
The Influence of Carbon Nature on the Catalytic Performance of Ru/C in Levulinic Acid Hydrogenation with Internal Hydrogen Source

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Textural Properties of Carbon Supports
2.2. Scanning Electron Microscopy
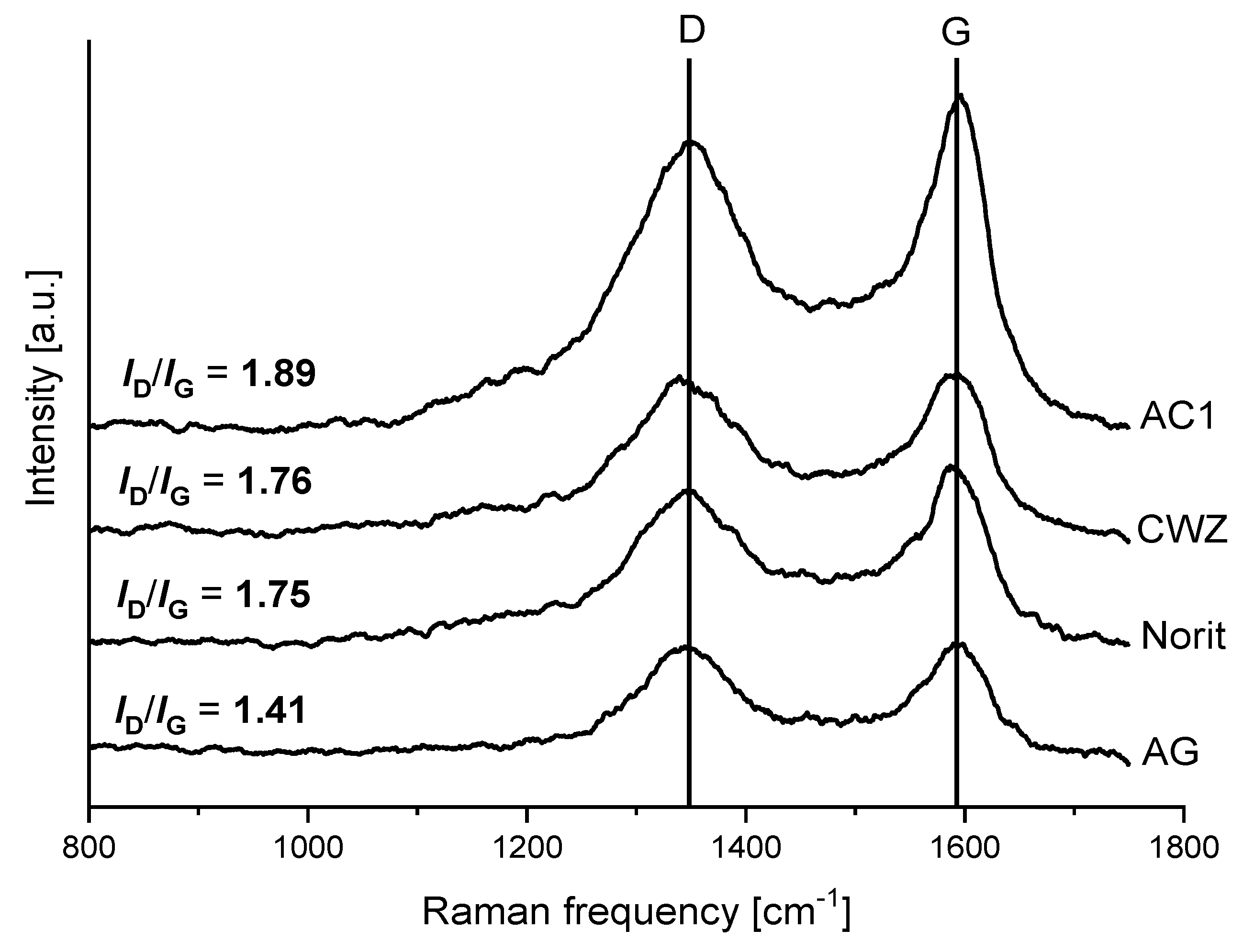
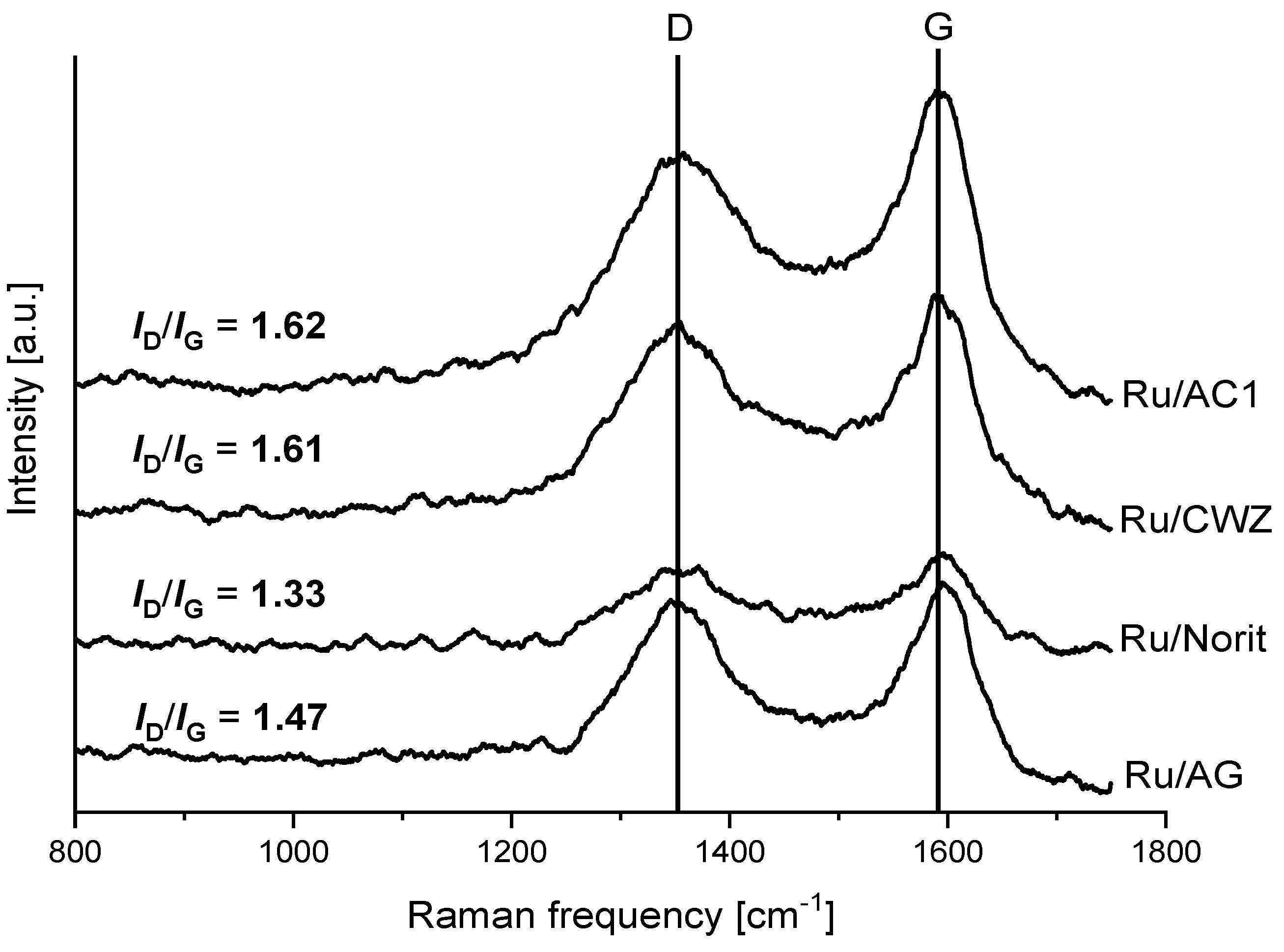
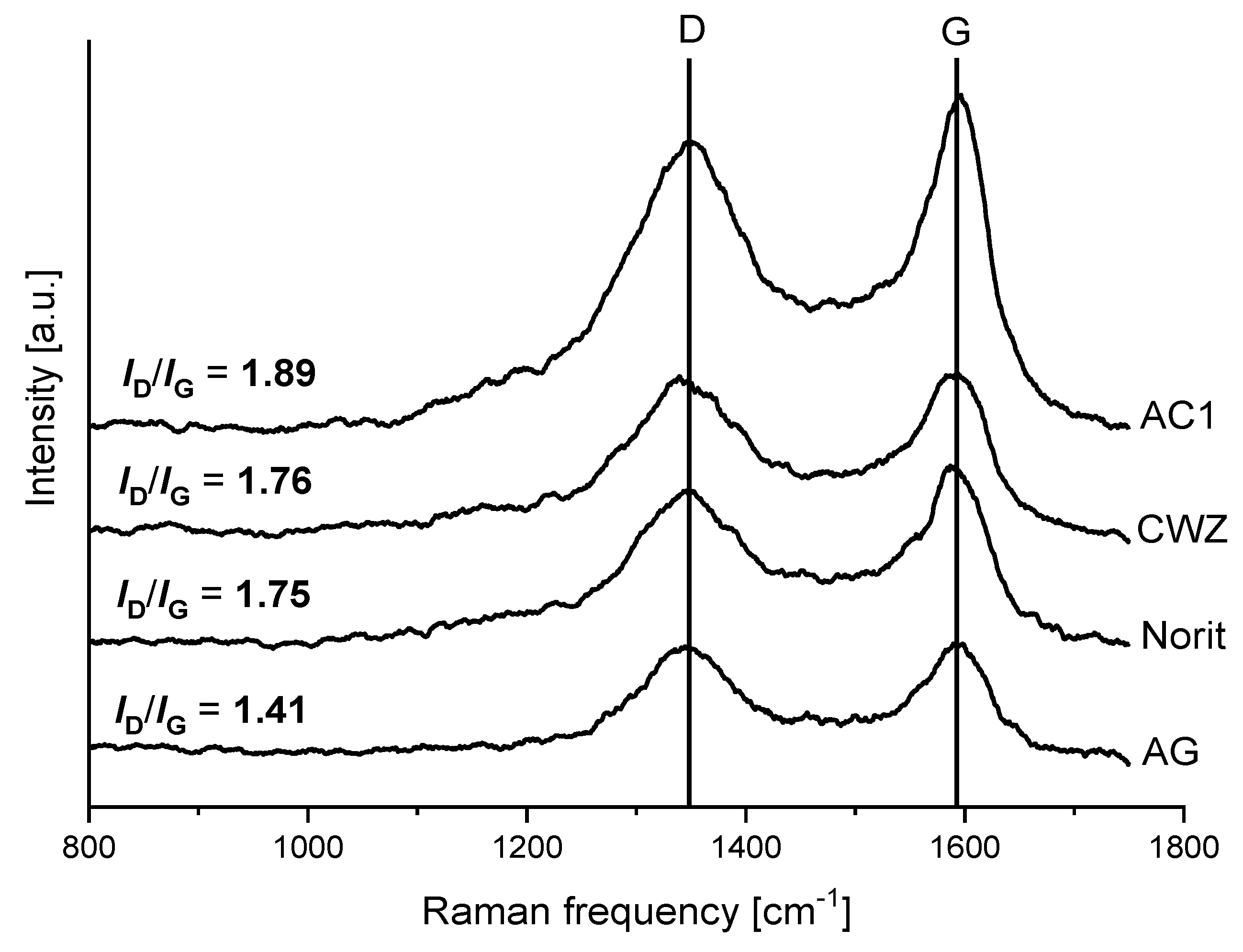
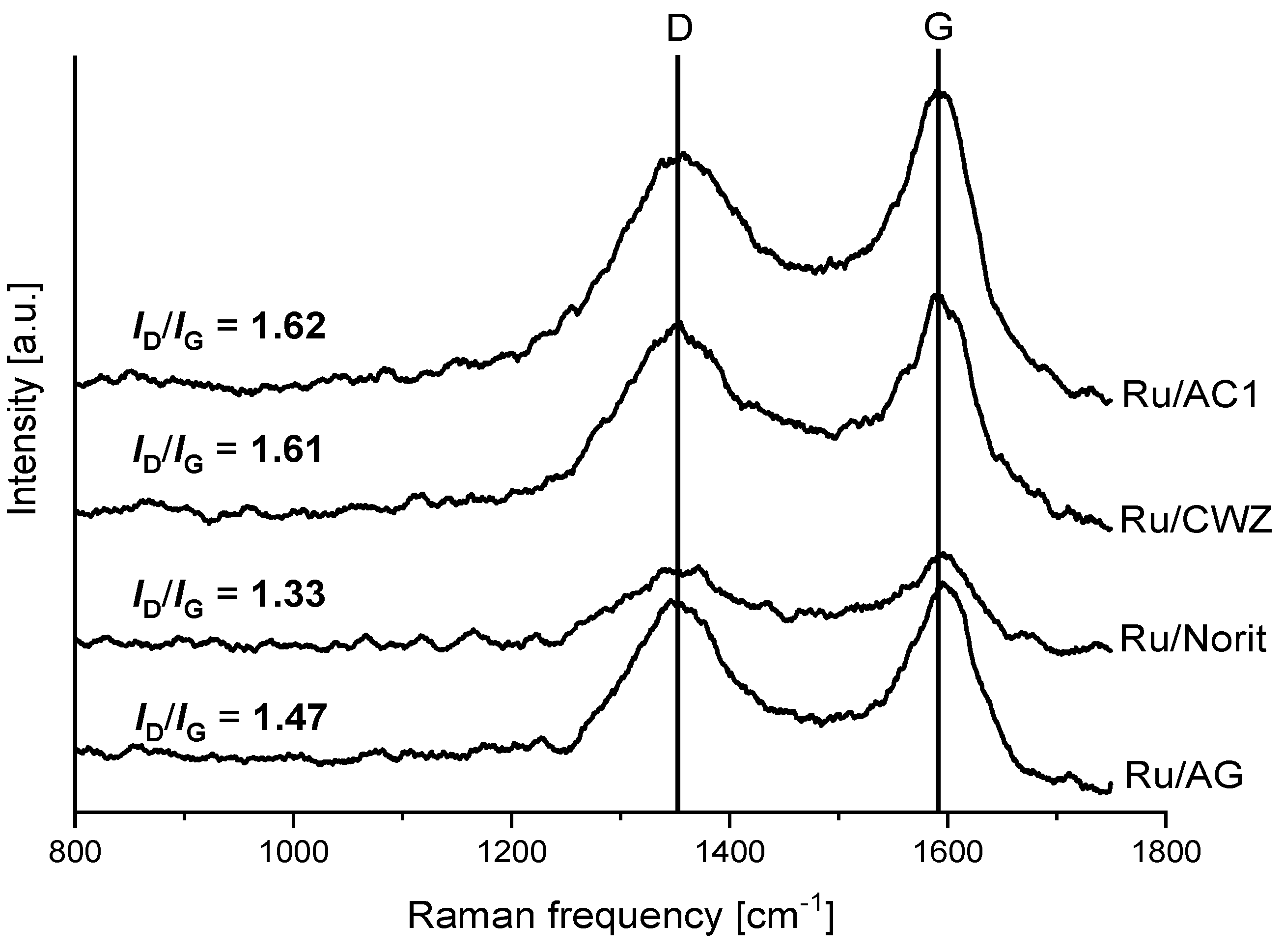
2.3. Raman Spectroscopy
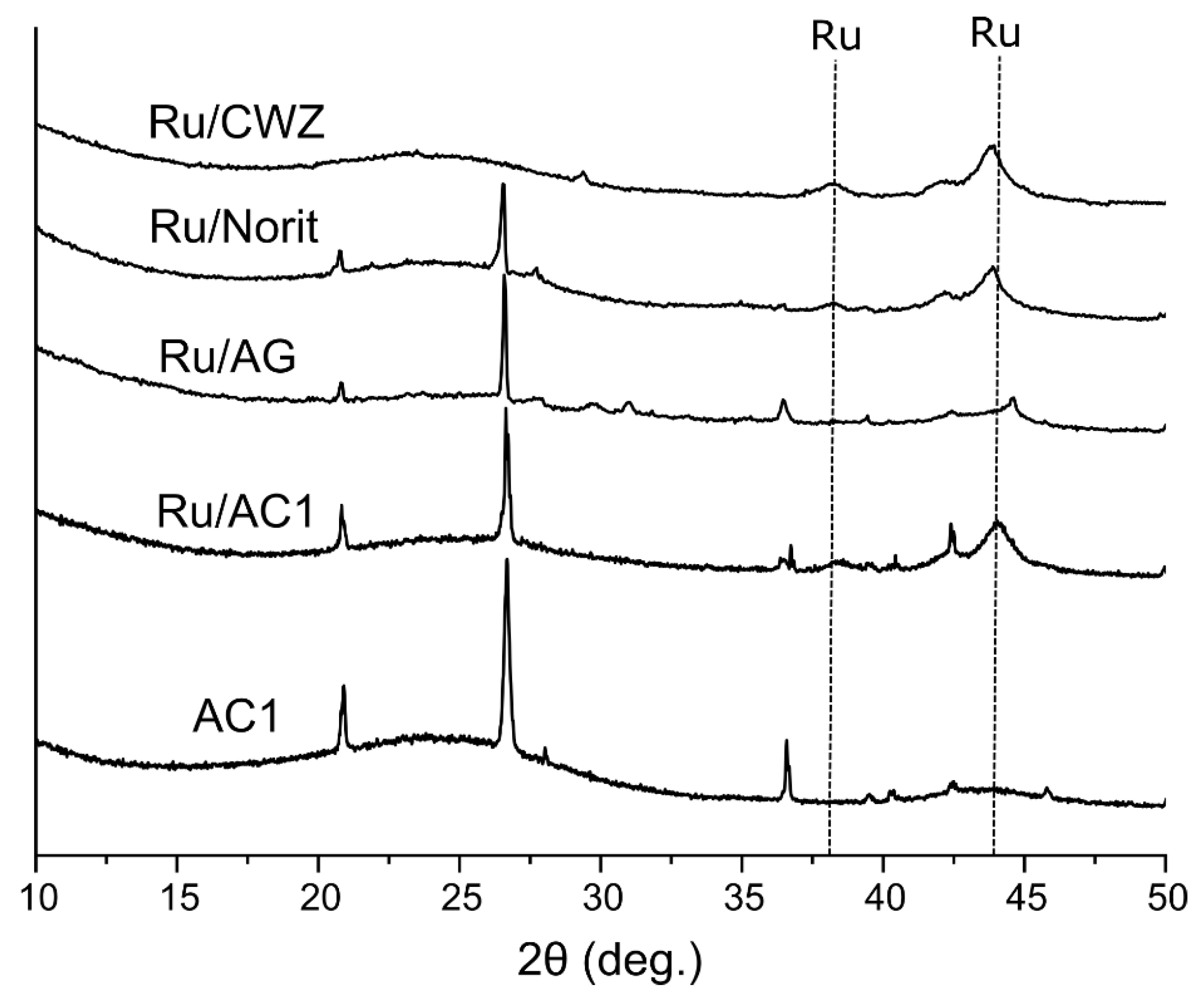
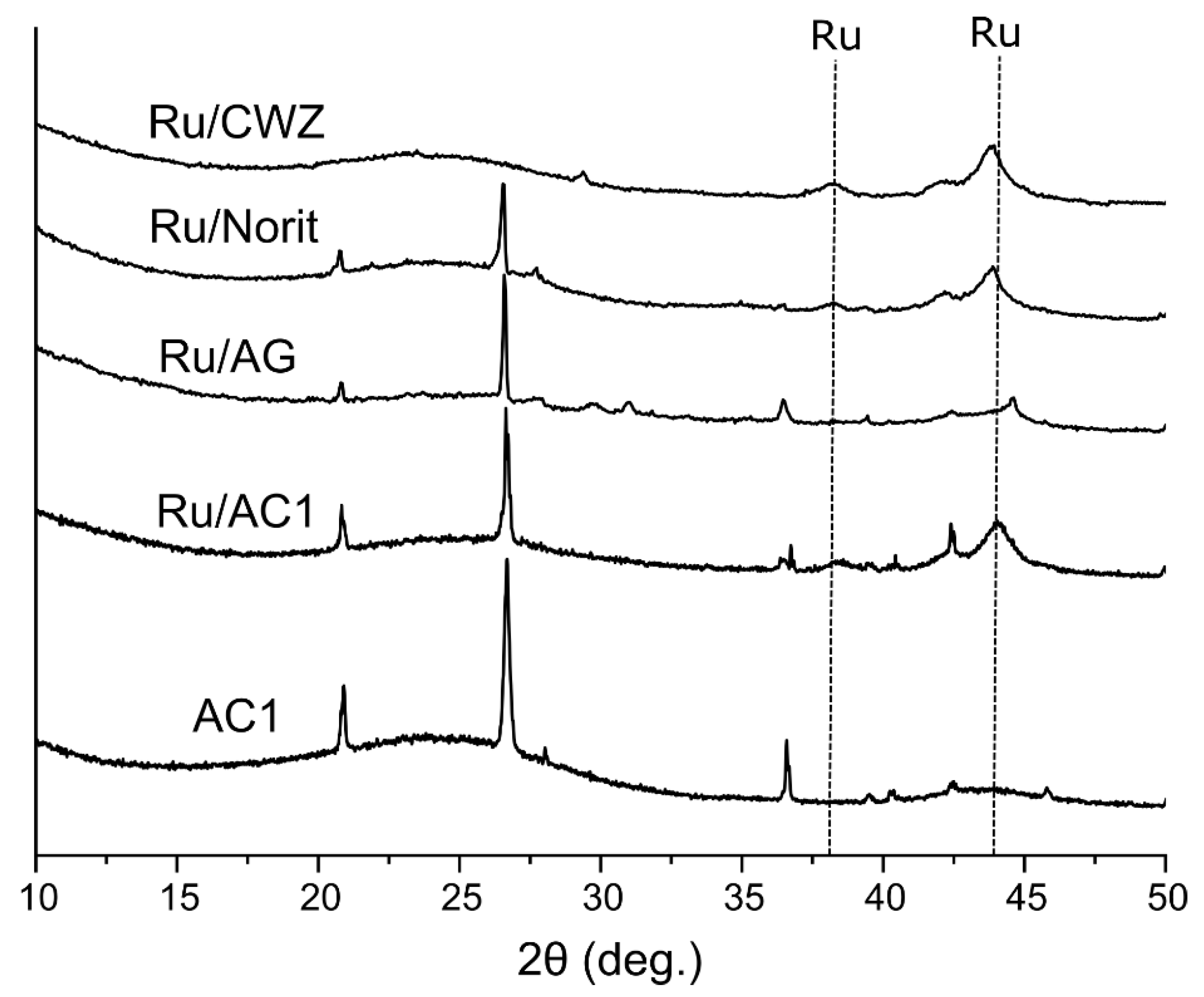
2.4. Phase Analysis of Catalysts
2.5. Temperature-Programmed Desorption of NH3 and CO2
2.6. Chemisorption of CO
2.7. Temperature-Programmed Reduction
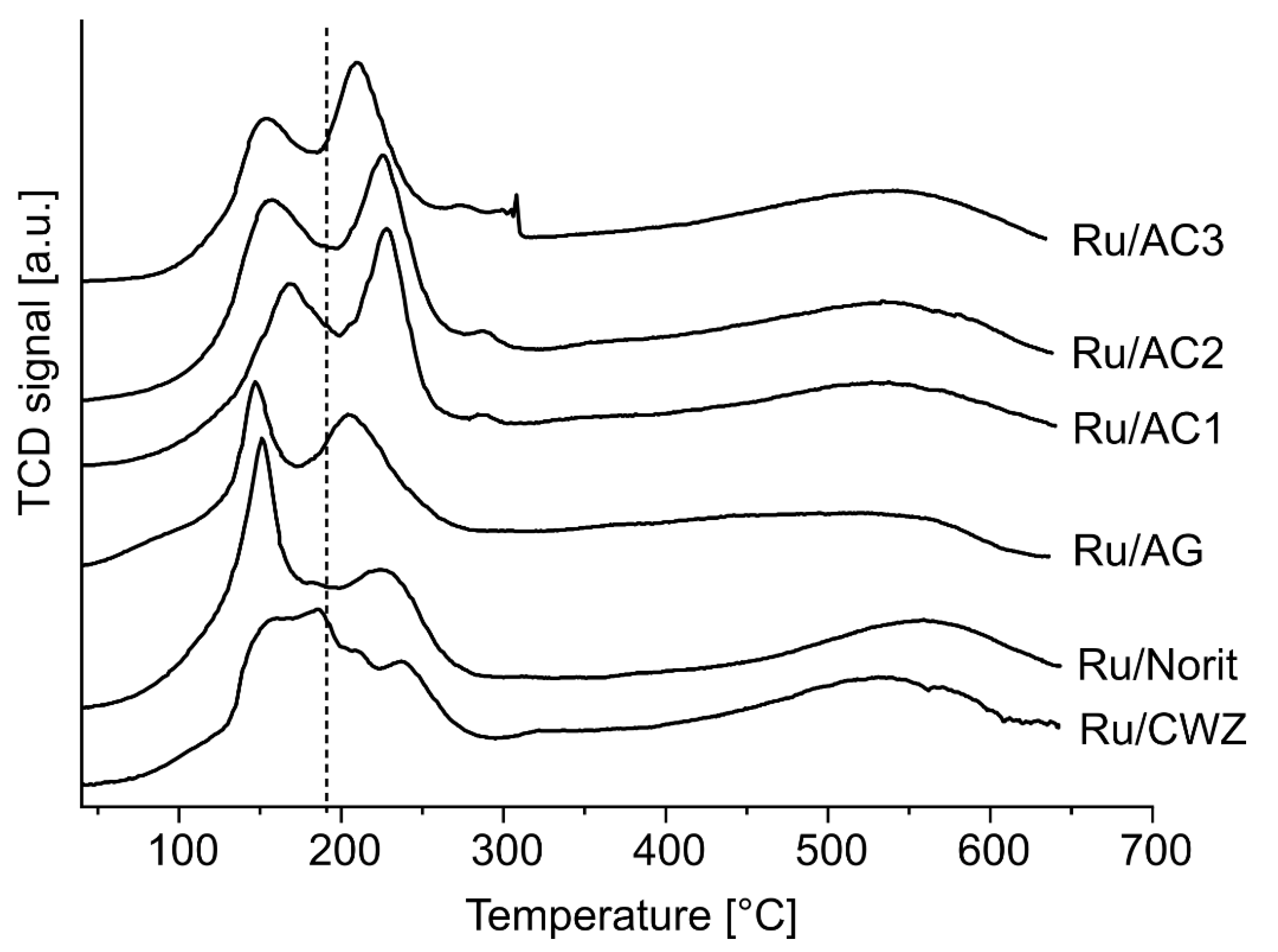
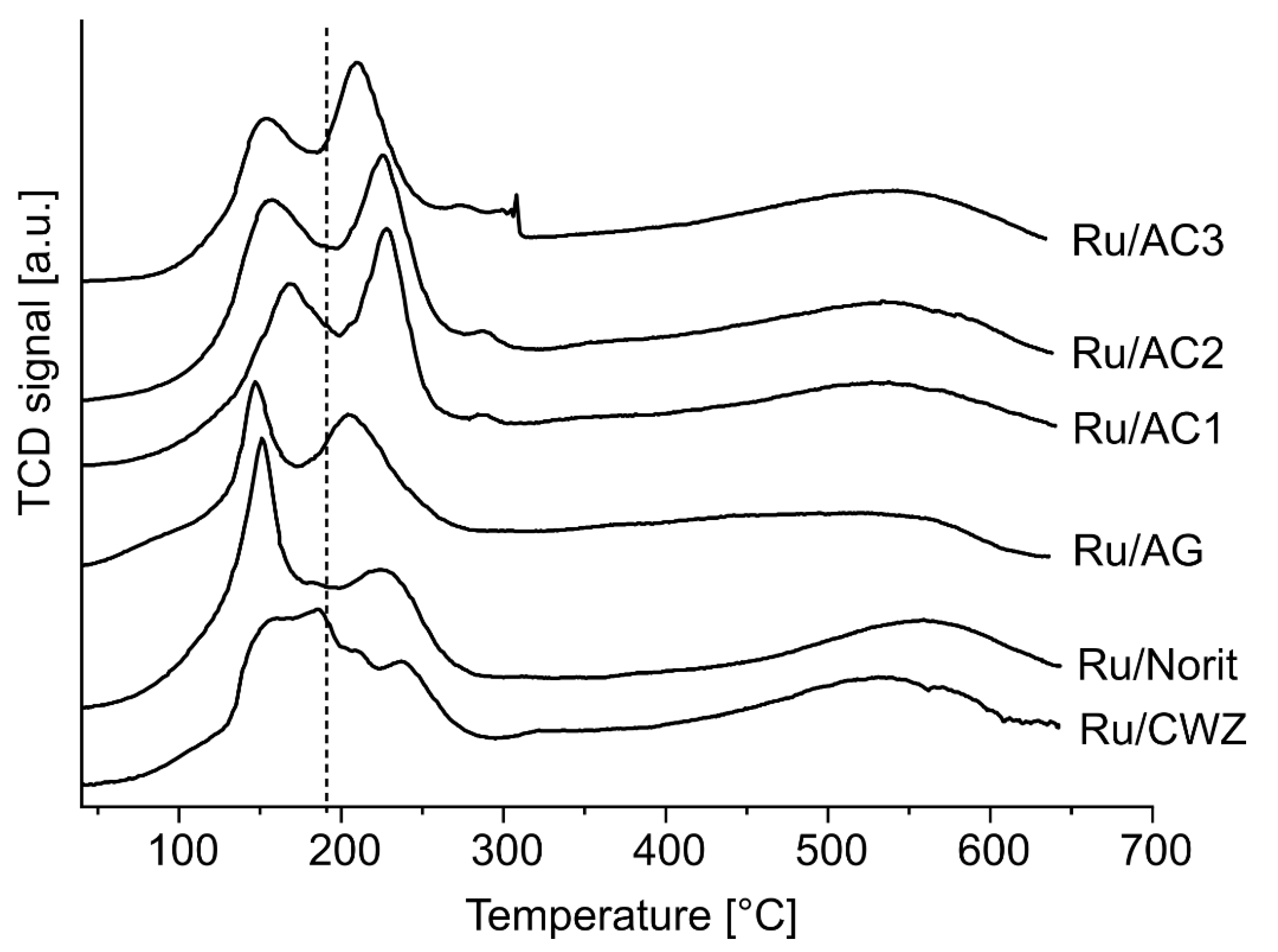
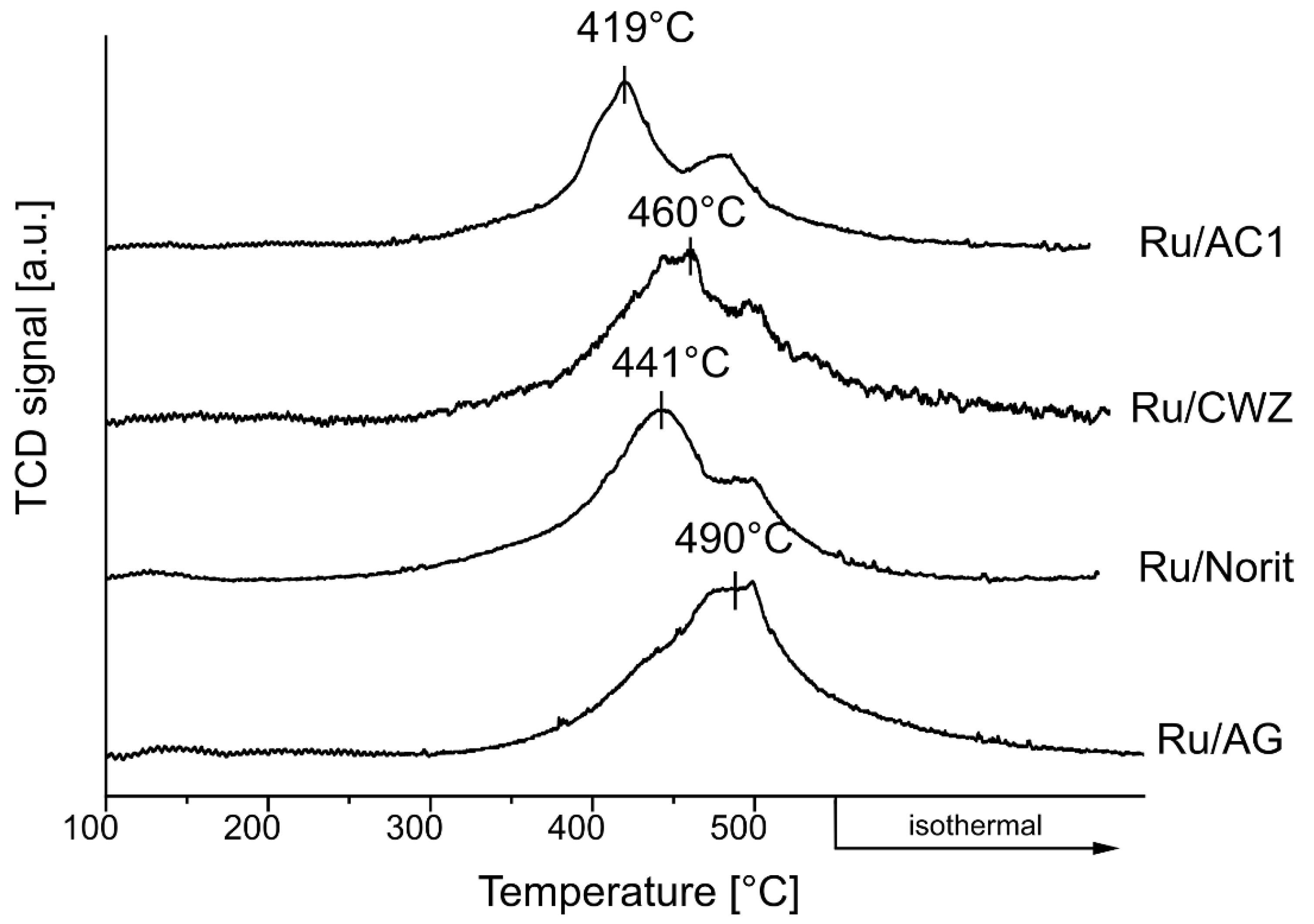
2.8. Temperature-Programmed Desorption of H2
2.9. Catalytic Activity
3. Discussion
4. Materials and Methods
4.1. Catalysts Preparation
4.2. Catalytic Activity Tests
4.3. Materials Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef] [Green Version]
- Werpy, T.A.; Holladay, J.; White, J.F. Top Value Added Chemicals from Biomass Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas. Natl. Renew. Energy Lab. 2004. [Google Scholar] [CrossRef] [Green Version]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Vasiliu, M.; Guynn, K.; Dixon, D.A. Prediction of the Thermodynamic Properties of Key Products and Intermediates from Biomass. J. Phys. Chem. C 2011, 115, 15686–15702. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Ruiz, J.C.; Luque, R.; Sepúlveda-Escribano, A. Transformations of biomass-derived platform molecules: From high added-value chemicals to fuels via aqueous-phase processing. Chem. Soc. Rev. 2011, 40, 5266–5281. [Google Scholar] [CrossRef]
- Yan, L.; Yao, Q.; Fu, Y. Conversion of levulinic acid and alkyl levulinates into biofuels and high-value chemicals. Green Chem. 2017, 19, 5527–5547. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Galletti, A.M.R.; Antonetti, C.; De Luise, V.; Martinelli, M. A sustainable process for the production of γ-valerolactone by hydrogenation of biomass-derived levulinic acid. Green Chem. 2012, 14, 688–694. [Google Scholar] [CrossRef]
