
Mechanochemical P-derivatization of 1,3,5-Triaza-7-Phosphaadamantane (PTA) and Silver-Based Coordination Polymers Obtained from the Resulting Phosphabetaines

 ,
, 
 , and
, and
Abstract
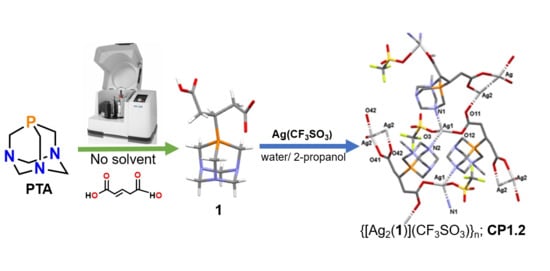
:
1. Introduction
2. Results and Discussions
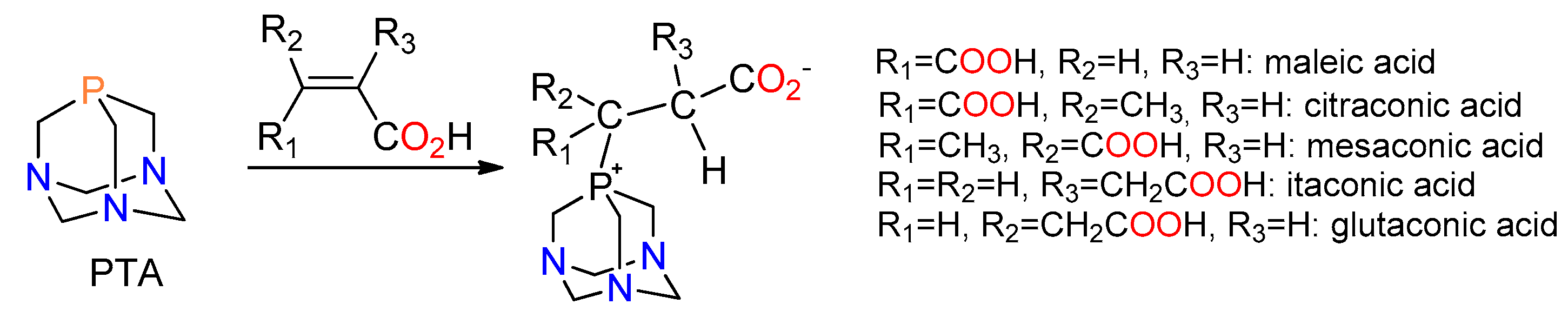
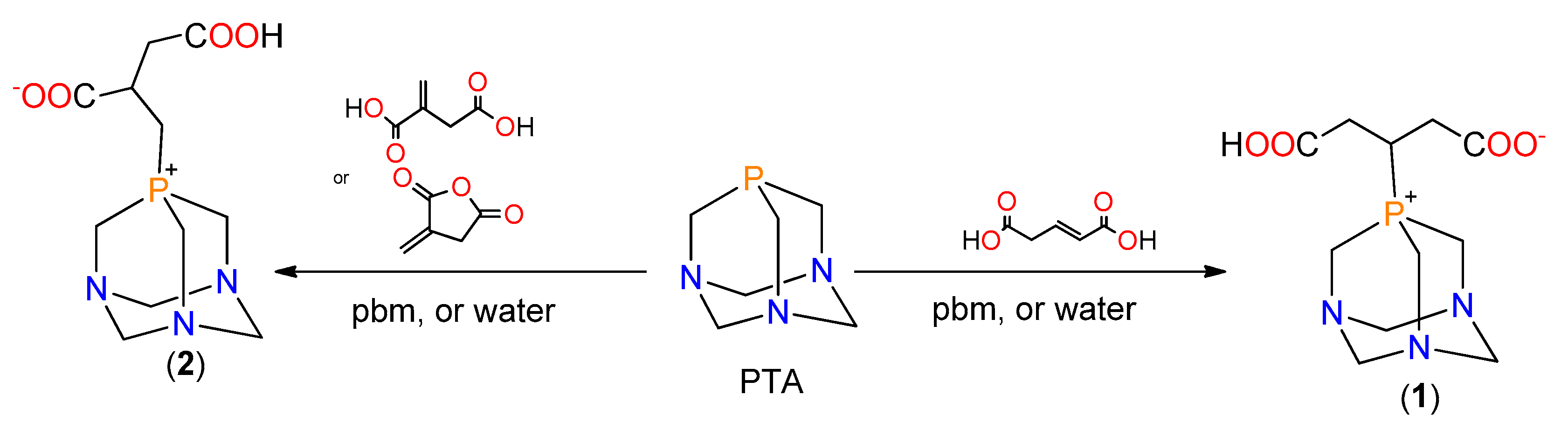
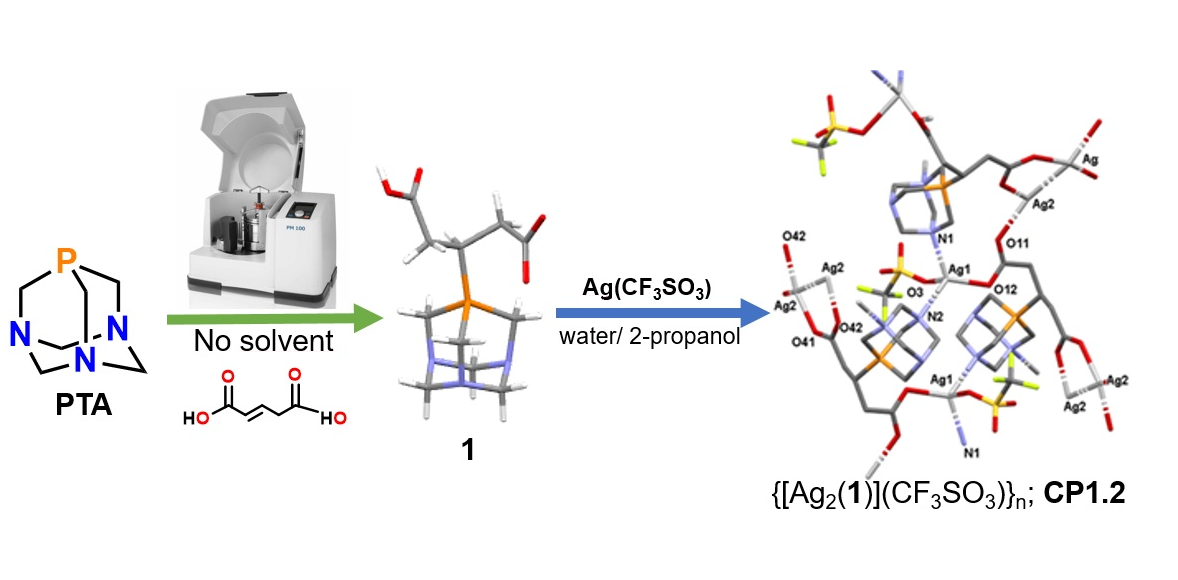
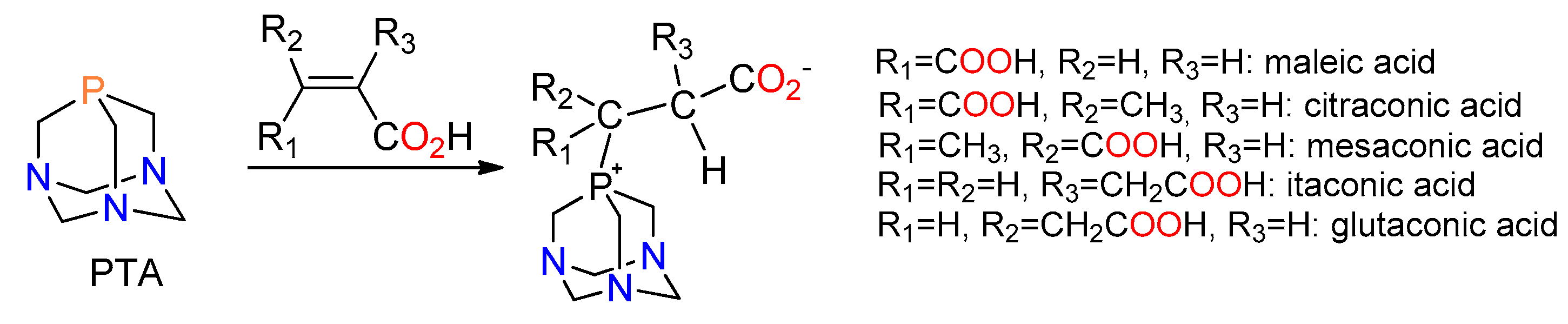
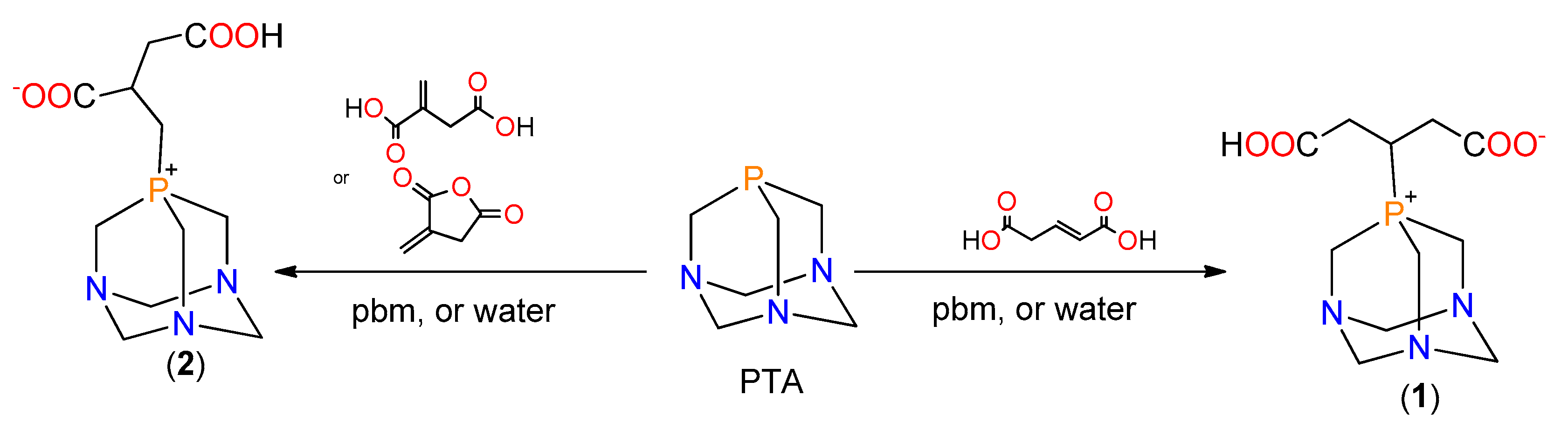
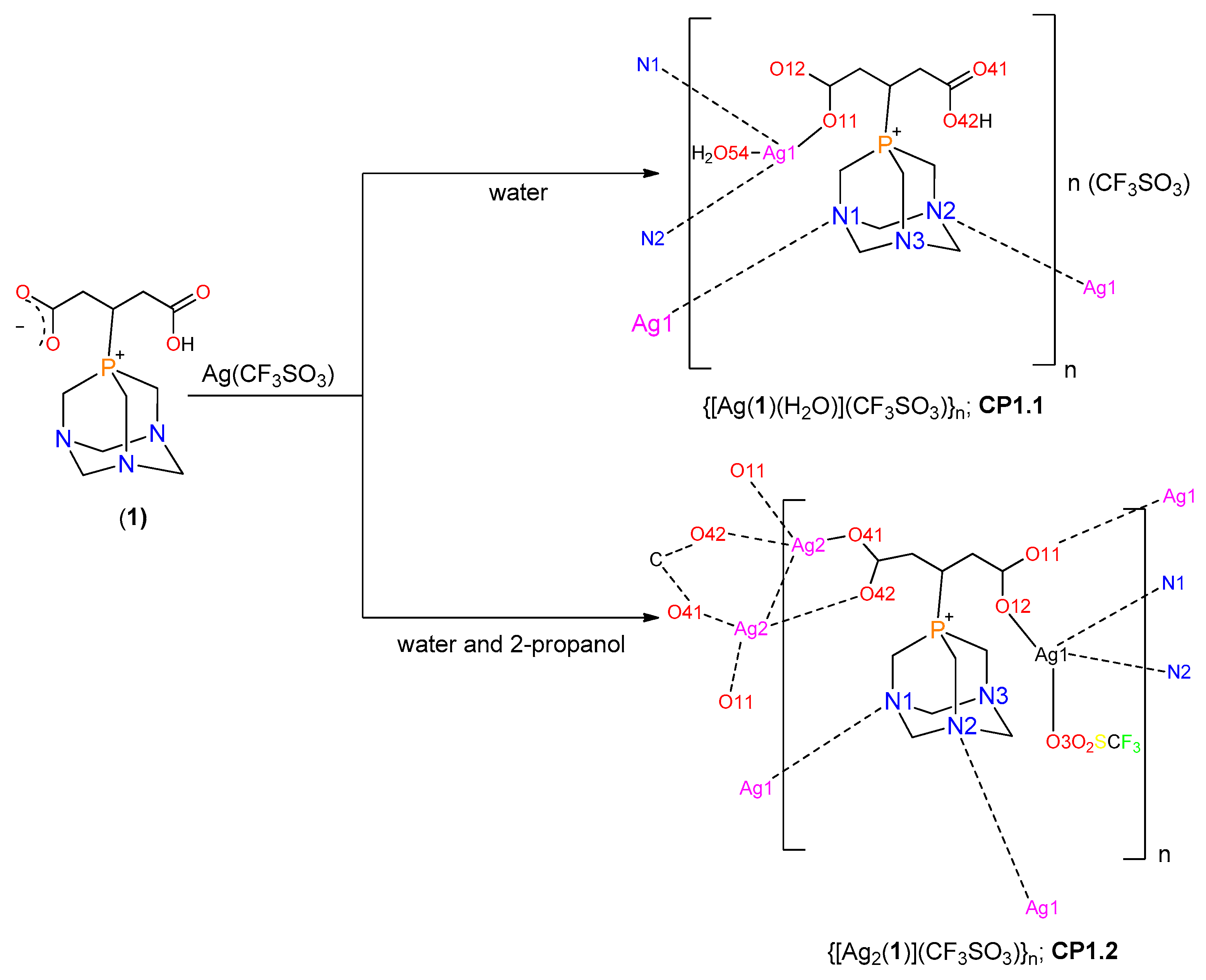
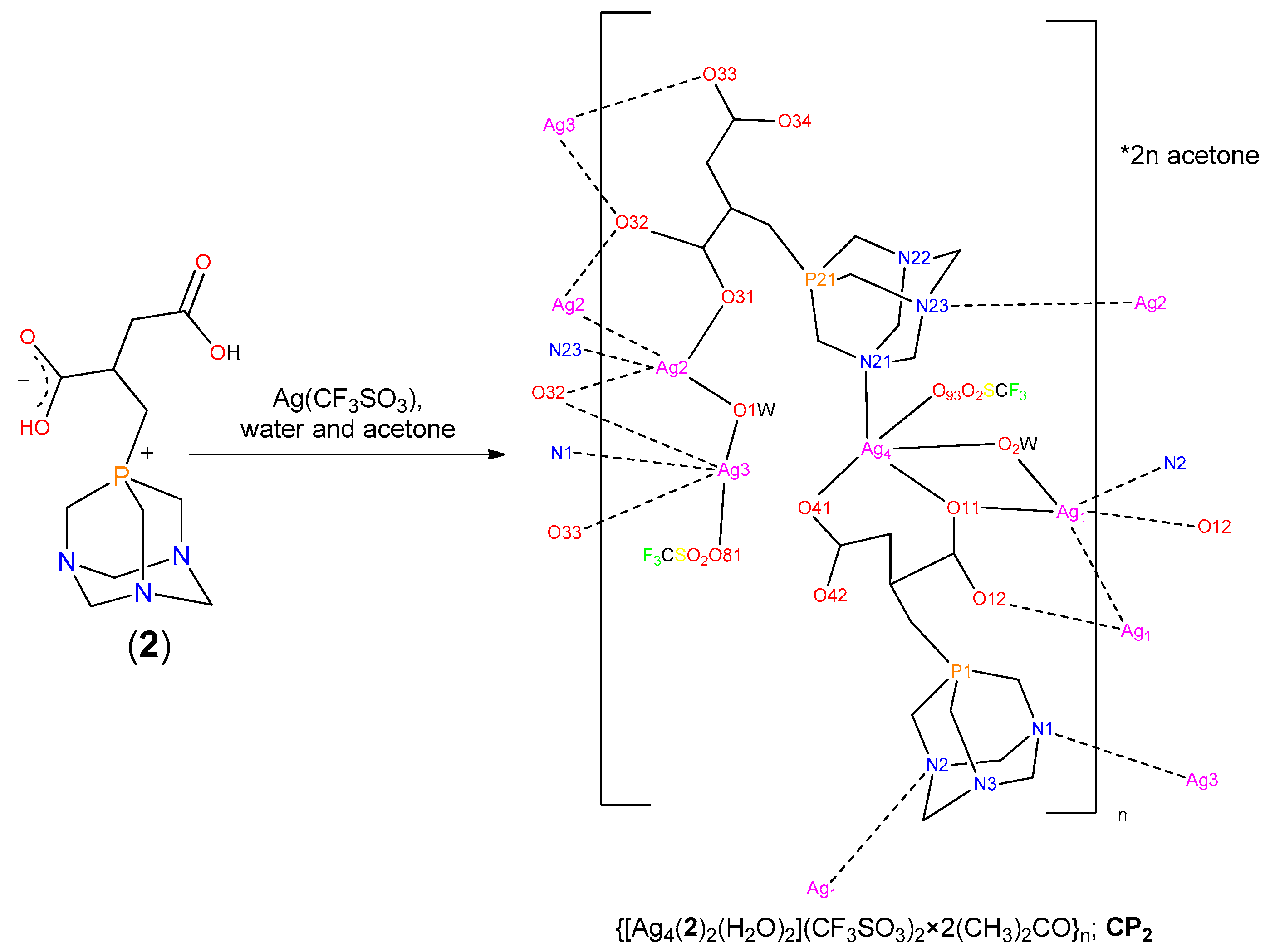
2.1. Synthesis of the Phosphabetaines 1 and 2
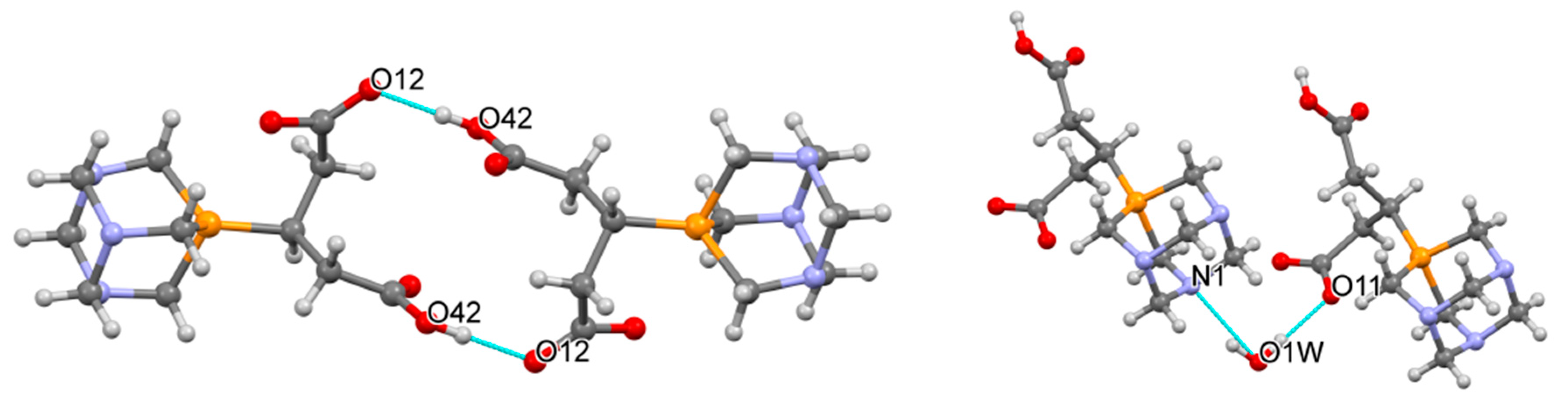
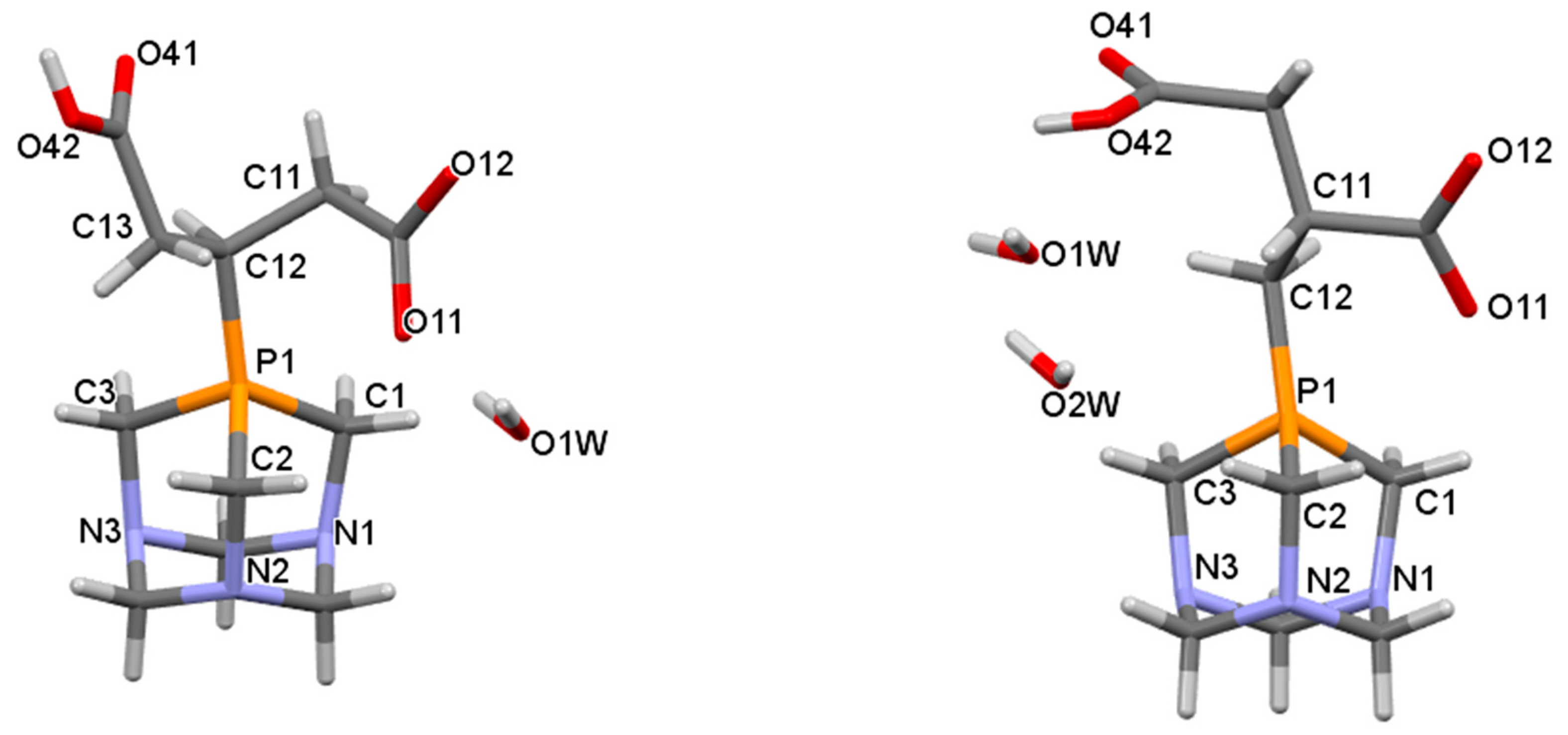
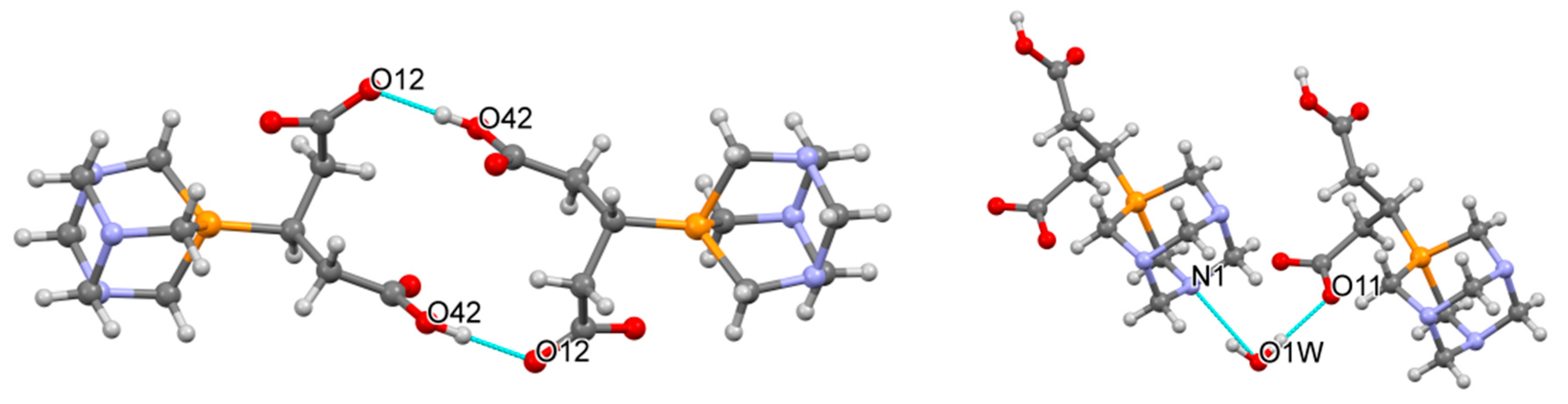
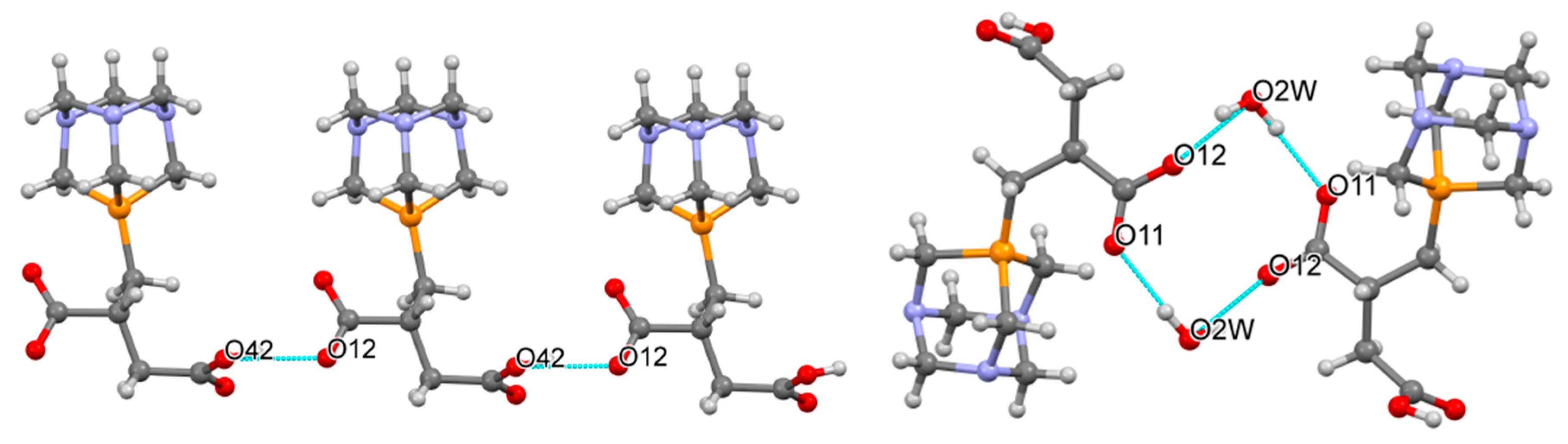
2.2. Structural Characterization of the Phosphabetaines 1 and 2
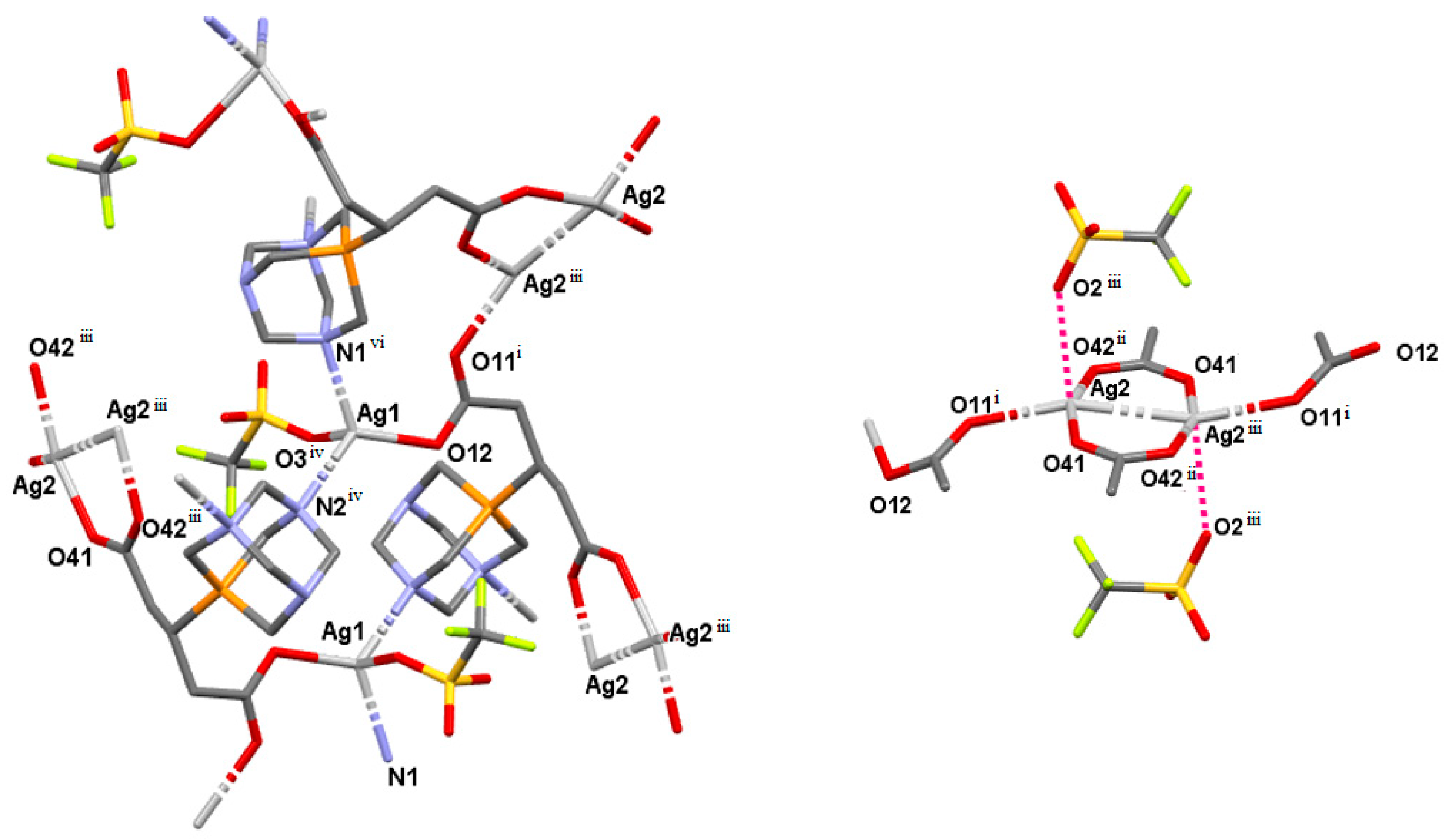
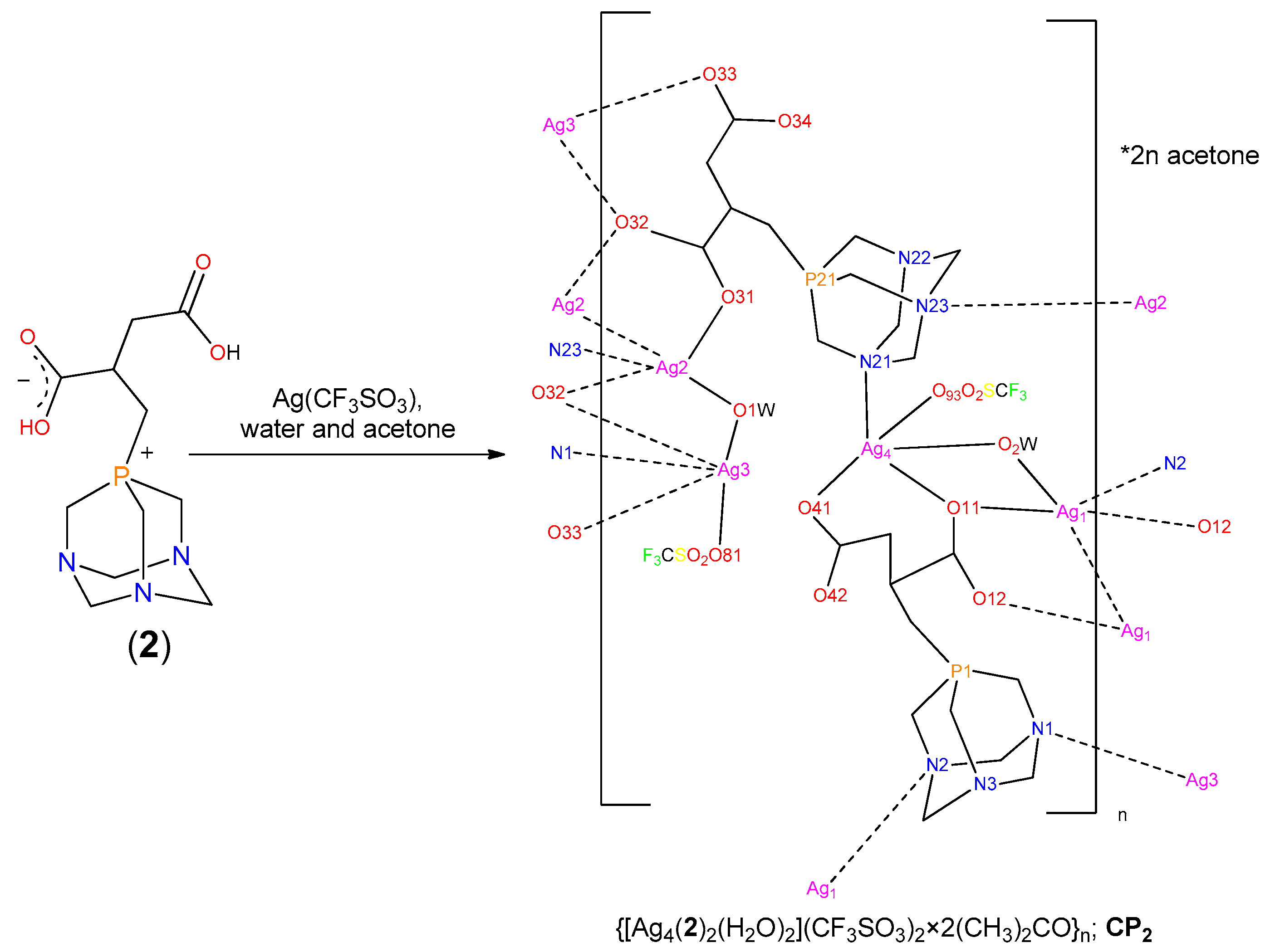
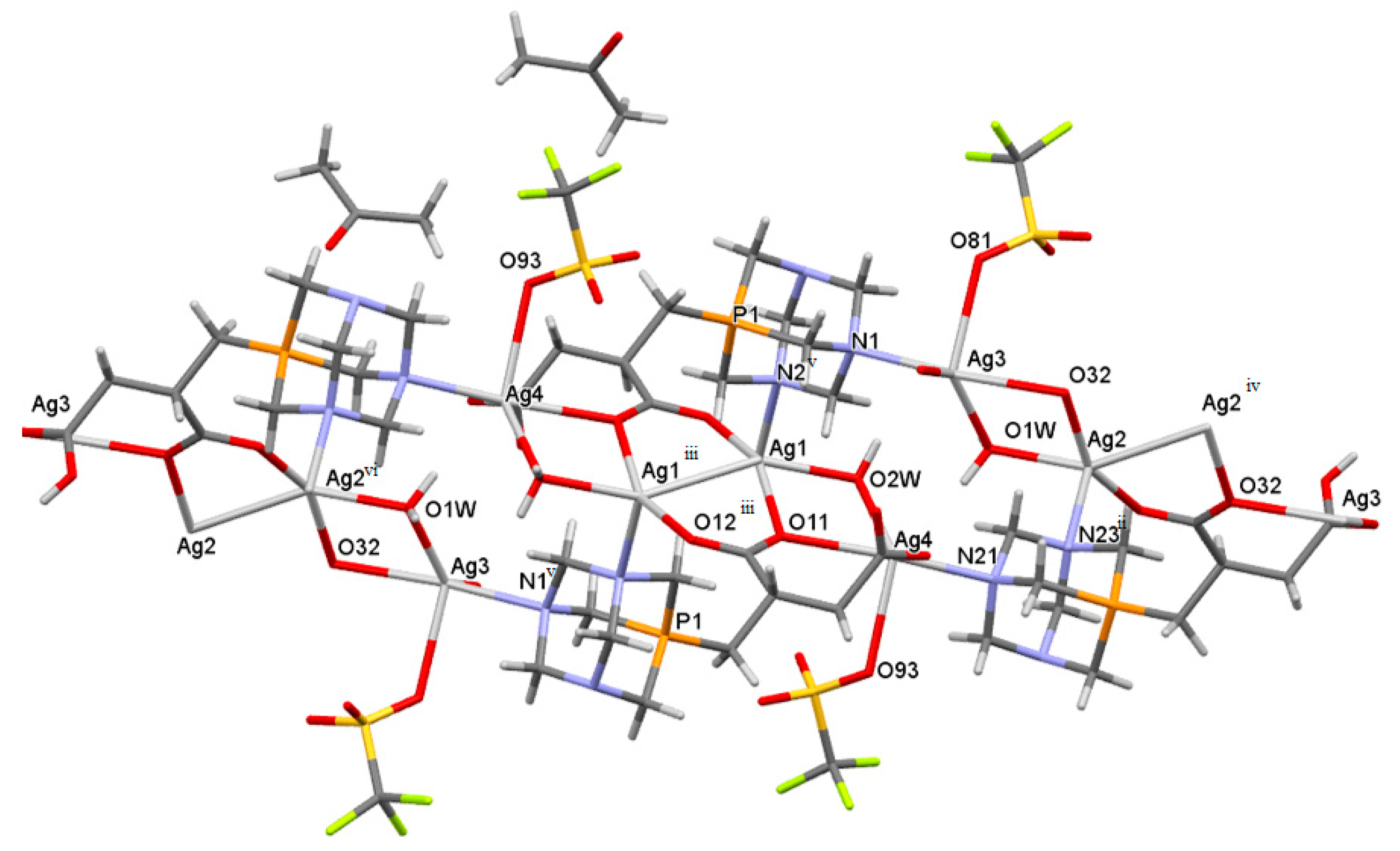
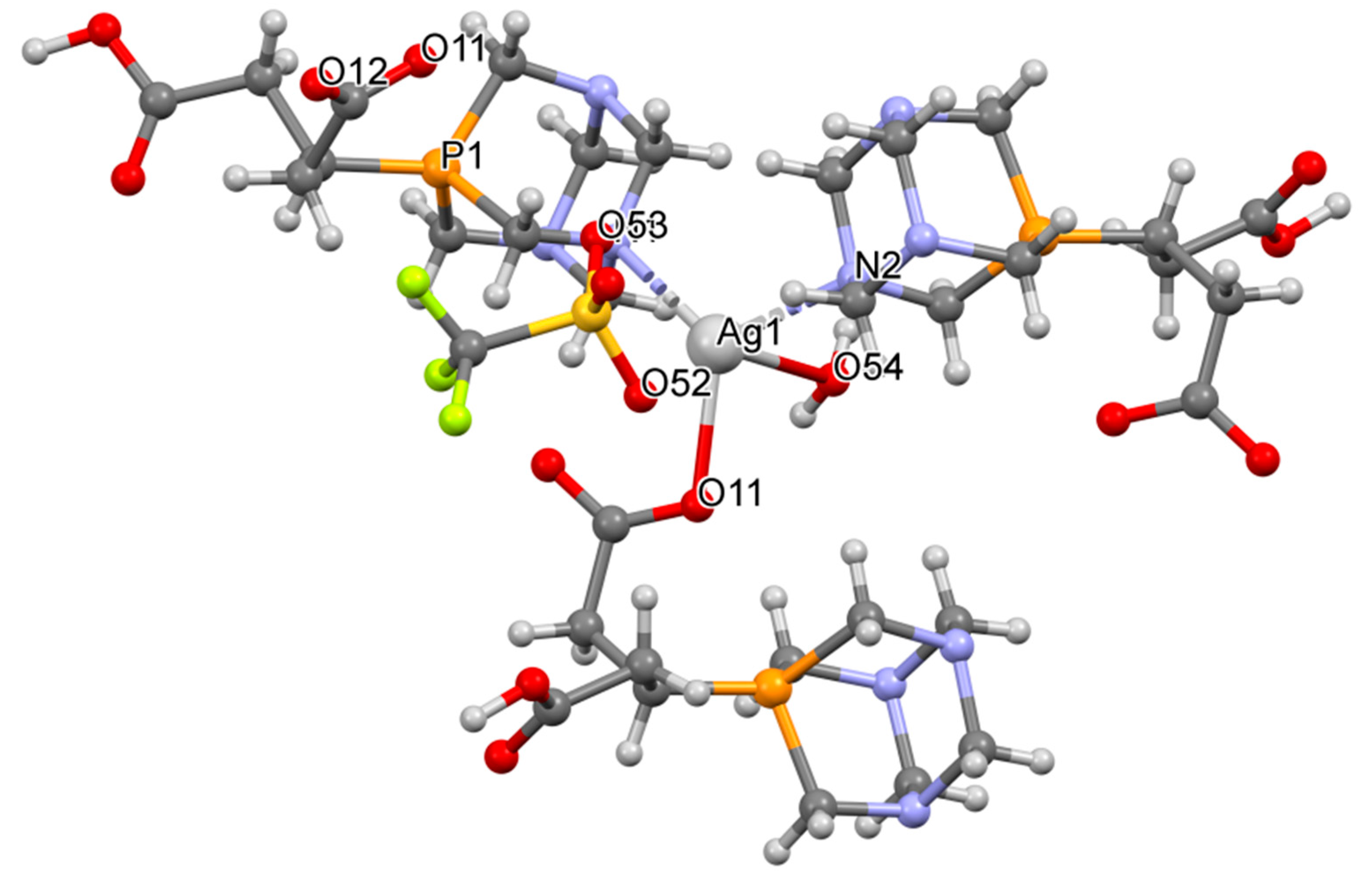
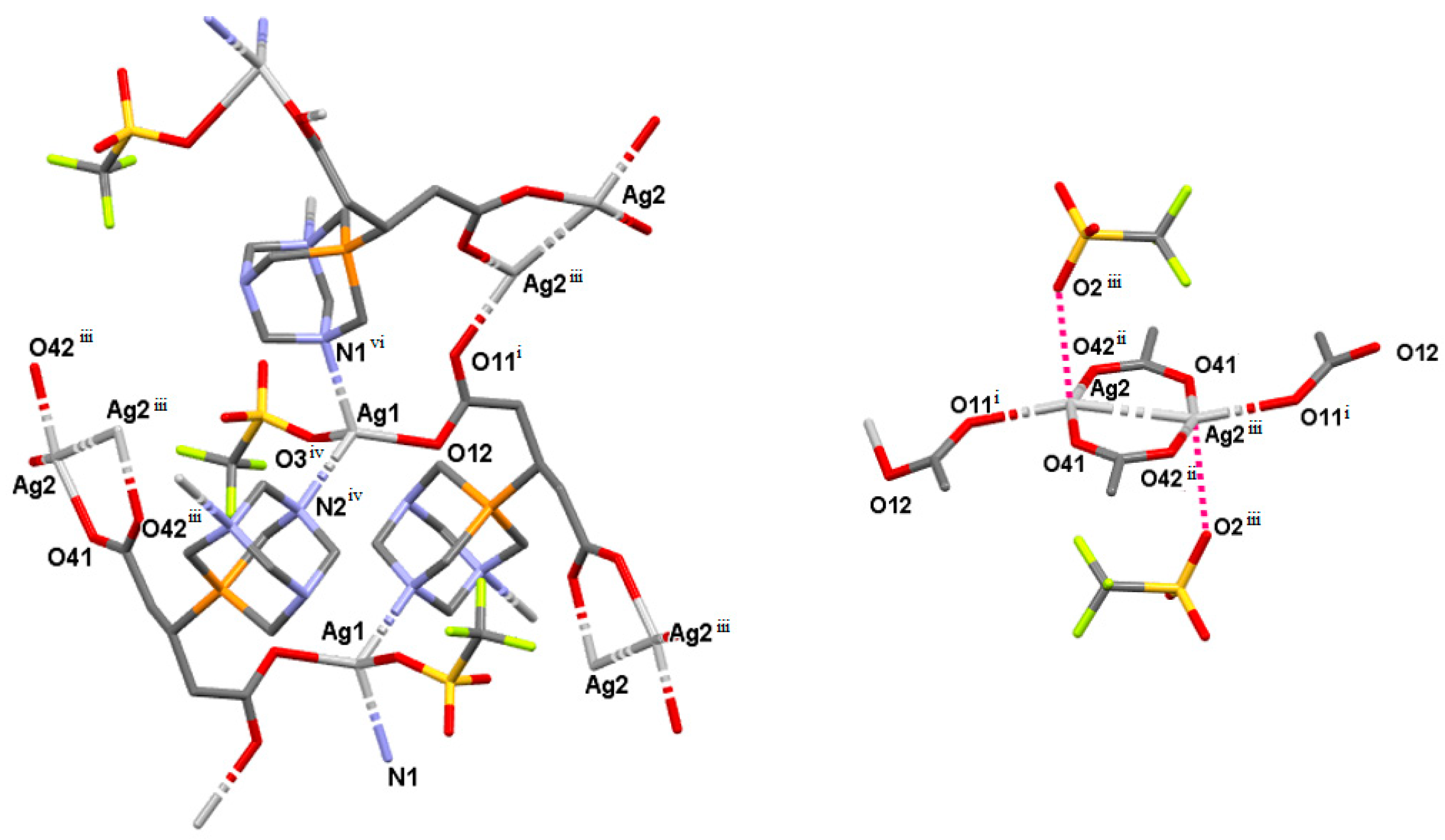
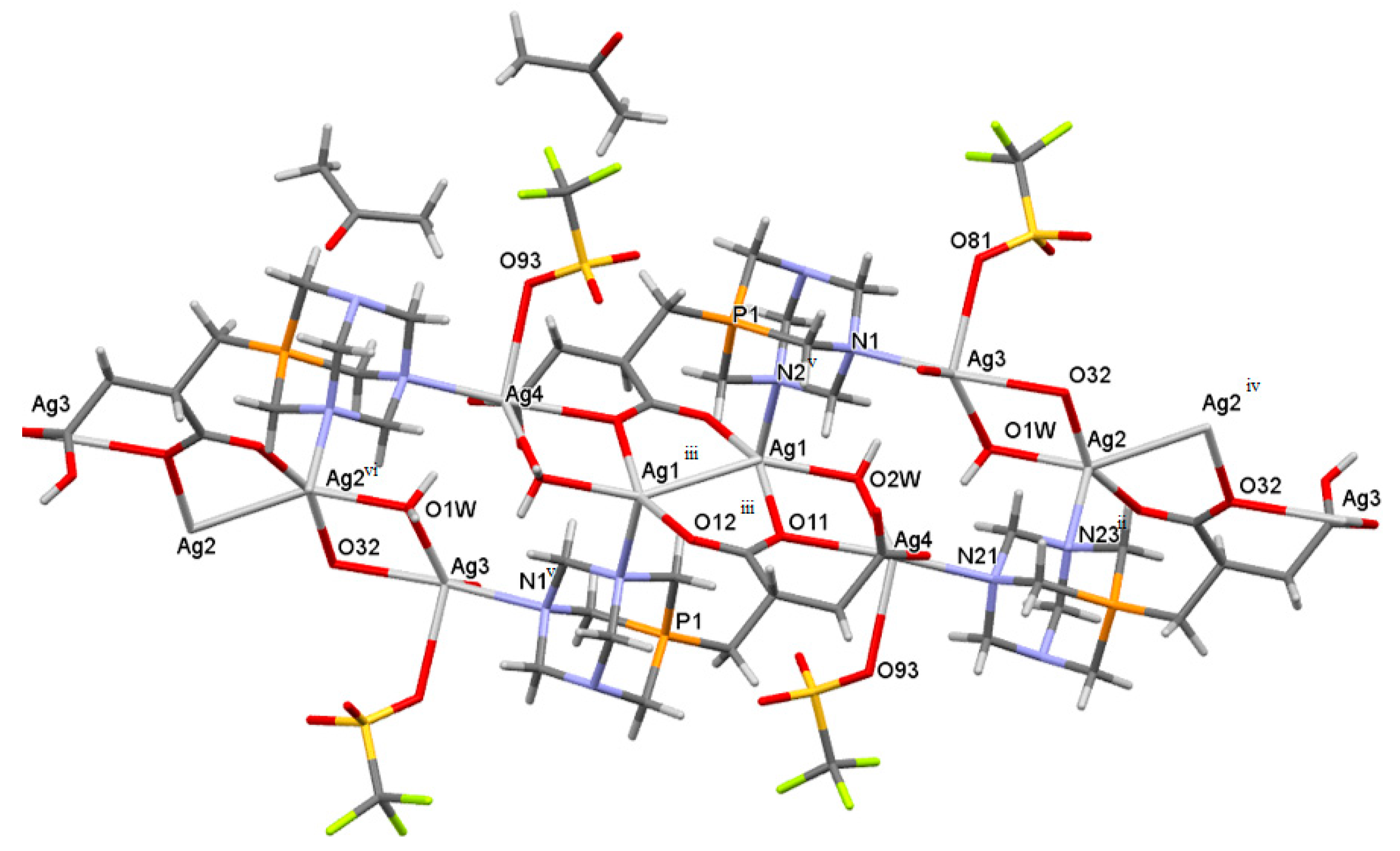
2.3. Structure of the Silver-Based Coordination Polymers
2.4. Antimicrobial Activity of the Phosphabetaines 1 and 2, and the Silver-Based Coordination Polymer CP1.2
3. Materials and Methods
3.1. Synthesis of the New Phosphabetaines 1 and 2
3.1.1. Synthesis of 4-carboxy-3-(1,3,5-triaza-7-phosphoniatricyclo-[3.3.1.13,7]dec-7-yl)butanoate (1)
3.1.2. Synthesis of 3-carboxy-2-(1,3,5-triaza-7-phosphoniatricyclo-[3.3.1.13,7]dec-7-ylmethyl)-propanoate (2)
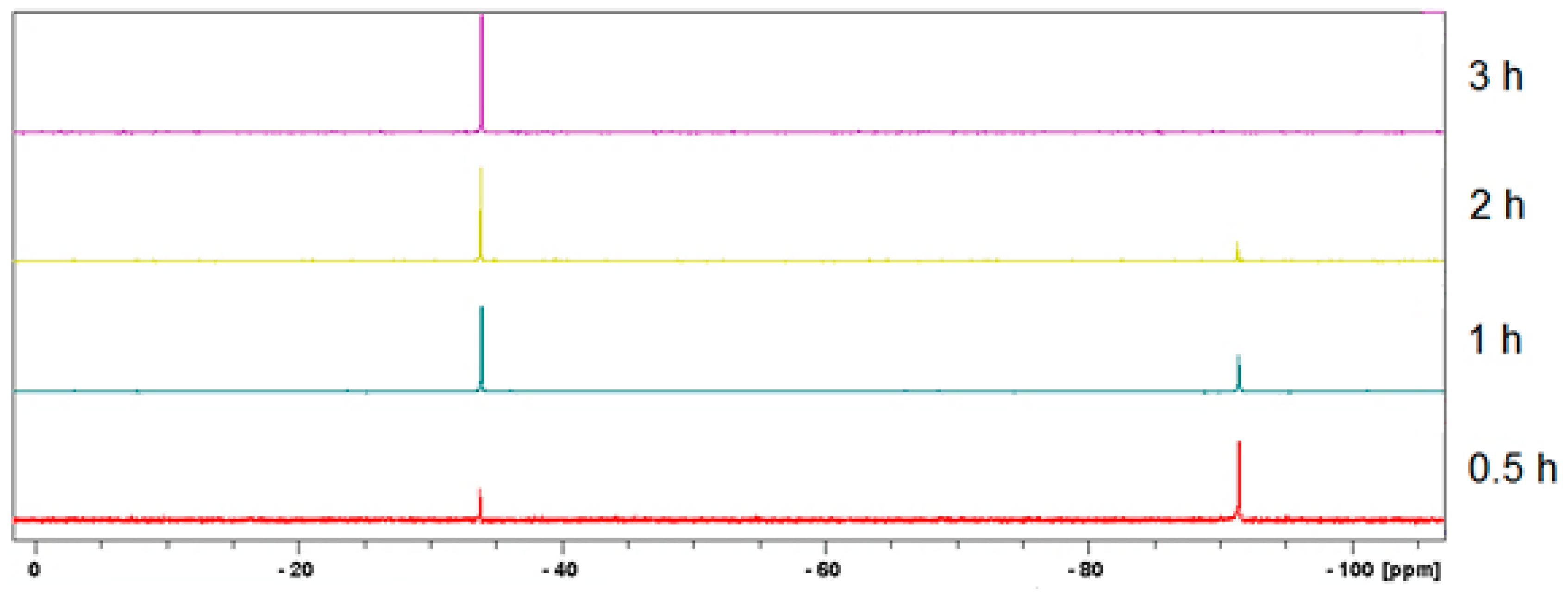
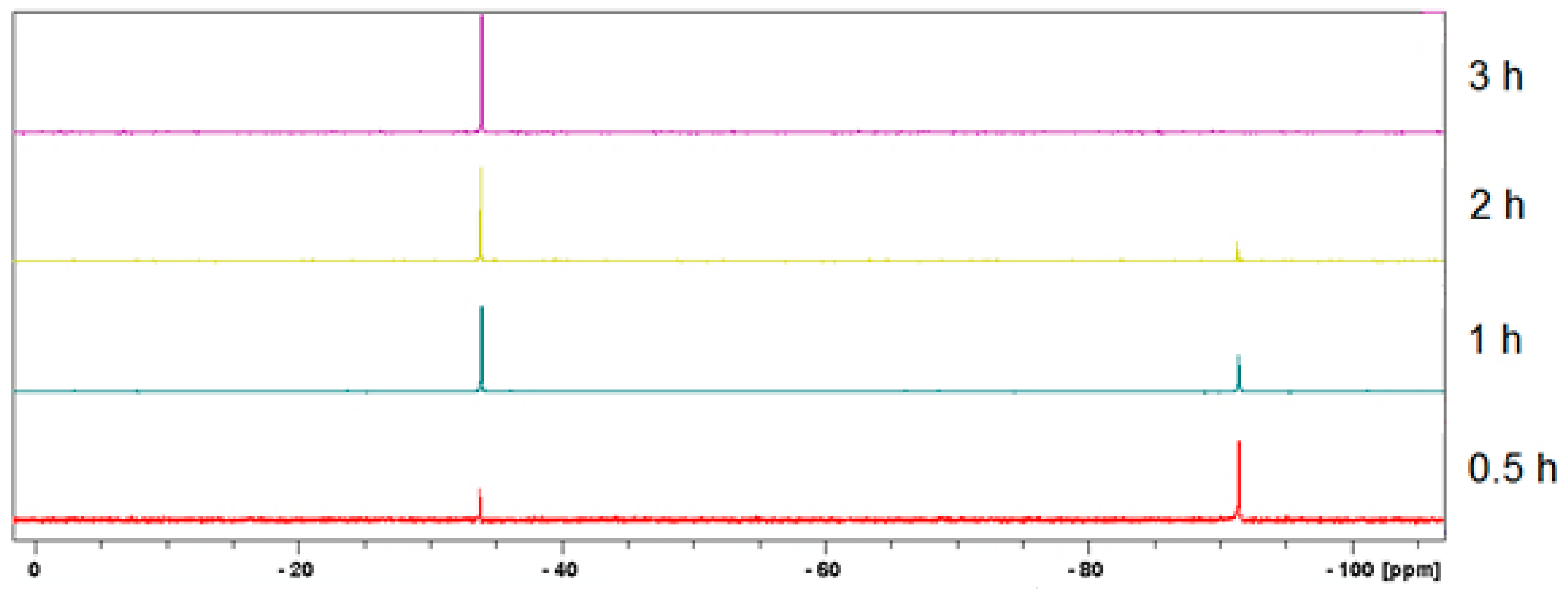
3.2. Mechanochemical Synthesis of 1, 2, and 3 in a Planetary Ball Mill
3.3. Synthesis of Silver(I)-Based Coordination Polymer
3.3.1. Synthesis of CP1.1
3.3.2. Synthesis of CP1.2
3.3.3. Synthesis of CP2
