholar] [CrossRef]
- Mohanty, S.; Rashid, M.H.A.; Mridul, M.; Mohanty, C.; Swayamsiddha, S. Application of Artificial Intelligence in COVID-19 drug repurposing. Diabetes Metab. Syndr. 2020, 14, 1027–1031. [Google Scholar] [CrossRef]
- Kowalewski, J.; Ray, A. Predicting novel drugs for SARS-CoV-2 using machine learning from a >10 million chemical space. Heliyon 2020, 6, e04639. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Databases | Specific Information | Ref. |
|---|---|---|
| BRENDA http://www.brenda-enzymes.org | Enzyme and enzyme-ligand information source. | [33] |
| KEGG http://www.genome.jp/kegg | Database containing genomic information for functional interpretation and practical application. | [33] |
| PubChem https://pubchem.ncbi.nlm.nih.gov | Database for encompassing information on chemicals and biological activities. | [33] |
| TTD http://bidd.nus.edu.sg/group/ttd/ttd.asp | Therapeutic Target Database containing encompassing information about the drug resistance mutations, gene expressions, and target combinations data. | [33] |
| DrugBank http://www.drugbank.ca | Detailed drug data and drug-target information database. | [33] |
| SuperTarget http://bioinfapache.charite.de/supertarget | Drug-related information databases with more than >300,000 compound-target protein relations. | [33] |
| TDR targets http://tdrtargets.org | Database containing chemogenomic information for neglected tropical diseases. | [33] |
| STITCH http://stitch-beta.embl.de | Chemical-Protein interaction networks. | [28] |
| SMD http://genome-www5.stanford.edu | Database of raw microarray datasets. | [34] |
| Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo | Database of raw microarray datasets. | [34] |
| caArray http://array.nci.nih.gov/caarray | Database of cancer-related microarray datasets. | [34] |
| CGAP database http://cgap.nci.nih.gov | Database of cancer-related microarray datasets. | [34] |
| Oncomine http://www.oncomine.org | Database of cancer-related microarray datasets. | [34] |
| UniHI http://www.unihi.org | Database of human molecular interaction networks. | [34] |
| Pathguide http://www.pathguide.org | Database of 702 biological pathway related resources and molecular interactions. | [34] |
| UniProt http://www.uniprot.org | Encompassing protein information center. | [34] |
| InterPro http://www.ebi.ac.uk/interpro | Database of protein domain information. | [34] |
| Web-Tools/Software Used for Target Discovery | Specific Information | Ref. |
|---|---|---|
| GoPubMed http://www.gopubmed.org | PubMed search engine utilized as a text-mining tool. | [34] |
| Textpresso http://www.textpresso.org | Full-text engine used in text mining, classification, and search. | [34] |
| BioRAT http://bioinfadmin.cs.ucl.ac.uk/biorat/docs/index | Full-text search engine used for text mining. | [34] |
| ABNER http://pages.cs.wisc.edu/~bsettles/abner | Molecular biology text analyzer and entity tagger tool. | [34] |
| PPICurator https://ppicurator.hupo.org.cn | Tool used for mining comprehensive protein-protein interaction. | [34] |
| GeneWays http://geneways.genomeleft.columbia.edu | Biological pathway extracting tool. | [34] |
| Database | Specific Information | Ref. |
|---|---|---|
| ADReCS http://bioinf.xmu.edu.cn/ADReCS | Database of toxicology information with 137,619 Drug-ADR pairs. | [35] |
| ChEMBL https://www.ebi.ac.uk/chembl | Database of drug-like small molecules with predicated bioactive properties. | [35] |
| ChemSpider http://www.chemspider.com | Encompassing database of over 64 million chemical structures. | [35] |
| DrugCentral http://drugcentral.org | Database containing relevant drug information of activity, chemical identity, mode of action, etc. | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. https://doi.org/10.3390/molecules25225277
Patel L, Shukla T, Huang X, Ussery DW, Wang S. Machine Learning Methods in Drug Discovery. Molecules. 2020; 25(22):5277. https://doi.org/10.3390/molecules25225277
Chicago/Turabian StylePatel, Lauv, Tripti Shukla, Xiuzhen Huang, David W. Ussery, and Shanzhi Wang. 2020. "Machine Learning Methods in Drug Discovery" Molecules 25, no. 22: 5277. https://doi.org/10.3390/molecules25225277
APA StylePatel, L., Shukla, T., Huang, X., Ussery, D. W., & Wang, S. (2020). Machine Learning Methods in Drug Discovery. Molecules, 25(22), 5277. https://doi.org/10.3390/molecules25225277