Application of Lanthanide Shift Reagent to the 1H-NMR Assignments of Acridone Alkaloids

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
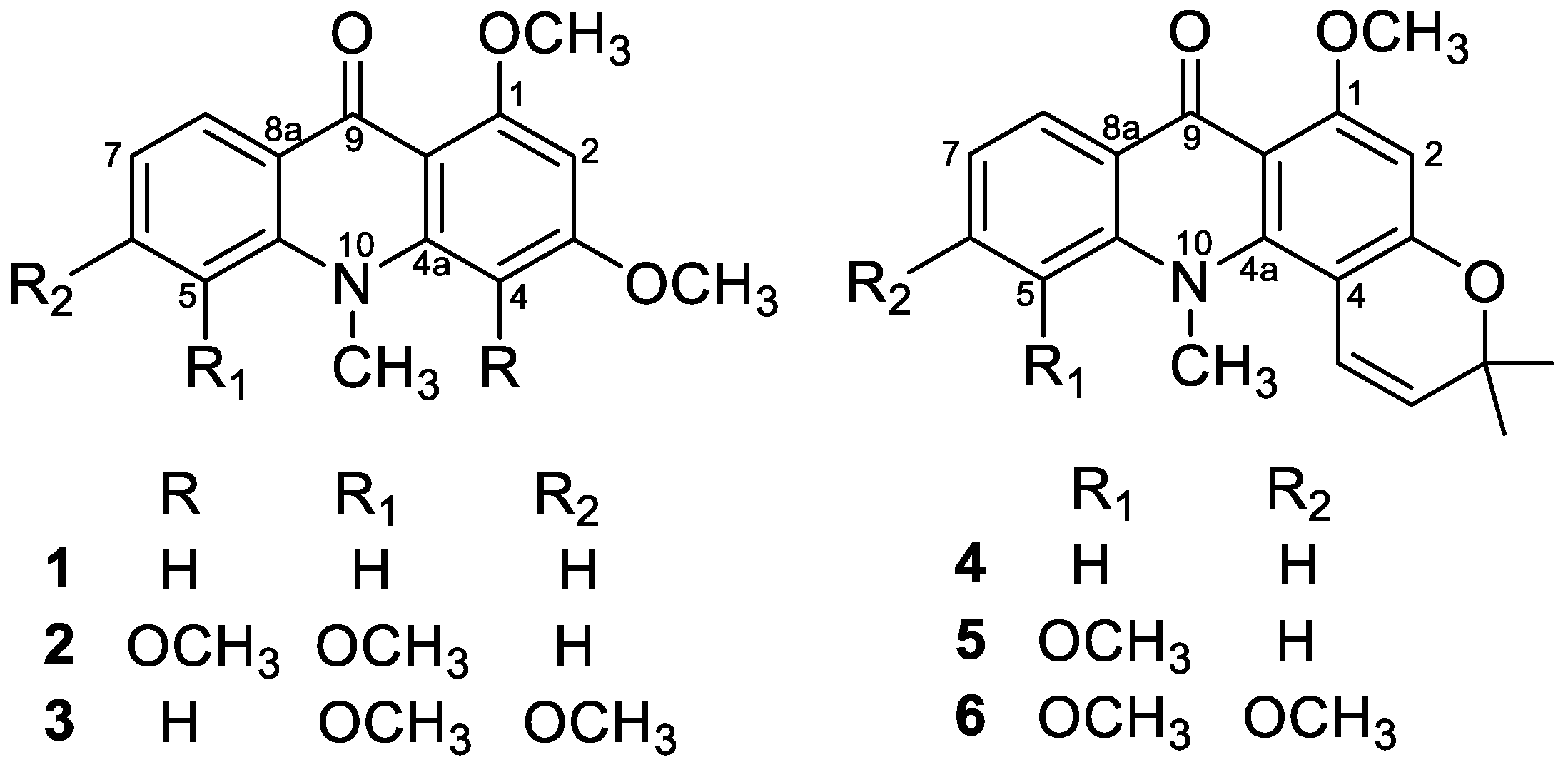
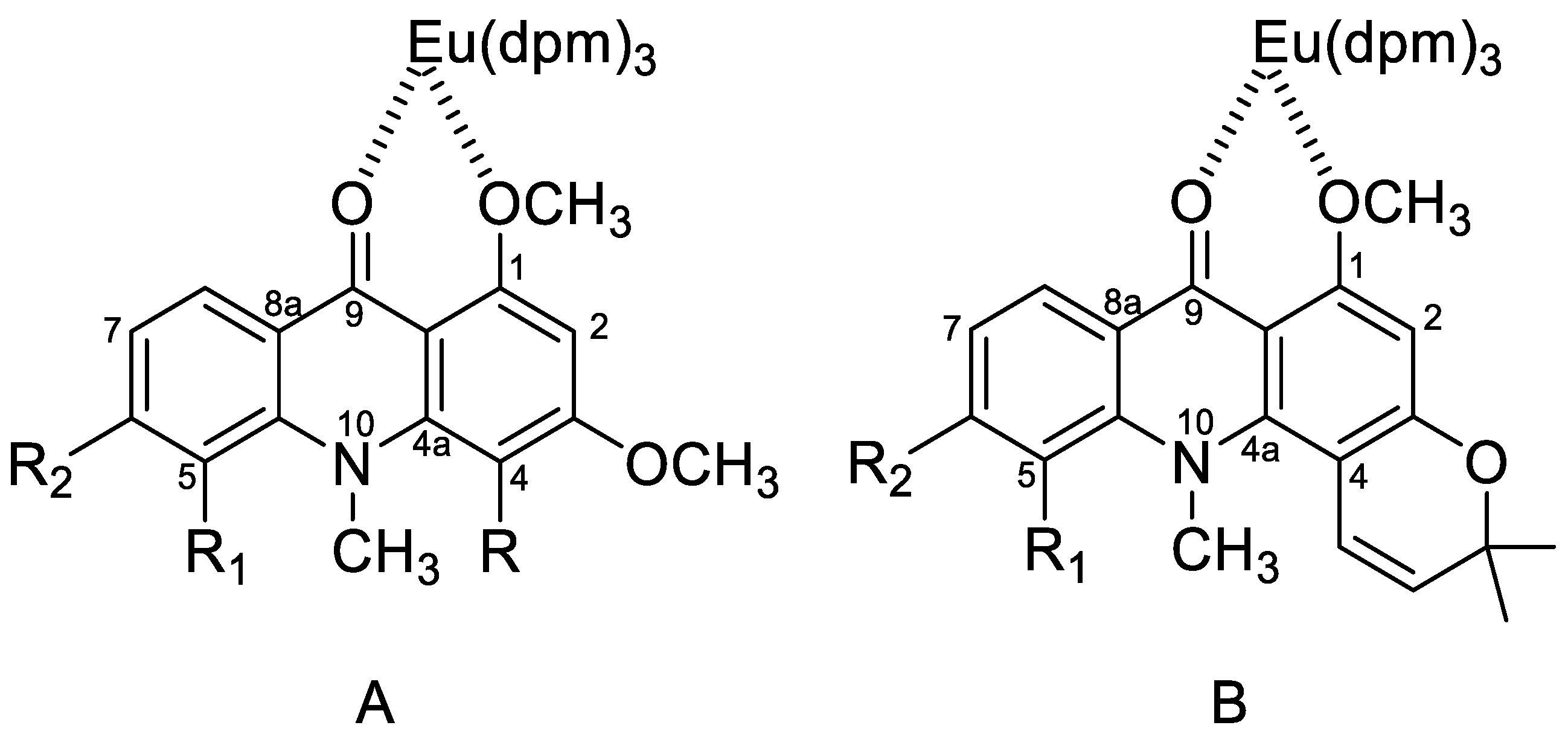
2.1. Eu(dpm)3-Induced 1H-Chemical Shifts Difference of Permethoxyacridone (1–3)
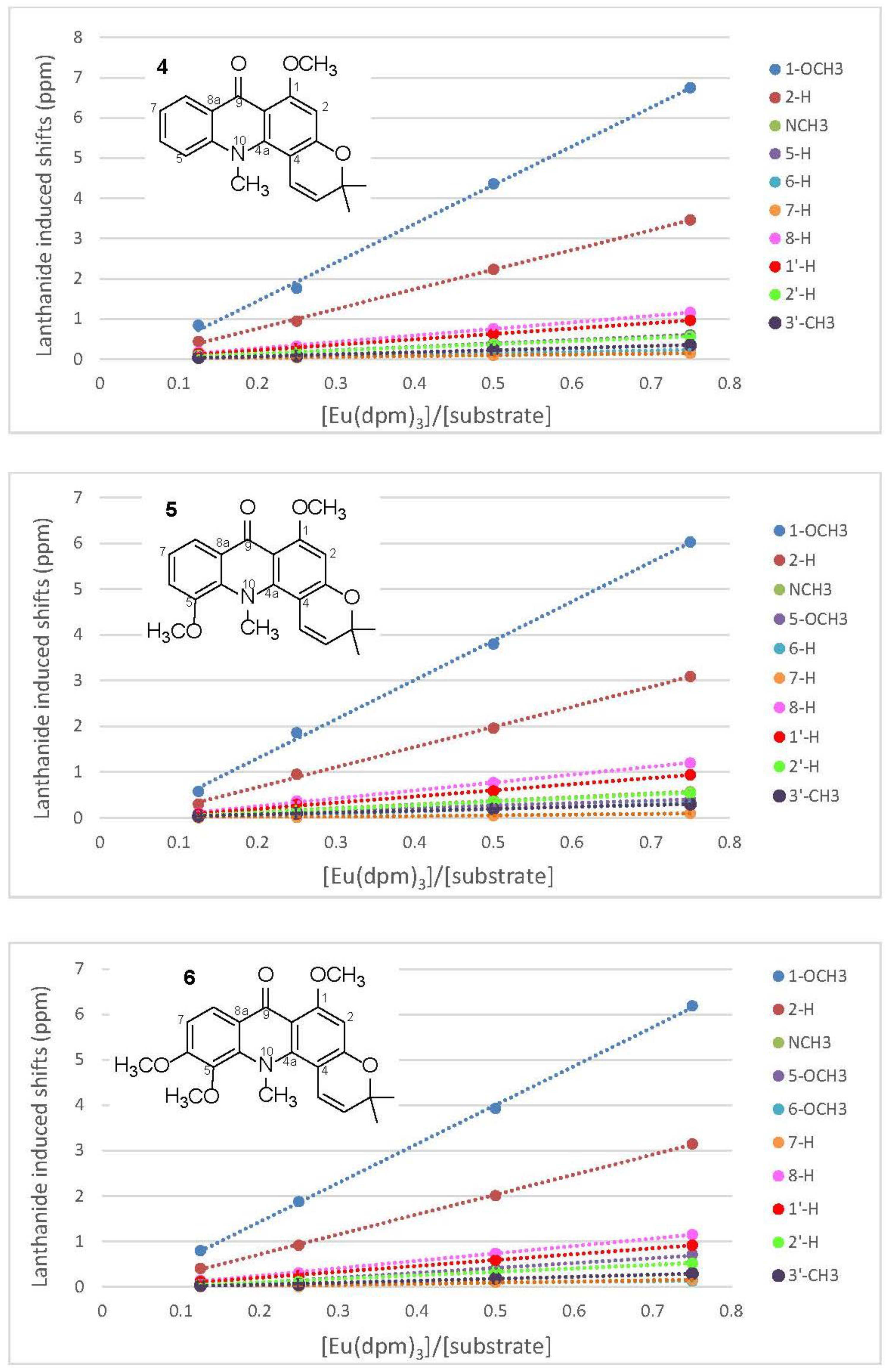
2.2. Eu(dpm)3-Induced 1H Chemical Shifts Difference of Pyranoacridone (4–6)
3. Experimental Section
3.1. General
3.2. Permethylation of Acridone Alkaloids
3.2.1. 1,3-Dimethoxy-N-methylacridone (1)
3.2.2. 1,5-O,O-Dimethylcitrusinine-I (2)
3.2.3. 1-O-Methylcitpressine-II (3)
3.2.4. Acronycine (4)
3.2.5. 5-Methoxyacronycine (5)
3.2.6. 1-O-Methylcitracridone-II (6)
3.3. Lanthanide Shift Reagent Preparation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mohammadi-Khanaposhtani, M.; Shabani, M.; Faizi, M.; Aghaei, I.; Jahani, R.; Sharafi, Z.; Zafarghandi, N.S.; Mahdavi, M.; Akbarzadeh, T.; Emami, S.; et al. Design, synthesis, pharmacological evaluation, and docking study of new acridone-based 1,2,4-oxadiazoles as potential anticonvulsant agents. Eur. J. Med. Chem. 2016, 112, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Murahari, M.; Prakash, K.V.; Peters, G.J.; Mayur, Y.C. Acridone-pyrimidine hybrids-design, synthesis, cytotoxicity studies in resistant and sensitive cancer cells and molecular docking studies. Eur. J. Med. Chem. 2017, 139, 961–981. [Google Scholar] [CrossRef]