3.4. General Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Welton, T. Solvents and sustainable chemistry. Proc. R. Soc. 2015, 471, 20150502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Friščić, T.; Mottillo, C.; Titi, H.M. Mechanochemistry for Synthesis. Angew. Chem. Int. Ed. 2020, 59, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Solvent-Free Organic Synthesis, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2009. [Google Scholar]
- Margetic, D.; Štrukil, V. Mechanochemical Organic Synthesis, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Ranu, B.; Stolle, A. (Eds.) Ball Milling towards Green Synthesis: Applications, Projects, Challenges; RSC Green Chemistry Series, No. 31; Royal Society of Chemistry: Cambridge, UK, 2015. [Google Scholar]
- Gomollón-Bell, F. Ten chemical innovations that will change our world. Chem. Int. 2019, 41, 12–17. [Google Scholar] [CrossRef]
- Wang, G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Tan, D.; Friščić, T. Mechanochemistry for organic chemists: An update. Eur. J. Org. Chem. 2018, 18–33. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Peters, D.; Axelsson, O. The Suzuki reaction under solvent-free conditions. Synth. Commun. 2000, 30, 3501–3509. [Google Scholar] [CrossRef]
- Tullberg, E.; Peters, D.; Frejd, T. The Heck reaction under ball-milling conditions. J. Organomet. Chem. 2004, 689, 3778–3781. [Google Scholar] [CrossRef]
- Fulmer, D.A.; Shearouse, W.C.; Medonza, S.T.; Mack, J. Solvent-free Sonogashira coupling reaction viahigh speed ball milling. Green Chem. 2009, 11, 1821–1825. [Google Scholar] [CrossRef]
- Schumacher, C.; Crawford, D.E.; Raguz, B.; Glaum, R.; James, S.L.; Bolm, C.; Hernandez, J.G. Mechanochemical dehydrocoupling of dimethylamine borane and hydrogenation reactions using Wilkinson’s catalyst. Chem. Commun. 2018, 54, 8355–8358. [Google Scholar] [CrossRef]
