
Mitocanic Di- and Triterpenoid Rhodamine B Conjugates




Abstract
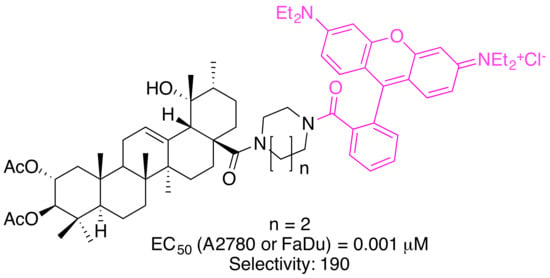
:
1. Introduction
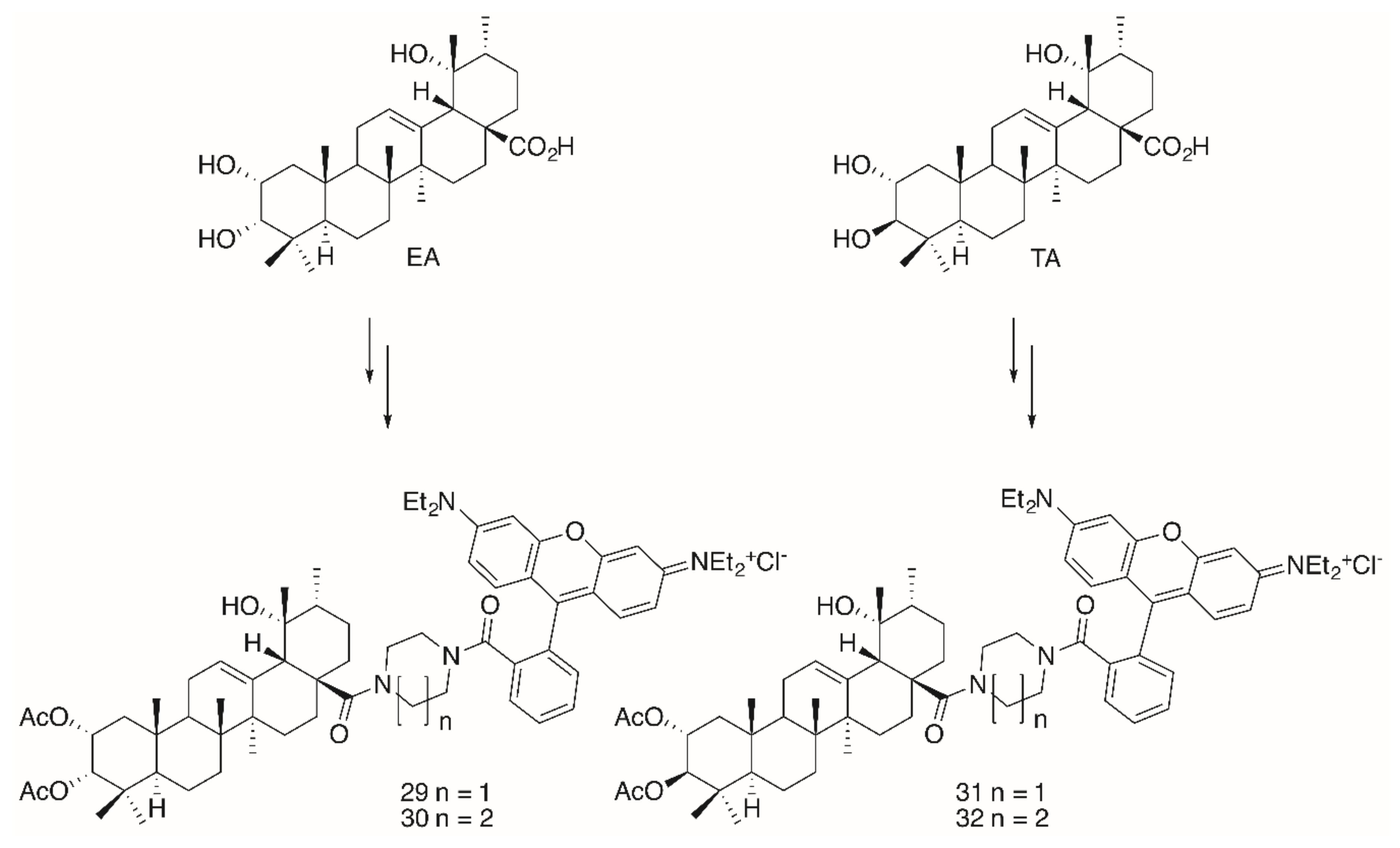
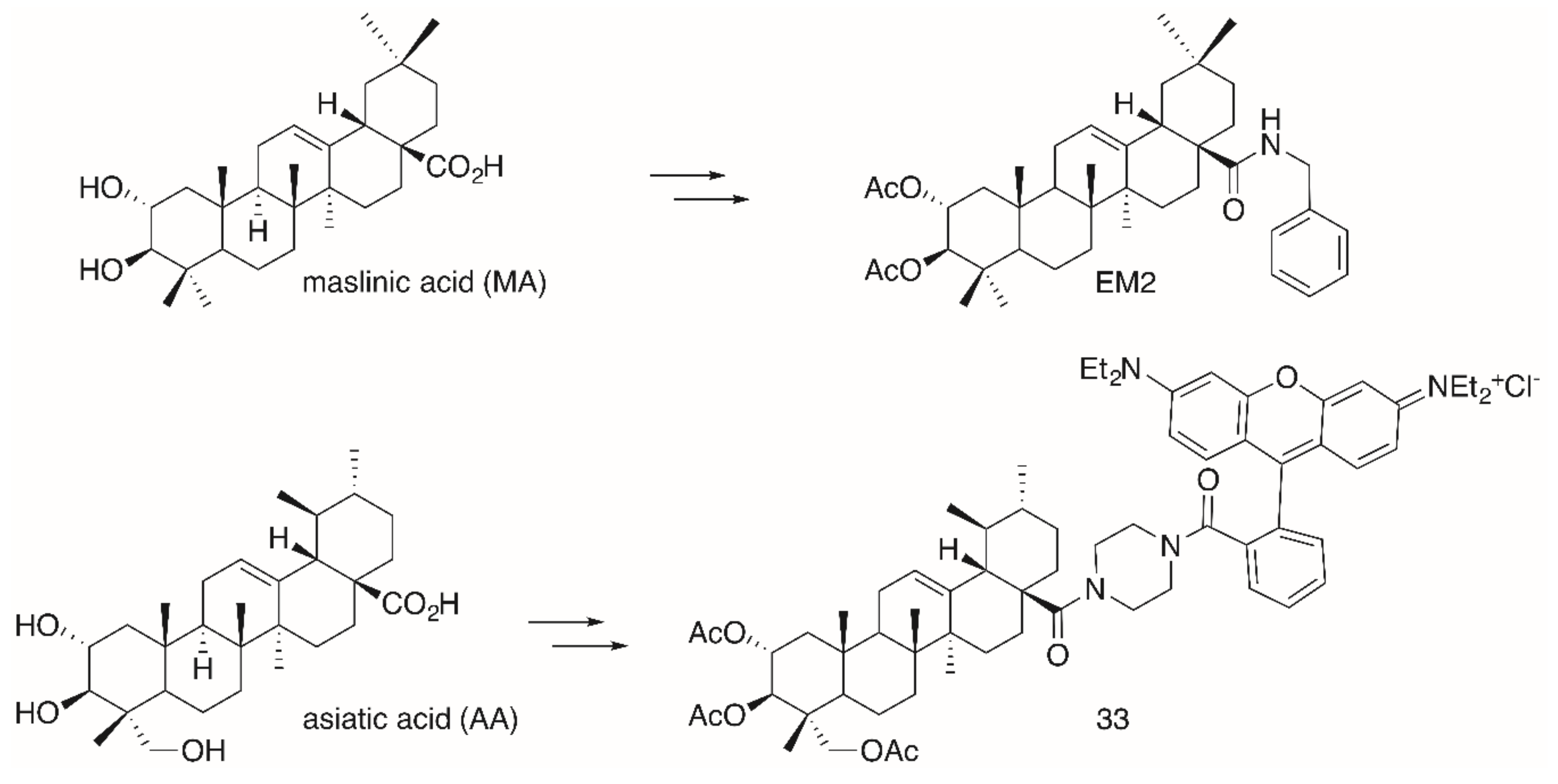
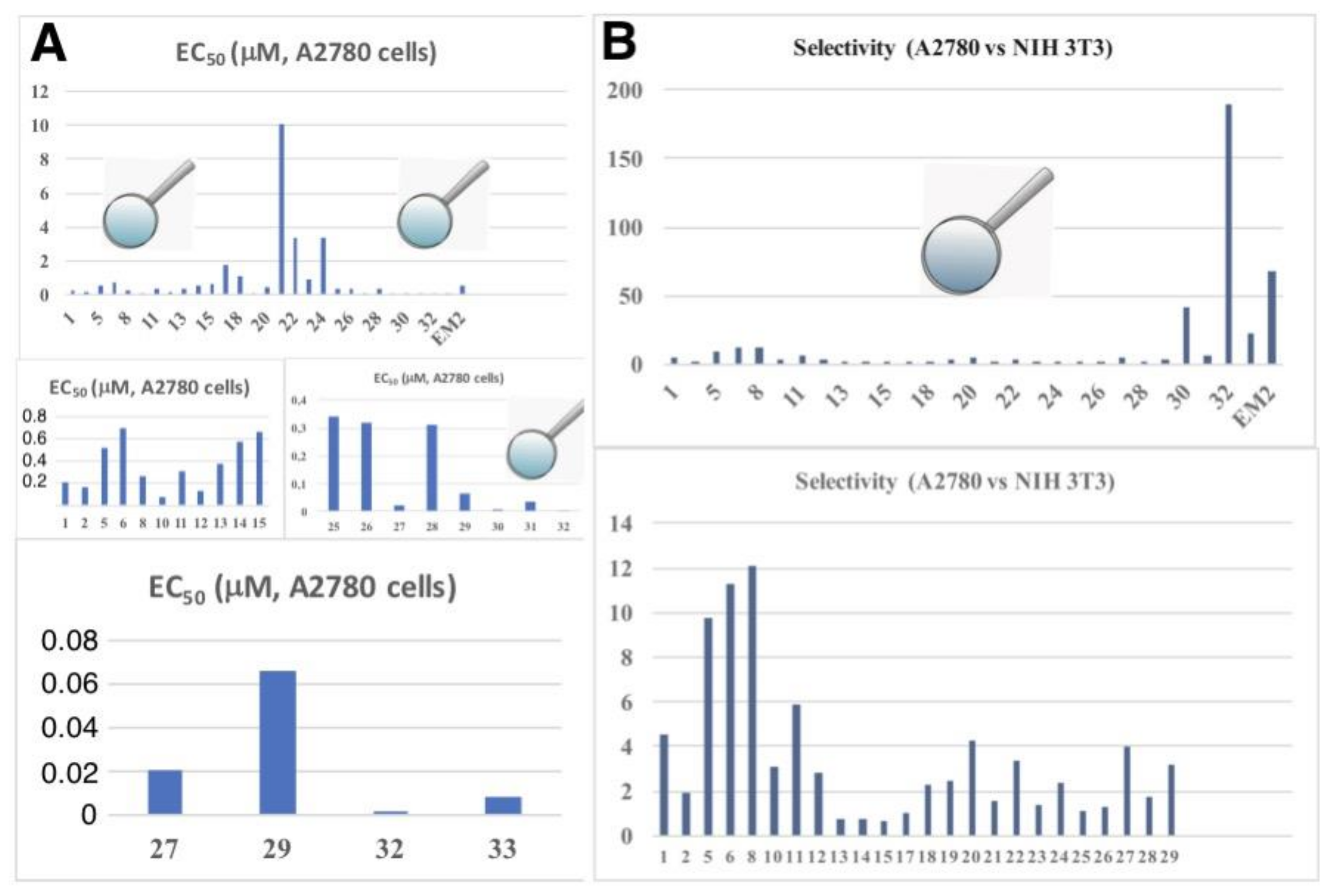