- Hengne, A.M.; Kadu, B.S.; Biadar, N.S.; Chikate, R.C.; Rode, C.V. Transfer hydrogenation of biomass-derived levulinic acid to γ-valerolactone over supported Ni catalysts. RSC Adv. 2016, 6, 59753–59761. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Beloshapkin, S.; Ross, J.R.H. Hydrogen from formic acid decomposition over Pd and Au catalysts. Catal. Today 2010, 154, 7–12. [Google Scholar] [CrossRef]
- Eppinger, J.; Huang, K. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Morikawa, Y.; Norskov, J.K. CO Chemisorption at Metal Surfaces and Overlayers. Phys. Rev. Lett. 1996, 76, 2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellay, C.; Yan, N.; Dyson, P.J.; Lautenczy, G. Selective Formic Acid Decomposition for High-Pressure Hydrogen Generation: A Mechanistic Study. Chem. Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lu, X.; Xiong, J.; Li, X.; Bai, H.; Ji, N. Heterogeneous Catalytic Hydrogenation of Levulinic Acid to γ-Valerolactone with Formic Acid as Internal Hydrogen Source: Key Issues and Their Effects. ChemSusChem 2020, 11, 2916–2930. [Google Scholar] [CrossRef]
- Michel, C.; Zaffran, J.; Ruppert, A.M.; Matras-Michalska, J.; Jędrzejczyk, M.; Grams, J.; Sautet, P. Role of water in metal catalyst performance for ketone hydrogenation: A joint experimental and theoretical study on levulinic acid conversion into gamma-valerolactone. Chem. Commun. 2014, 50, 12450–12453. [Google Scholar] [CrossRef] [Green Version]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Seretis, A.; Diamantopoulou, P.; Thanou, I.; Tzevelekidis, P.; Fakas, C.; Lilas, P.; Papadogianakis, G. Recent Advances in Ruthenium-Catalyzed Hydrogenation Reactions of Renewable Biomass-Derived Levulinic Acid in Aqueous Media. Front. Chem. 2020, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Goh, T.-W.; Qi, Z.; Goes, S.; Brashler, K.; Perez, C.; Huang, W. Conversion of Levulinic Acid toγ Valerolactone over Few-Layer Graphene-Supported Ruthenium Catalysts. ACS Catal. 2016, 6, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Mai, E.F.; Machado, M.A.; Davies, T.E.; Lopez-Sanchez, J.A.; da Silva, V.T. Molybdenum carbide nanoparticles within carbon nanotubes as superior catalysts for γ-valerolactone production via levulinic acid hydrogenation. Green Chem. 2014, 16, 4092–4097. [Google Scholar] [CrossRef]