- Portada, T.; Margetić, D.; Štrukil, V. Mechanochemical catalytic transfer hydrogenation of aromatic nitro derivatives. Molecules 2018, 23, 3163. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D.L. Controlling reactivity through liquid assisted grinding: The curious case of mechanochemical fluorination. Green Chem. 2017, 19, 2798–2802. [Google Scholar] [CrossRef]
- Collom, S.L.; Anastas, P.T.; Beach, E.S.; Crabtree, R.H.; Hazari, N.; Sommer, T.J. Differing selectivities in mechanochemical versus conventional solution oxidation using Oxone. Tetrahedron Lett. 2013, 54, 2344–2347. [Google Scholar] [CrossRef]
- Abdulwahaab, B.H.; Burke, B.P.; Domarkas, J.; Silversides, J.D.; Prior, T.J.; Archibald, S.J. Mono- and bis-alkylation of glyoxal-bridged tetraazamacrocycles using mechanochemistry. J. Org. Chem. 2016, 81, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Balema, V.P.; Wiench, J.W.; Pruski, M.; Pecharsky, V.K. Solvent-free mechanochemical synthesis of phosphonium salts. Chem. Commun. 2002, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Ravnsbæk, J.B.; Swager, T.M. Mechanochemical synthesis of poly(phenylene vinylenes). ACS Macro Lett. 2014, 3, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Thorwirth, R.; Stolle, A.; Ondruschka, B.; Wild, A.; Schubert, U.S. Fast, ligand- and solvent-free copper-catalyzed click reactions in a ball mill. Chem. Commun. 2011, 47, 4370–4372. [Google Scholar] [CrossRef]
- Beillard, A.; Métro, T.-X.; Bantreil, X.; Martinez, J.; Lamaty, F. Cu(0), O2 and mechanical forces: A saving combination for efficient production of Cu–NHC complexes. Chem. Sci. 2017, 8, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Beillard, A.; Golliard, E.; Gillet, V.; Bantreil, X.; Metro, T.-X.; Martinez, J.; Lamaty, F. Expedient mechanosynthesis of N,N-dialkyl imidazoliums and silver(I)−carbene complexes in a Ball-Mill. Chem. Eur. J. 2015, 21, 17614–17617. [Google Scholar] [CrossRef]
- Egbert, J.D.; Slawin, A.M.Z.; Nolan, S.P. Synthesis of NHeterocyclic carbene gold complexes using solution-phase and solid-state protocols. Organometallics 2013, 32, 2271–2274. [Google Scholar] [CrossRef]