- Xia, Y.F.; Chu, H.J.; Kuang, G.F.; Jiang, G.J.; Che, Y.C. Inhibition effects of acridone on the growth of breast cancer cells in vivo. Eur. Rev. Med. Pharmacol. 2018, 22, 2356–2363. [Google Scholar]
- Segun, P.A.; Ismail, F.M.D.; Ogbole, O.O.; Nahar, L.; Evans, A.R.; Ajaiyeoba, E.O.; Sarker, S.D. Acridone alkaloids alkaloids from the stem bark of Citrus aurantium display selective cytotoxicity against breast, liver, lung and prostate human carcinoma cells. J. Ethnopharmacol. 2018, 227, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.R.F.; Bittar, C.; Jardim, A.C.G.; Shimizu, J.F.; Batista, M.N.; Paganini, E.R.; de Assis, L.R.; Bartlett, C.; Harris, M.; da Silva Bolzani, V.; et al. Hepatitis C virus in vitro replication is efficiently inhibited by acridone Fac4. J. Gen. Virol. 2017, 98, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Mazzucco, M.B.; Talarico, L.B.; Vatansever, S.; Carro, A.C.; Fascio, M.L.; D’Accorso, N.B.; Garcia, C.C.; Damonte, E.B. Antiviral activity of an N-allyl acridone against dengue virus. J. Biomed. Sci. 2015, 22, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chansriniyom, C.; Ruangrungsi, N.; Lipipun, V.; Kumamoto, T.; Ishikawa, T. Isolation of acridone alkaloids and N-[(4-monoterpenyloxy)phenylethyl]-substituted sulfur-containing propanamide derivatives from Glycosmis parva and their anti-herpes simplex virus activity. Chem. Pharm. Bull. 2009, 57, 1246–1250. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi-Khanaposhtani, M.; Saeedi, M.; Zafarghandi, N.S.; Mahdavi, M.; Sabourian, R.; Razkenari, E.K.; Alinezhad, H.; Khanavi, M.; Foroumadi, A.; Shafiee, A.; et al. Potent acetylcholinesterase inhibitors: Design, synthesis, biological evaluation, and docking study of acridone linked to 1,2,3-triazole derivatives. Eur. J. Med. Chem. 2015, 92, 799–806. [Google Scholar] [CrossRef]
- Chakraborty, B.; Sengupta, C.; Pal, U.; Basu, S. Acridone in a biological nanocavity: Detailed spectroscopic and docking analyses of probing both the tryptophan residues of bovine serum albumin. New J. Chem. 2017, 41, 12520–12534. [Google Scholar] [CrossRef]
- Atangana, A.F.; Toze, F.A.A.; Langat, M.K.; Happi, E.N.; Mbaze, L.L.M.; Mulholland, D.A.; Waffo, A.F.K.; Sewald, N.; Wansi, J.D. Acridone alkaloids from Vepris verdoorniana (Excell & Mendonca) Mziray (Rutaceae). Phytochem. Lett. 2017, 19, 191–195. [Google Scholar]
- Bissim, S.M.; Kenmogne, S.B.; Tcho, A.T.; Lateef, M.; Ahmed, A.; Happi, E.N.; Wansi, J.D.; Ali, M.S.; Waffo, A.F.K. Bioactive acridone alkaloids and their derivatives from Citrus aurantium (Rutaceae). Phytochem. Lett. 2019, 29, 148–153. [Google Scholar] [CrossRef]
- Ono, T.; Ito, C.; Furukawa, H.; Wu, T.S.; Kuoh, C.S.; Hsu, K.S. Two new acridone alkaloids from Glycosmis species. J. Nat. Prod. 1995, 58, 1629–1631. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Bandara, B.M.R.; Gunatilaka, A.A.L.; Wijeratne, E.M.K.; Macleod, J.K. Acridone alkaloids and coumarins from Pleiospermium alatum. Phytochemistry 1990, 29, 297–301. [Google Scholar] [CrossRef]