- Cao, W.; Luo, W.; Ge, H.; Su, Y.; Wang, A.; Zhang, T. UiO-66 derived Ru/ZrO2@C as a highly stable catalyst for hydrogenation of levulinic acid to γ-valerolactone. Green Chem. 2017, 19, 2201–2211. [Google Scholar] [CrossRef]
- Al-Shall, M.G.; Wright, W.R.H.; Palkovits, R. Exploring the ruthenium catalysed synthesis of γ-valerolactone in alcohols and utilisation of mild solvent-free reaction conditions. Green Chem. 2012, 14, 1260–1263. [Google Scholar] [CrossRef]
- Simakova, I.L.; Demidova, Y.S.; Simonov, M.N.; Niphadkar, P.S.; Bokade, V.V.; Devi, N.; Dhepe, P.L.; Murzin, D.Y. Mesoporous carbon and microporous zeolite supported Ru catalysts for selective levulinic acid hydrogenation into γ-valerolactone. Catal. Sustain. Energy 2019, 6, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Deka, U.; Beale, A.M.; van Eck, E.R.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Ruthenium-catalyzed hydrogenation of levulinic acid: Influence of the support and solvent on catalyst selectivity and stability. J. Catal. 2013, 301, 175–186. [Google Scholar] [CrossRef]
- Wojciechowska, J.; Jędrzejczyk, M.; Grams, J.; Keller, N.; Ruppert, A.M. Enhanced Production of γ-Valerolactone with an Internal Source of Hydrogen on Ca-Modified TiO2 Supported Ru Catalysts. ChemSusChem 2018, 12, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruppert, A.M.; Grams, J.; Jędrzejczyk, M.; Matras-Michalska, J.; Keller, N.; Ostojska, K.; Sautet, P. Titania-Supported Catalysts for Levulinic Acid Hydrogenation: Influence of Support and its Impact on γ-Valerolactone Yield. ChemSusChem 2015, 8, 1538–1547. [Google Scholar] [CrossRef]
- Ftouni, J.; Munoz-Murillo, A.; Goryachev, A.; Hofmann, J.P.; Hensen, E.J.M.; Lu, L.; Kiely, C.J.; Bruijnincx, P.C.A.; Weckhuysen, B.M. ZrO2 Is Preferred over TiO2 as Support for the Ru-Catalyzed Hydrogenation of Levulinic Acid to γ-Valerolactone. ACS Catal. 2016, 6, 5462–5472. [Google Scholar] [CrossRef] [Green Version]