- Juribasic, M.; Uzarevic, K.; Gracin, D.; Curic, M. Mechanochemical C-H bond activation: Rapid and regioselective double cyclopalladation monitored by In Situ Raman spectroscopy. Chem. Commun. 2014, 50, 10287–10290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.-H.; Yang, L.-F.; Cao, M.-L.; Wu, J.-J.; Ye, B.-H. Liquid-assisted solid-state reaction: Assembly of (6,3) and (10,3) hydrogen-bonded networks based on [M(Hbiim)3] by oxidation of [M(H2biim)3]2+ complexes in the presence of acetate anion. CrystEngComm 2011, 13, 4512–4518. [Google Scholar] [CrossRef]
- Beillard, A.; Bantreil, X.; Métro, T.-X.; Martinez, J.; Lamaty, F. Alternative technologies that facilitate access to discrete metal complexes. Chem. Rev. 2019, 119, 7529–7609. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Joó, F.; Horváth, H.; Udvardy, A.; Czégéni, C.E. Stirring or milling? First synthesis of Rh(I)-(di-N-heterocyclic carbene) complexes both in solution and in a ball mill. J. Organomet. Chem. 2020, 918, 121308. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA): Transition metal complexes and related catalytic, medicinal and photoluminescent applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Bravo, J.; Bolaño, S.; Gonsalvi, L.; Peruzzini, M. Coordination chemistry of 1,3,5-triaza-7-phosphaadamantane (PTA) and derivatives. Part II. The quest for tailored ligands, complexes and related applications. Coord. Chem. Rev. 2010, 254, 555–607. [Google Scholar] [CrossRef]
- Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Coordination chemistry of 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane(PTA) and derivatives. Part III. Variations on a theme: Novel architectures, materials and applications. Coord. Chem. Rev. 2018, 355, 328–361. [Google Scholar] [CrossRef]