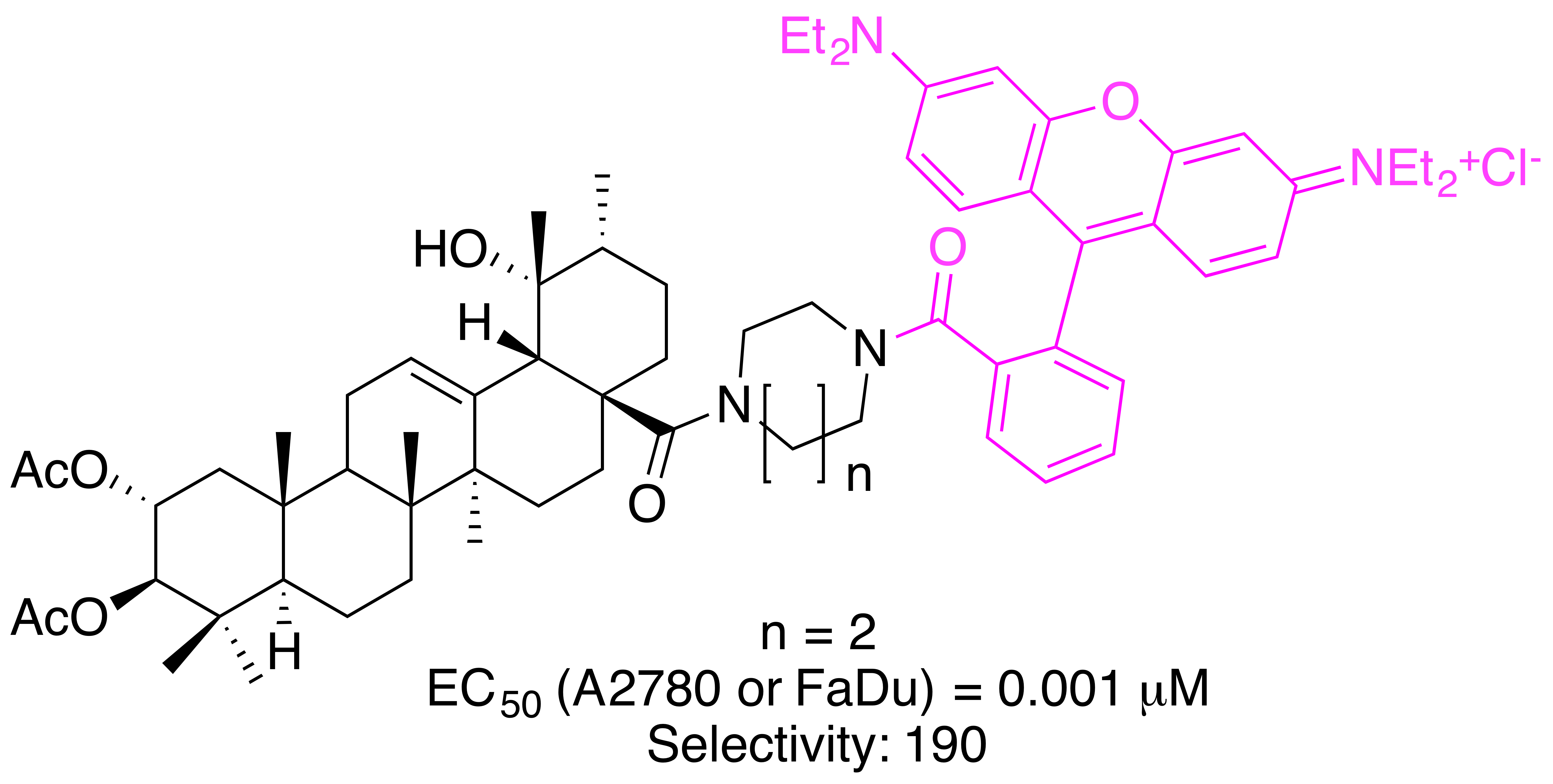
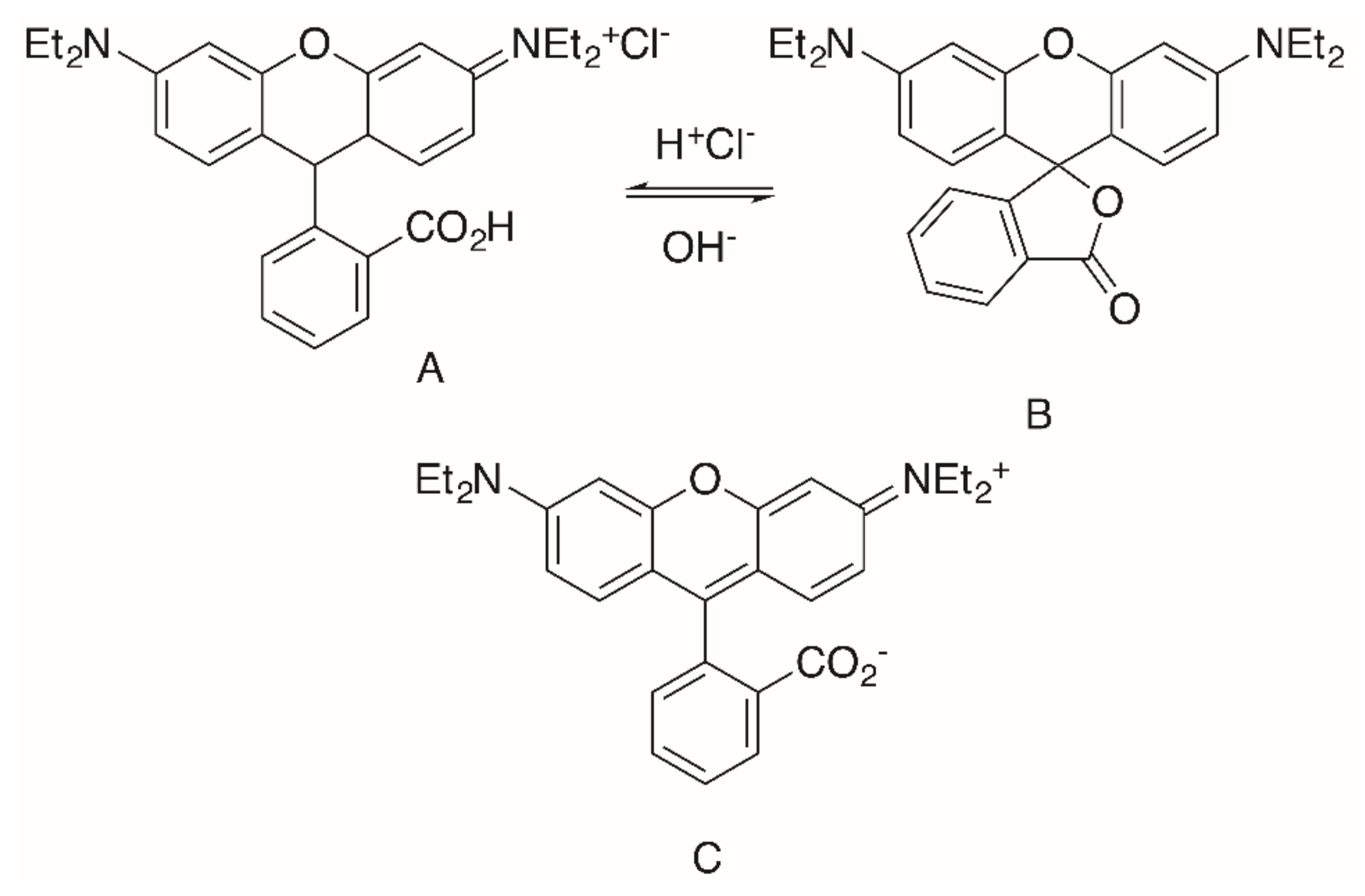
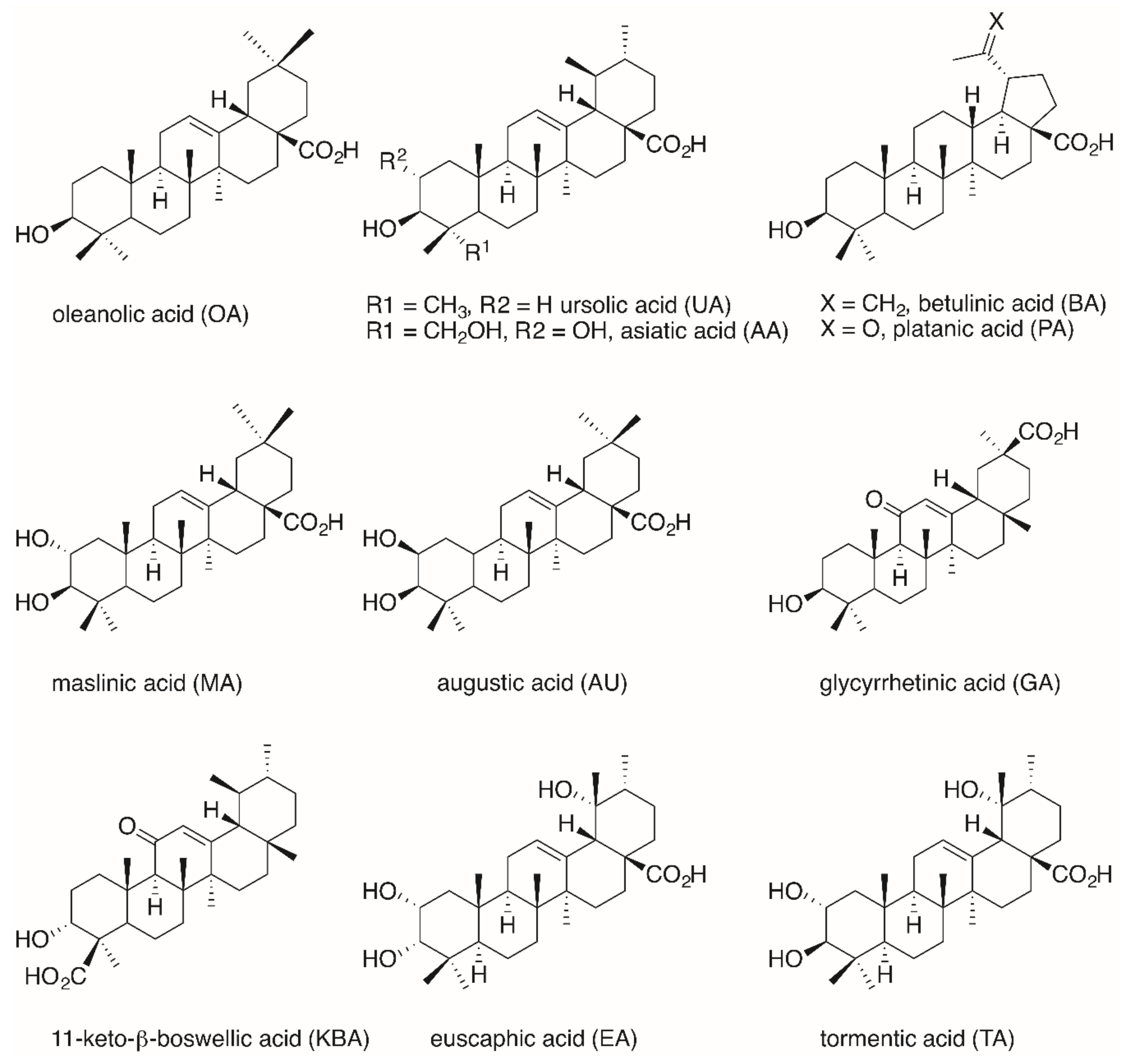