- Hengne, A.M.; Biradar, N.S.; Rode, C.V. Surface Species of Supported Ruthenium Catalysts in Selective Hydrogenation of Levulinic Esters for Bio-Refinery Application. Catal. Lett. 2012, 142, 779–787. [Google Scholar] [CrossRef]
- Yan, Z.; Lin, L.; Liu, S. Synthesis of γ-Valerolactone by Hydrogenation of Biomass-derived Levulinic Acid over Ru/C Catalyst. Energy Fuels 2009, 23, 3853–3858. [Google Scholar] [CrossRef]
- Piskun, A.S.; van de Bovenkamp, H.H.; Rasrendra, C.B.; Winkelman, J.G.M.; Heeres, H.J. Kinetic modeling of levulinic acid hydrogenation to γ-valerolactone in water using a carbon supported Ru catalyst. App. Catal. A Gen. 2016, 525, 158–167. [Google Scholar] [CrossRef]
- Abdelrahman, O.A.; Heyden, A.; Bond, J.Q. Analysis of Kinetics and Reaction Pathways in the Aqueous-Phase Hydrogenation of Levulinic Acid to Form γ-Valerolactone over Ru/C. ACS Catal. 2014, 4, 1171–1181. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Jędrzejczyk, M.; Sneka-Płatek, O.; Keller, N.; Dumon, A.S.; Michel, C.; Sautet, P.; Grams, J. Ru catalysts for levulinic acid hydrogenation with formic acid as a hydrogen source. Green Chem. 2016, 18, 2014–2028. [Google Scholar] [CrossRef]
- Wu, M.; Liao, J.; Yu, L.; Lv, R.; Li, P.; Sun, W.; Tan, R.; Duan, X.; Zhang, L.; Li, F.; et al. 2020 Roadmap on Carbon Materials for Energy Storage and Conversion. Chem. Asian J. 2020, 15, 995–1013. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Song, J.; Zhou, B.; Wu, T.; Jiang, T.; Han, B. Preparation of Ru/Graphene using Glucose as Carbon Source and Hydrogenation of Levulinic Acid to g-Valerolactone. Chem. Asian J. 2016, 11, 2792–2796. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Lou, J.; Su, C.; Guo, D.; Liu, Y.; Deng, S. An Efficient and Reusable Embedded Ru Catalyst for the Hydrogenolysis of Levulinic Acid to g-Valerolactone. ChemSusChem 2017, 10, 1720–1732. [Google Scholar] [CrossRef]
- Zanutelo, C.; Landers, R.; Carvalho, W.A.; Cobo, A.J.G. Carbon support treatment effect on Ru/C catalyst performance for benzene partial hydrogenation. App. Catal. A Gen. 2011, 15, 409–410. [Google Scholar] [CrossRef]