- Mohr, F.; Falvello, L.R.; Laguna, M. A Silver(I) coordination polymer containing tridentate N- and P-coordinating 1,3,5-triaza-7-phosphaadamantane (PTA) ligands. Eur. J. Inorg. Chem. 2006, 3152–3154. [Google Scholar] [CrossRef]
- Jaros, S.W.; Guedes da Silva, M.F.C.G.; Florek, M.; Smoleński, P.; Pombeiro, A.J.L.; Kirillov, A.M. Silver(I) 1,3,5-triaza-7-phosphaadamantane coordination polymers driven by substituted glutarate and malonate building blocks: Self-assembly synthesis, structural features, and antimicrobial properties. Inorg. Chem. 2016, 55, 5886–5894. [Google Scholar] [CrossRef]
- Jaros, S.W.; Guedes da Silva, M.F.C.G.; Florek, M.; Oliveira, M.C.; Smoleński, P.; Pombeiro, A.J.L.; Kirillov, A.M. Aliphatic dicarboxylate directed assembly of silver(I) 1,3,5-triaza-7-phosphaadamantane coordination networks: Topological versatility and antimicrobial activity. Cryst. Growth. Des. 2014, 14, 5408–5417. [Google Scholar] [CrossRef]
- Udvardy, A.; Szolnoki, C.T.; Joó, F.; Kathó, Á. Solvent-free N-alkylation of 1,3,5-triaza-7: Phospha-adamantane (PTA). Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 469–470. [Google Scholar] [CrossRef]
- He, Z.; Tang, X.; Chen, Y.; He, Z. The first air-stable and efficient nucleophilic trialkylphosphine organocatalyst for the Baylis–Hillman reaction. Adv. Synth. Catal. 2006, 348, 413–417. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, B.; He, Z.; Gao, R.; He, Z. 1,3,5-Triaza-7-phosphaadamantane (PTA): A practical and versatile nucleophilic phosphine organocatalyst. Adv. Synth. Catal. 2007, 349, 2007–2017. [Google Scholar] [CrossRef]
- Udvardy, A.; Purgel, M.; Szarvas, T.; Joó, F.; Kathó, Á. Synthesis and structure of stable water-soluble phosphonium alkanoate zwitterions derived from 1,3,5-triaza-7-phosphaadamantane. Struct. Chem. 2015, 26, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Teimouri, M.B. Reaction between phosphines and itaconic anhydride in the presence of water: An efficient one-pot synthesis of 5-oxo-1,2λ5-oxaphospholanes. J. Chem. Res. 2006, 2006, 98–100. [Google Scholar] [CrossRef]