- Bowen, I.H.; Patel, Y.N. Acridone alkaloids from Pleiospermium alatum (Rutaceae). Phytochemistry 1986, 25, 429–431. [Google Scholar] [CrossRef]
- Qin, D.K. N acridone alkaloid from the Chinese drug “tung-feng-jie” (Atalantia buxifolia). Yao Xue Xue Bao 1986, 21, 683–685. [Google Scholar]
- Furukawa, H.; Yogo, M.; Wu, T.S. Acridone alkaloids. 10. C-13-nuclear magnetic-resonance spectra of acridone alkaloids. Chem. Pharm. Bull. 1983, 31, 3084–3090. [Google Scholar] [CrossRef] [Green Version]
- Geraldes, C.F.G.C. Lanthanide shift-reagents. Method. Enzymol. 1993, 227, 43–78. [Google Scholar]
- Adams, C.M.; Ghosh, I.; Kishi, Y. Validation of lanthanide chiral shift reagents for determination of absolute configuration: Total synthesis of glisoprenin A. Org. Lett. 2004, 6, 4723–4726. [Google Scholar] [CrossRef]
- Baldovini, N.; Tomi, F.; Casanova, J. Enantiomeric differentiation of bornyl acetate by C13 NMR using a chiral lanthanide shift reagent. Phytochem. Anal. 2003, 14, 241–244. [Google Scholar] [CrossRef]
- Yamamoto, N.; Furukawa, H.; Ito, Y.; Yoshida, S.; Maeno, K.; Nishiyama, Y. Anti-herpesvirus activity of citrusinine-I, a new acridone alkaloid, and related-compounds. Antivir. Res. 1989, 12, 21–36. [Google Scholar] [CrossRef]