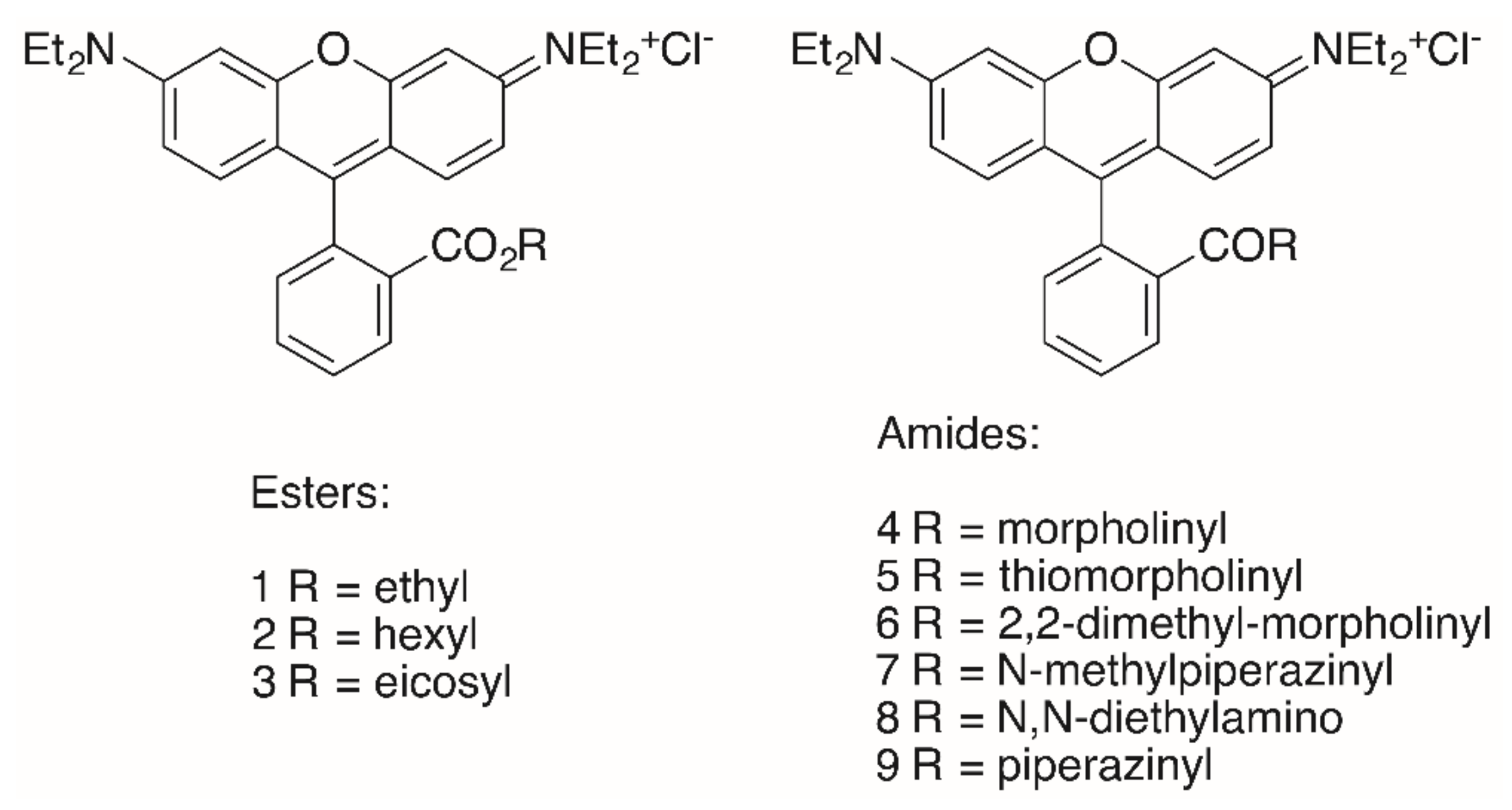
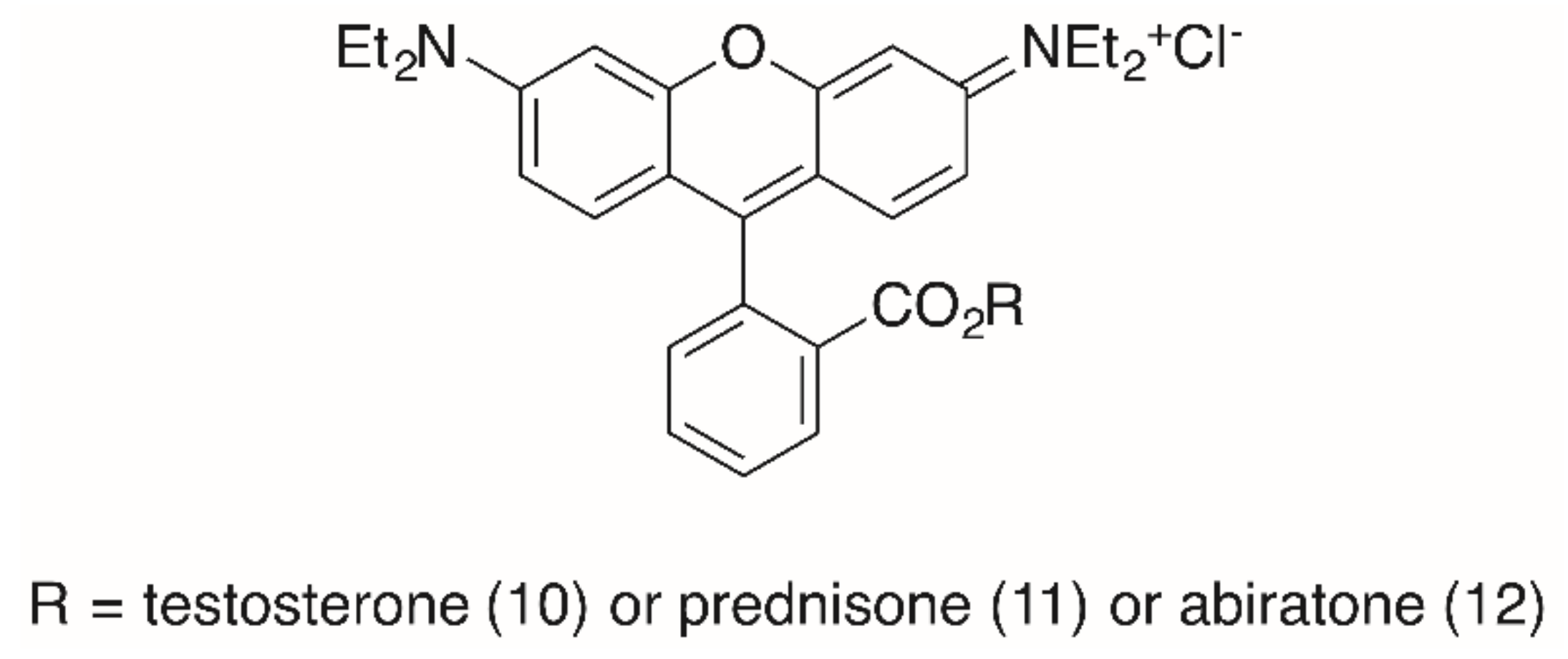
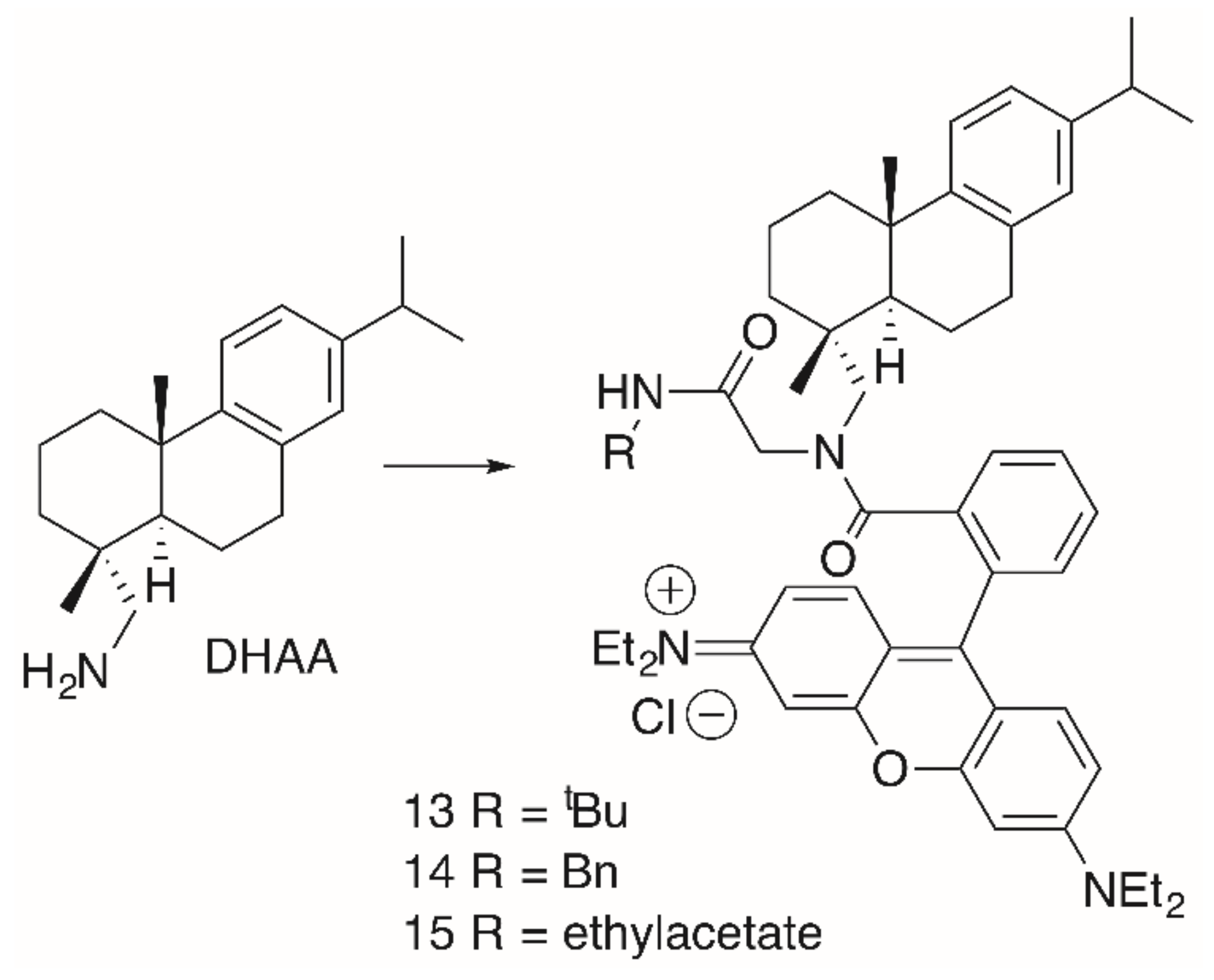