- Larpent, C.; Patin, H. Nucleophilic addition of water-soluble phosphines on activated olefins. Tetrahedron 1988, 44, 6107–6116. [Google Scholar] [CrossRef]
- Bényei, A.; Stafford, J.N.W.; Kathó, Á.; Darensbourgb, D.J.; Joó, F. The effect of phosphonium salt formation on the kinetics of homogeneous hydrogenations in water utilizing a rhodium meta-sulfonatophenyl-diphenylphosphine complex. J. Mol. Catal. 1993, 84, 157–163. [Google Scholar] [CrossRef]
- Budavari, S. (Ed.) The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 12th ed.; CRC Press: Cambridge, UK, 1996. [Google Scholar]
- Fluck, E.; Forster, J.E.; Weidlein, J.; Hadicke, E. 1.3.5-Triaza-7-phosphaadamantan (Monophospha-urotropin)/1,3,5-Triaza-7-phosphaadamantane (Monophospha-urotropine). Z. Nat. B Chem. Sci. 1977, 32, 499–501. [Google Scholar] [CrossRef]
- Thomas, L.; Srikrishnan, T. Crystal and molecular structure of glutaconic acid. J. Chem. Crystal. 2003, 33, 689–693. [Google Scholar] [CrossRef]
- Harlow, R.L.; Pfluger, C.E. Itaconic acid. Acta Cryst. 1973, B29, 2965–2966. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Pyykko, P. Strong Closed-shell interactions in inorganic chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef] [PubMed]
- Jaros, S.W.; Haukka, M.; Florek, M.; Guedes da Silva, M.; Pombeiro, A.; Kirillov, A.M.; Smoleński, P. New microbe killers: Self-assembled silver(I) coordination polymers driven by a cagelike aminophosphine. Materials 2019, 12, 3353. [Google Scholar] [CrossRef] [Green Version]
- Daigle, D.J. 1,3,5-Triaza-7-Phosphatricyclo[3.3.1.13,7]decane and derivatives. Inorg. Synth. 1998, 32, 40–41. [Google Scholar] [CrossRef]
- Nonius, B.V. CAD-Express Software, Ver. 5.1/1.2.; Enraf Nonius: Delft, The Netherlands, 1994. [Google Scholar]
- Harms, K.; Wocaldo, S. XCAD4; University of Marburg: Marburg, Germany, 1995. [Google Scholar]