- Zhu, H.; Han, W.; Liu, H. Influence of oxidation on heat-treated activated carbon support properties and metallic dispersion of Ru/C catalyst. Catal. Lett. 2007, 115, 13–18. [Google Scholar] [CrossRef]
- Gallegos-Suarez, E.; perez-Cadenas, M.; Guerrero-Ruiz, A.; Rodriguez-Ramos, I.; Arcoya, A. Effect of the functional groups of carbon on the surface and catalytic properties of Ru/C catalysts for hydrogenolysis of glycerol. Appl. Surf. Sci. 2013, 287, 108–116. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095. [Google Scholar] [CrossRef] [Green Version]
- Dillon, R.O.; Woollam, J.A.; Katkanant, V. Use of Raman scattering to investigate disorder and crystallite formation in as-deposited and anriealed carbon films. Phys. Rev. B 1984, 29, 3482. [Google Scholar] [CrossRef]
- Wang, J.; Yin, G.; Shao, Y.; Wang, Z.; Gao, Y. Investigation of Further Improvement of Platinum Catalyst Durability with Highly Graphitized Carbon Nanotubes Support. J. Phys. Chem. C 2008, 112, 5784–5789. [Google Scholar] [CrossRef]
- Casiraghi, C.; Ferrari, A.C.; Robertson, J. Raman spectroscopy of hydrogenated amorphous carbons. Phys. Rev. B 2005, 72, 085401. [Google Scholar] [CrossRef] [Green Version]
- Stankovich, S.; Dikin, D.A.; Piner, R.D.; Kohlhaas, K.A.; Kleinhammes, A.; Jia, Y.; Wu, Y.; Nguyen, S.T.; Ruoff, R.S. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007, 45, 1558–1565. [Google Scholar] [CrossRef]
- Luukkonen, T.; Tolonen, E.; Runtti, H.; Pellinen, J.; Hu, T.; Ramo, J.; Lassi, U. Removal of total organic carbon (TOC) residues from power plant make-up water by activated carbon. J. Water Process. Eng. 2014, 3, 46–52. [Google Scholar] [CrossRef]
- Nevskaia, D.M.; Santianes, A.; Munoz, V.; Guerrero-Ruiz, A. Interaction of aqueous solutions of phenol with commercial activated carbons: An adsorption and kinetic study. Carbon 1999, 37, 1065–1074. [Google Scholar] [CrossRef]
- Zhang, X.; Chan, K. Microemulsion synthesis and electrocatalytic properties of platinum–cobalt nanoparticles. J. Mater. Chem. 2002, 12, 1203–1206. [Google Scholar] [CrossRef]