- Wu, T.S.; Kuoh, C.S.; Furukawa, H. Acridone Alkaloids. 6. The constituents of Citrus depressa—Isolation and structure elucidation of new acridone alkaloids from Citrus genus. Chem. Pharm. Bull. 1983, 31, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Ito, C.; Kondo, Y.; Wu, T.S.; Furukawa, H. Structures of four new acridones and three new quinolone alkaloids. Chem. Pharm. Bull. 2000, 48, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.S.; Chen, C.M. Acridone alkaloids from the root bark of Severinia buxifolia in Hainan. Chem. Pharm. Bull. 2000, 48, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Sample Availability: Samples of all the isolated compounds are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, S.-H.; Hung, H.-Y.; Kuo, P.-C.; Kuo, D.-H.; Chen, F.-A.; Wu, T.-S. Application of Lanthanide Shift Reagent to the 1H-NMR Assignments of Acridone Alkaloids. Molecules 2020, 25, 5383. https://doi.org/10.3390/molecules25225383
Lam S-H, Hung H-Y, Kuo P-C, Kuo D-H, Chen F-A, Wu T-S. Application of Lanthanide Shift Reagent to the 1H-NMR Assignments of Acridone Alkaloids. Molecules. 2020; 25(22):5383. https://doi.org/10.3390/molecules25225383
Chicago/Turabian StyleLam, Sio-Hong, Hsin-Yi Hung, Ping-Chung Kuo, Daih-Huang Kuo, Fu-An Chen, and Tian-Shung Wu. 2020. "Application of Lanthanide Shift Reagent to the 1H-NMR Assignments of Acridone Alkaloids" Molecules 25, no. 22: 5383. https://doi.org/10.3390/molecules25225383