- North, A.C.T.; Phillips, D.C.; Mathews, F.S. A semi-empirical method of absorption correction. Acta Cryst. 1968, A24, 351–359. [Google Scholar] [CrossRef]
- Bruker. APEX3 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2017. [Google Scholar]
- CrysAlisPro, Agilent Technologies, Version 1.171.37.35 (Release 13-08-2014 CrysAlis171 .NET) (Compiled Aug 13 2014). CrysAlisPro Version 1.171.3 Agilent Technologies, UK, 2014.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Cryst. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Cryst. 2020, E76, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrip, S.P. publCIF: Software for editing, validating and formatting crystallographic information files. J. Appl. Cryst. 2010, 43, 920–925. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.D.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Grove, D.C.; Randall, W.A. Assay Methods of Antibiotic. A Laboratory Manual. In Medical Encyclopedia; Antibiotics monographs, no. 2; Intercsience Publishers Inc.: New York, NY, USA, 1955. [Google Scholar]
- Wackett, L.P.; Gibson, D.T. Degradation of trichloroethylene by toluene dioxygenase in whole-cell studies with Pseudomonas putida F1. Appl. Enviro. Microbiol. 1988, 54, 1703–1708. [Google Scholar] [CrossRef] [Green Version]
- Mortimer, R.K.; Johnston, J.R. Genealogy of principal strains of the yeast genetic stock center. Genetics 1986, 113, 35–43. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution Synthesis | Mechanochemical Synthesis | |||
|---|---|---|---|---|
| Dicarboxylic acid (# of phosphabetaine) | Conversion,% (reaction time, h) | Yield (%) [Reference] | Conversion,% (milling time, h) | Yield (%) |
| Glutaconic (1) | 100 (3) | 87 a [this work] | 100 (4) | 74 |
| Itaconic (2) | 100 (2) | 71 a [this work] | 100 (4) | 77 |
| Maleic (3) | 100 (3) | 74 b [38] | 100(4) | 91 |
| Citraconic (4) | n.d. | 40 c [38] | 5 (8) | n.d. |