- Boehm, H.P. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, Z.H.; Coomes, A.; Haghseresht, F.; Lu, G.Q. The physical and surface chemical characteristics of activated carbons and the adsorption of methylene blue from wastewater. J. Colloid Interface Sci. 2005, 284, 440–446. [Google Scholar] [CrossRef]
- Chen, J.P.; Wu, S.N. Acid/Base-Treated Activated Carbons: Characterization of Functional Groups and Metal Adsorptive Properties. Langmuir 2004, 20, 2233–2242. [Google Scholar] [CrossRef]
- Perez-Cadenas, A.F.; Maldonado-Hodar, J.; Moreno-Castilla, C. On the nature of surface acid sites of chlorinated activated carbons. Carbon 2003, 41, 473–478. [Google Scholar] [CrossRef]
- Newman, C.; Zhou, X.; Goundie, B.; Ghampson, I.T.; Pollock, R.A.; Ross, Z.; Wheeler, M.C.; Meulenberg, R.W.; Austin, R.N.; Frederick, B.G. Effects of support identity and metal dispersion in supported ruthenium hydrodeoxygenation catalysts. Appl. Catal. A Gen. 2014, 477, 64–74. [Google Scholar] [CrossRef]
- Peng, G.; Gramm, F.; Ludwig, C.; Vogel, F. Effect of carbon surface functional groups on the synthesis of Ru/C catalysts for supercritical water gasification. Catal. Sci. Technol. 2015, 5, 3658–3666. [Google Scholar] [CrossRef] [Green Version]
- Prado-Burguete, C.; Linares-Solano, A.; Rodriguez-Reinoso, F.; de Lecea, C.S. The Effect of Oxygen Surface Groups of the Support on Platinum Dispersion in Pt/Carbon Catalysts. J. Catal. 1989, 115, 98–106. [Google Scholar] [CrossRef]
- Mazzieri, V.A.; L’Argentiere, P.C.; Coloma-Pascual, F.; Figoli, N.S. Effect of Chlorine on the Properties of Ru/Al2O3. Ind. Eng. Chem. Res. 2003, 42, 2269–2272. [Google Scholar] [CrossRef]
- Mazzieri, V.; Coloma-Pascual, F.; Arcoya, A.; L’Argentiere, P.C.; Figoli, N.S. XPS, FTIR and TPR characterization of Ru/Al2O3 catalysts. Appl. Surf. Sci. 2003, 210, 222–230. [Google Scholar] [CrossRef]
- Wang, X.; Lan, G.; Liu, H.; Zhu, Y.; Li, Y. Effect of acidity and ruthenium species on catalytic performance of ruthenium catalysts for acetylene hydrochlorination. Catal. Sci. Technol. 2018, 8, 6143–6149. [Google Scholar] [CrossRef]
- Liu, X.; Yao, K.X.; Meng, C.; Han, Y. Graphene substrate-mediated catalytic performance enhancement of Ru nanoparticles: A first-principles study. Dalton Trans. 2012, 41, 1289–1296. [Google Scholar] [CrossRef]