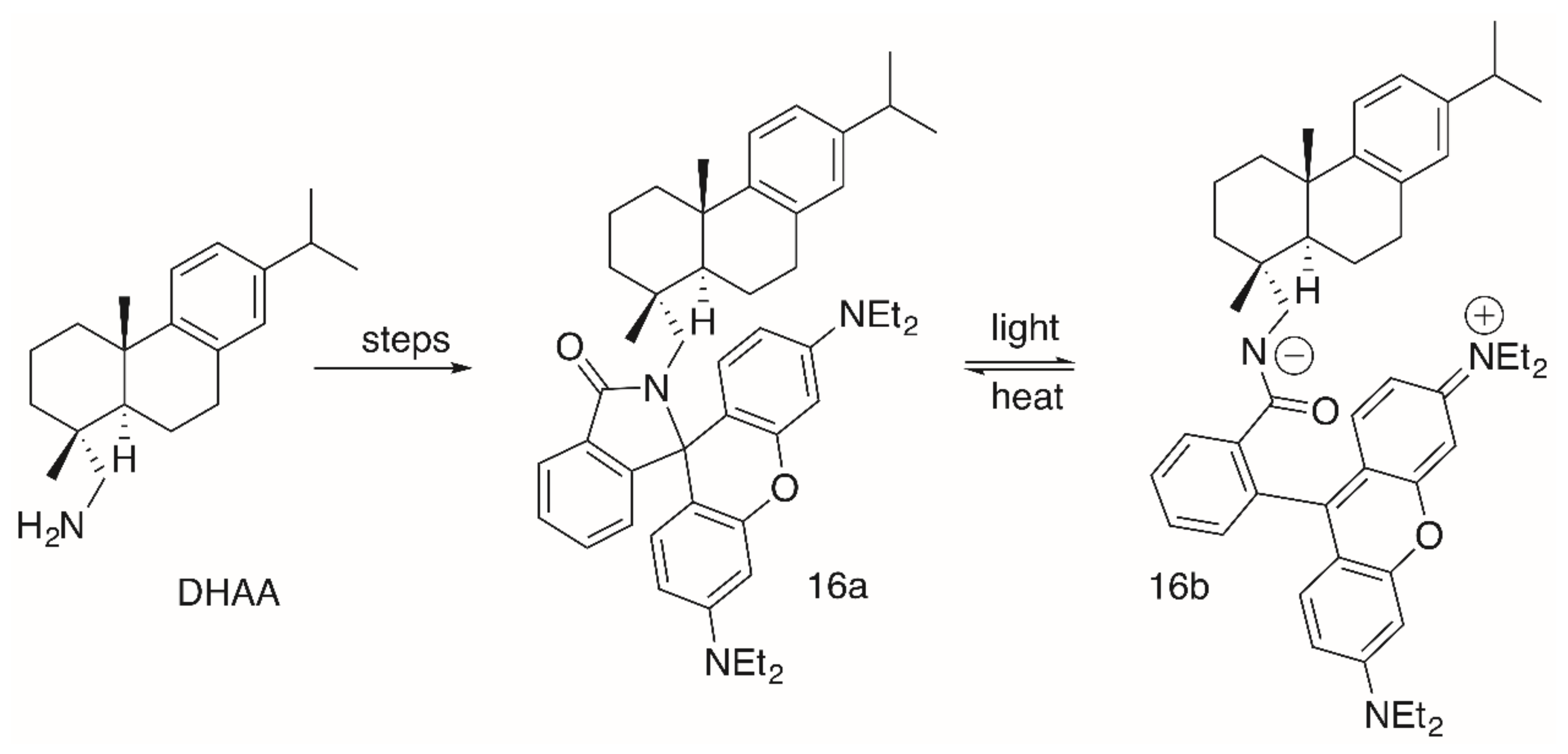
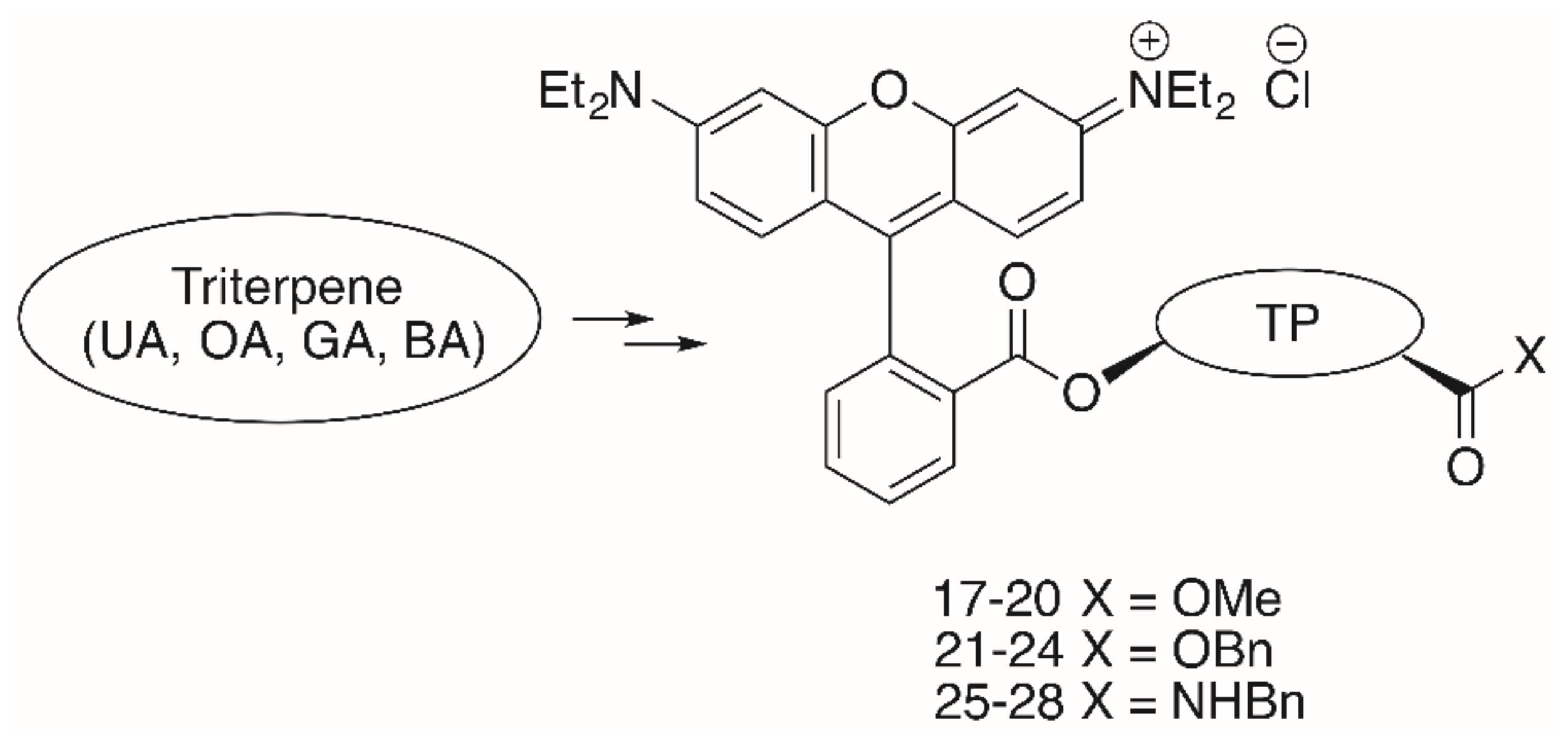
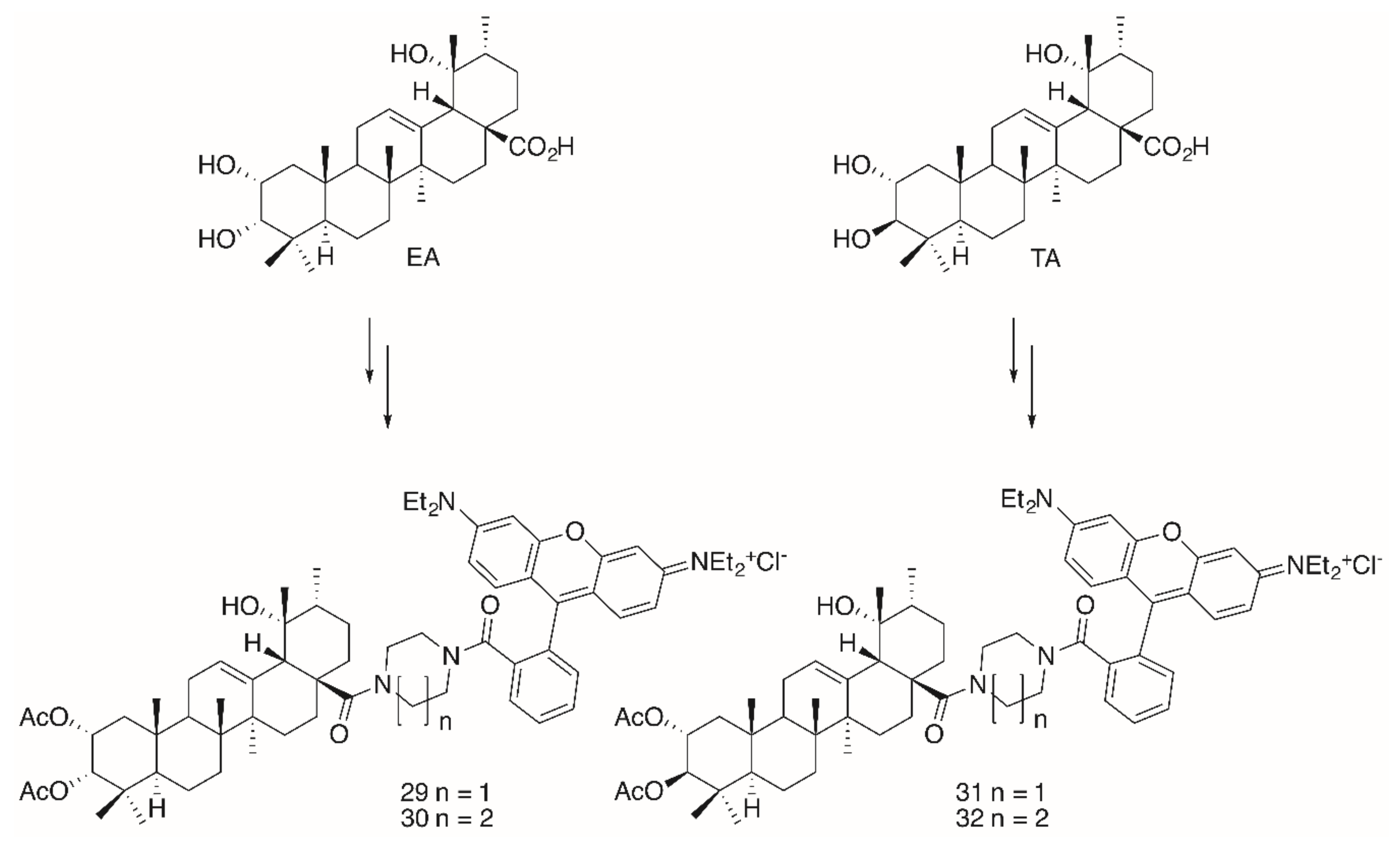