| Antimicrobial Agent | MIC a | ||
|---|---|---|---|
| Pseudomonas putida F1 | Bacillus subtilis | Saccharomyces cerevisiae S288C | |
| PTA | 723 | 647 | 286 |
| 114 b | 102 b | 45 b | |
| 1 | 422 | 264 | 385 |
| 122 b | 76 b | 111 b | |
| 2 | 443 | 269 | 348 |
| 128 b | 77 b | 100 b | |
| CP1.2 | 86 | 23 | 55 |
| 56 b | 15 b | 36 b | |
| Ag(CF3SO3) + 1 | 77 | 100 | 135 |
| 51 b | 66 b | 89 b | |
| AgNO3 | 77 | 100 | 135 |
| 13 b | 17 b | 23 b | |
| Ag(CF3SO3) | 77 | 100 | 143 |
| 20 b | 26 b | 37 b | |
Sample Availability: Samples of the compounds 1–3 are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udvardy, A.; Szolnoki, C.T.; Gombos, R.; Papp, G.; Kováts, É.; Joó, F.; Kathó, Á. Mechanochemical P-derivatization of 1,3,5-Triaza-7-Phosphaadamantane (PTA) and Silver-Based Coordination Polymers Obtained from the Resulting Phosphabetaines. Molecules 2020, 25, 5352. https://doi.org/10.3390/molecules25225352
Udvardy A, Szolnoki CT, Gombos R, Papp G, Kováts É, Joó F, Kathó Á. Mechanochemical P-derivatization of 1,3,5-Triaza-7-Phosphaadamantane (PTA) and Silver-Based Coordination Polymers Obtained from the Resulting Phosphabetaines. Molecules. 2020; 25(22):5352. https://doi.org/10.3390/molecules25225352
Chicago/Turabian StyleUdvardy, Antal, Csenge Tamara Szolnoki, Réka Gombos, Gábor Papp, Éva Kováts, Ferenc Joó, and Ágnes Kathó. 2020. "Mechanochemical P-derivatization of 1,3,5-Triaza-7-Phosphaadamantane (PTA) and Silver-Based Coordination Polymers Obtained from the Resulting Phosphabetaines" Molecules 25, no. 22: 5352. https://doi.org/10.3390/molecules25225352
APA StyleUdvardy, A., Szolnoki, C. T., Gombos, R., Papp, G., Kováts, É., Joó, F., & Kathó, Á. (2020). Mechanochemical P-derivatization of 1,3,5-Triaza-7-Phosphaadamantane (PTA) and Silver-Based Coordination Polymers Obtained from the Resulting Phosphabetaines. Molecules, 25(22), 5352. https://doi.org/10.3390/molecules25225352