- Prudenziati, M.; Morten, B.; Travan, E. Reduction process of RuO2 powders and kinetics of their re-oxidation. Mater. Sci. Eng. B 2003, 98, 167–176. [Google Scholar] [CrossRef]
- Zhang, H.; Lancelot, C.; Chu, W.; Hong, J.; Khodakov, A.Y.; Chernavskii, P.A.; Zheng, J.; Tong, D. The nature of cobalt species in carbon nanotubes and their catalytic performance in Fischer–Tropsch reaction. J. Mater. Chem. 2009, 19, 9241–9249. [Google Scholar] [CrossRef]
- Lin, B.; Wang, R.; Yu, X.; Lin, J.; Xie, F.; Wei, K. Physicochemical Characterization and H2-TPD Study of Alumina Supported Ruthenium Catalysts. Catal. Lett. 2008, 124, 178–184. [Google Scholar] [CrossRef]
- Sneka-Płatek, O.; Kaźmierczak, K.; Jędrzejczyk, M.; Sautet, P.; Keller, N.; Michel, C.; Ruppert, A.M. Understanding the influence of the composition of the Ag[sbnd]Pd catalysts on the selective formic acid decomposition and subsequent levulinic acid hydrogenation. Int. J. Hydrog. Energ. 2020, 45, 17339–17353. [Google Scholar] [CrossRef]
- Kocemba, I. Apparatus for investigation of catalysts by temperature programmed methods—Peak4. Przem. Chem. 2003, 3, 142–145. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Support | Grain Size (mm) | Surface Area (m2 g−1) | Total Pore Volume (cm3 g−1) | Average Pore Diameter (nm) |
|---|---|---|---|---|
| Norit | <0.10 | 649 | 0.421 | 5.7 |
| CWZ | <0.10 | 789 | 0.211 | 4.7 |
| AG | <0.10 | 973 | 0.652 | 4.6 |
| AC1 | <0.10 | 691 | 0.092 | 3.6 |
| AC2 | 0.25–0.50 | 724 | 0.090 | 3.4 |
| AC3 | 0.75–1.00 | 882 | 0.116 | 3.4 |
| Carbon Support | Acidity (µmol/g) | Basicity (µmol/g) | Catalyst | Acidity (µmol/g) | Basicity (µmol/g) |
|---|---|---|---|---|---|
| Norit | 32 | 18 | Ru/Norit | 329 | 113 |
| CWZ | 154 | 86 | Ru/CWZ | 513 | 226 |
| AG | 58 | 28 | Ru/AG | 419 | 117 |
| AC1 | 118 | 60 | Ru/AC1 | 525 | 153 |
| Catalyst | Volume of CO Adsorbed (cm3 g−1) | Dispersion of Ru from CO Chemisorption (%) | Particle Size of Ru from CO Chemisorption (nm) |
|---|---|---|---|
| Ru/Norit | 1.797 | 16.2 | 2.6 |
| Ru/CWZ | 1.705 | 15.4 | 2.7 |
| Ru/AG | 1.651 | 14.9 | 2.8 |