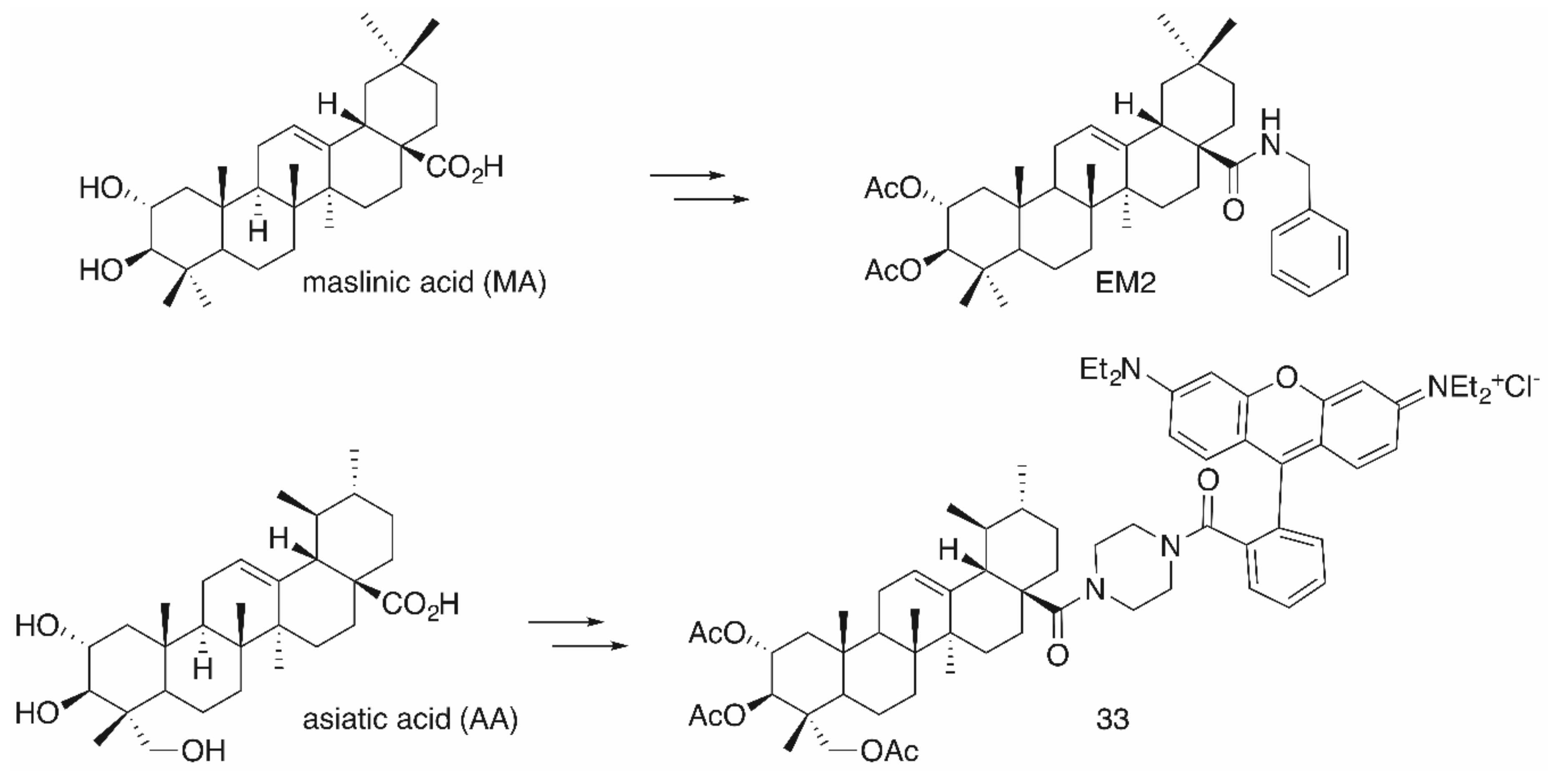
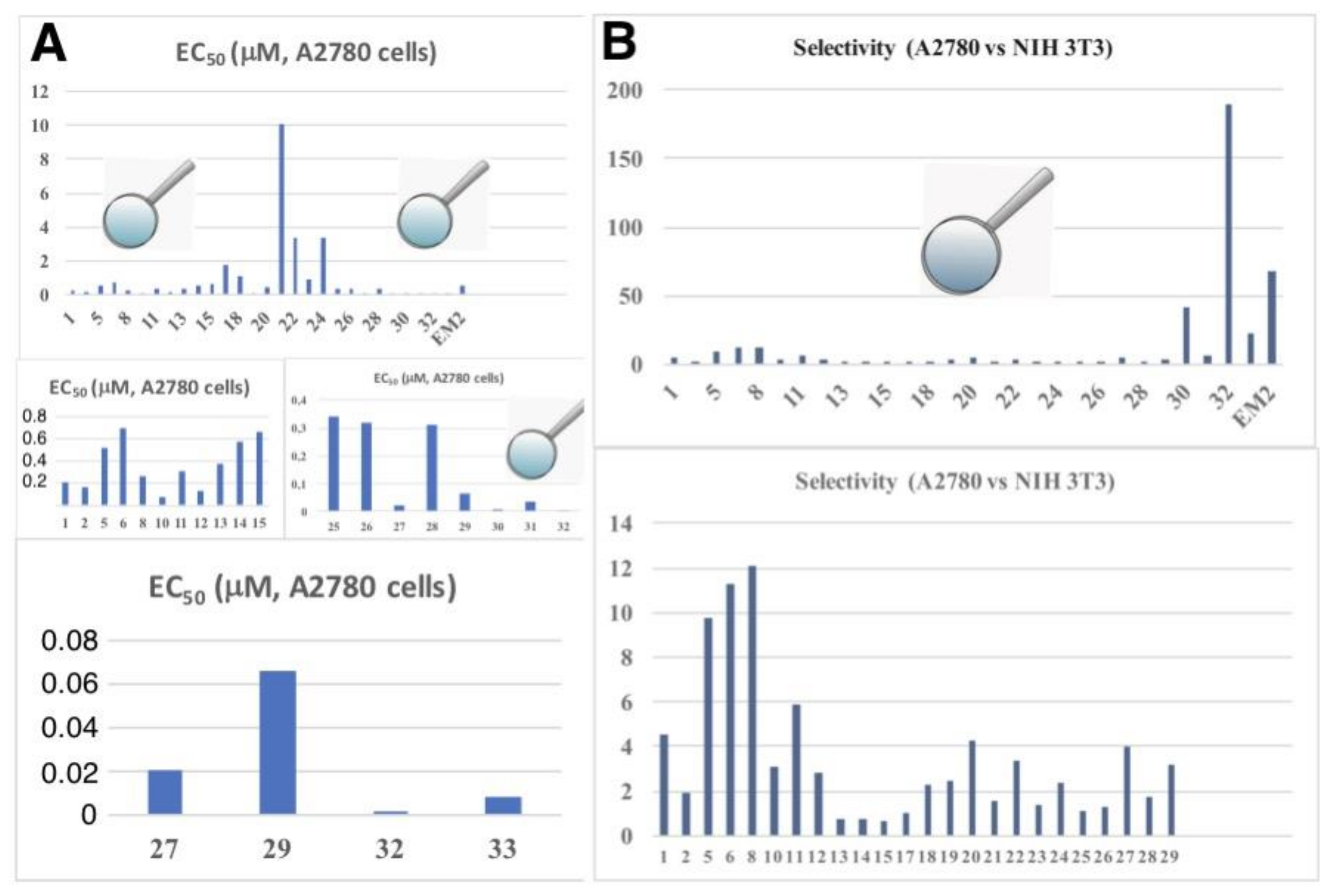
2. Results
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Available online: https://www.who.int/health-topics/cancer-tab=tab_1 (accessed on 20 October 2020).