| Ru/AC1 | 1.663 | 15.1 | 2.8 |
| Ru/AC2 | 0.702 | 6.4 | 6.7 |
| Ru/AC3 | 0.595 | 5.4 | 7.9 |
| Catalyst | FA Decomposition | LA Hydrogenation | |||||
|---|---|---|---|---|---|---|---|
| FA Conversion (%) | Gaseous Products (% vol) | LA Conversion (%) | GVL Yield (%) | ||||
| H2 | CO | CH4 | CO2 | ||||
| Ru/Norit | 84 | 47 | 3 | 0 | 50 | 82 | 71 |
| Ru/CWZ | 80 | 47 | 5 | 0 | 48 | 80 | 64 |
| Ru/AG | 77 | 49 | 1 | 0 | 50 | 81 | 70 |
| Ru/AC1 | 97 | 47 | 7 | 0 | 46 | 95 | 78 |
| Ru/AC2 | 89 | 46 | 8 | 0 | 46 | 76 | 63 |
| Ru/AC3 | 74 | 46 | 9 | 0 | 45 | 75 | 63 |
| Catalyst | FALA Reaction | ||
|---|---|---|---|
| FA Conversion (%) | LA Conversion (%) | GVL Yield (%) | |
| Ru/Norit | 100 | 58 | 38 |
| Ru/CWZ | 100 | 58 | 35 |
| Ru/AG | 100 | 38 | 18 |
| Ru/AC1 | 100 | 75 | 59 |
| Ru/AC2 | 100 | 46 | 25 |
| Ru/AC3 | 100 | 48 | 27 |
Sample Availability: Samples of the compounds are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jędrzejczyk, M.; Soszka, E.; Goscianska, J.; Kozanecki, M.; Grams, J.; Ruppert, A.M. The Influence of Carbon Nature on the Catalytic Performance of Ru/C in Levulinic Acid Hydrogenation with Internal Hydrogen Source. Molecules 2020, 25, 5362. https://doi.org/10.3390/molecules25225362
Jędrzejczyk M, Soszka E, Goscianska J, Kozanecki M, Grams J, Ruppert AM. The Influence of Carbon Nature on the Catalytic Performance of Ru/C in Levulinic Acid Hydrogenation with Internal Hydrogen Source. Molecules. 2020; 25(22):5362. https://doi.org/10.3390/molecules25225362
Chicago/Turabian StyleJędrzejczyk, Marcin, Emilia Soszka, Joanna Goscianska, Marcin Kozanecki, Jacek Grams, and Agnieszka M. Ruppert. 2020. "The Influence of Carbon Nature on the Catalytic Performance of Ru/C in Levulinic Acid Hydrogenation with Internal Hydrogen Source" Molecules 25, no. 22: 5362. https://doi.org/10.3390/molecules25225362
APA StyleJędrzejczyk, M., Soszka, E., Goscianska, J., Kozanecki, M., Grams, J., & Ruppert, A. M. (2020). The Influence of Carbon Nature on the Catalytic Performance of Ru/C in Levulinic Acid Hydrogenation with Internal Hydrogen Source. Molecules, 25(22), 5362. https://doi.org/10.3390/molecules25225362