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Siegel, R.L.; Mph, K.D.M.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA: A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, M.J.; Wachter, K.; Agarwal, N.; Bertagnolli, M.M.; Chang, S.M.; Dale, W.; Diefenbach, C.S.M.; Rodriguez-Galindo, C.; George, D.J.; Gilligan, T.D.; et al. Clinical Cancer Advances 2020: Annual Report on Progress Against Cancer From the American Society of Clinical Oncology. J. Clin. Oncol. 2020, 38, 1081. [Google Scholar] [CrossRef] [PubMed]
- Oostveen, M.; Pritchard-Jones, K. Pharmacotherapeutic Management of Wilms Tumor: An Update. Pediatr. Drugs 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Di Paolo, A.; Arrigoni, E.; Luci, G.; Cucchiara, F.; Danesi, R.; Galimberti, S. Precision Medicine in Lymphoma by Innovative Instrumental Platforms. Front. Oncol. 2019, 9, 1417. [Google Scholar] [CrossRef]
- Available online: https://www.paho.org/hq/index.php?option=com_docman&view=download&category_slug=4-cancer-country-profiles-2020&alias=51561-global-cancer-profile-2020&Itemid=270&lang=fr (accessed on 20 October 2020).
- Green, D.R. Cancer and Apoptosis: Who Is Built to Last? Cancer Cell 2017, 31, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Pretta, A.; Trevisi, E.; Bregni, G.; Deleporte, A.; Hendlisz, A.; Sclafani, F. Treatment compliance in early-stage anal cancer. Ann. Oncol. 2020, 31, 1282–1284. [Google Scholar] [CrossRef]
- Muzumder, S.; Srikantia, N.; Vashishta, G.D.; Udayashankar, A.H.; Raj, J.M.; Sebastian, M.G.J.; Kainthaje, P.B. Compliance, toxicity and efficacy in weekly versus 3-weekly cisplatin concurrent chemoradiation in locally advanced head and neck cancer. J. Radiother. Pr. 2018, 18, 21–25. [Google Scholar] [CrossRef]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X.-Y. Cancer cell reprogramming: A promising therapy converting malignancy to benignity. Cancer Commun. 2019, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef]
- Wang, C.X.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Fiers, W.; Beyaert, R.; Declercq, W.; Vandenabeele, P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 1999, 18, 7719–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pedro, J.M.B.-S.; Kroemer, G. Organelle-specific initiation of cell death. Nat. Cell Biol. 2014, 16, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Bing, Y.; Dong, L.; Neuzil, J. Mitochondria: An intriguing target for killing tumour-initiating cells. Mitochondrion 2016, 26, 86–93. [Google Scholar] [CrossRef]
- Pei, J.; Panina, S.B.; Kirienko, N.V. An Automated Differential Nuclear Staining Assay for Accurate Determination of Mitocan Cytotoxicity. J. Vis. Exp. 2020, 159, e61295. [Google Scholar] [CrossRef]
- Panina, S.B.; Pei, J.; Baran, N.; Konopleva, M.; Kirienko, N.V. Utilizing Synergistic Potential of Mitochondria-Targeting Drugs for Leukemia Therapy. Front. Oncol. 2020, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Neuzil, J. Mitochondria as Targets for Cancer Therapy. In Mitochondria and Cancer; Costello, L., Singh, K., Eds.; Springer: New York, NY, USA, 2009; pp. 211–249. [Google Scholar]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Moreno-Sánchez, R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol. Asp. Med. 2010, 31, 29–59. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation – Why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxidants Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Singh, Y.; Viswanadham, K.K.D.R.; Pawar, V.K.; Meher, J.; Jajoriya, A.K.; Omer, A.; Jaiswal, S.; Dewangan, J.; Bora, H.K.; Singh, P.; et al. Induction of Mitochondrial Cell Death and Reversal of Anticancer Drug Resistance via Nanocarriers Composed of a Triphenylphosphonium Derivative of Tocopheryl Polyethylene Glycol Succinate. Mol. Pharm. 2019, 16, 3744–3759. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Pandey, S. Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells. Cancers 2019, 11, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivas-Aguirre, M.; Pottosin, I.; Dobrovinskaya, O. Mitochondria as emerging targets for therapies against T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2019, 105, 935–946. [Google Scholar] [CrossRef]
- Sassi, N.; Biasutto, L.; Mattarei, A.; Carraro, M.; Giorgio, V.; Citta, A.; Bernardi, P.; Garbisa, S.; Szabò, I.; Paradisi, C.; et al. Cytotoxicity of a mitochondriotropic quercetin derivative: Mechanisms. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2012, 1817, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Sassi, N.; Mattarei, A.; Azzolini, M.; Bernardi, P.; Szabò, I.; Paradisi, C.; Zoratti, M.; Biasutto, L. Mitochondria-targeted resveratrol derivatives act as cytotoxic pro-oxidants. Curr. Pharm. Des. 2014, 20, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, F.; Trachootham, D.; Huang, P. Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion 2010, 10, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Villanueva, D.; Weissig, V. Mitochondria-Targeted Agents: Mitochondriotropics, Mitochondriotoxics, and Mitocans. Muscarinic Receptors 2016, 240, 423–438. [Google Scholar] [CrossRef]
- Rohlena, J.; Dong, L.-F.; Neuzil, J. Targeting the mitochondrial electron transport chain complexes for the induction of apoptosis and cancer treatment. Curr. Pharm. Biotechnol. 2013, 14, 377–389. [Google Scholar] [CrossRef]
- Rohlena, J.; Dong, L.; Ralph, S.J.; Neuzil, J. Anticancer Drugs Targeting the Mitochondrial Electron Transport Chain. Antioxidants Redox Signal. 2011, 15, 2951–2974. [Google Scholar] [CrossRef]
- Kornblihtt, L.I.; Carreras, M.C.A.; Blanco, G. Targeting Mitophagy in Combined Therapies of Haematological Malignancies. Autophagy Curr. Trends Cell. Physiol. Pathol. 2016, 19, 411–431. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Dyason, J.C.; Freeman, R.; Dong, L.; Prochazka, L.; Wang, X.-F.; Scheffler, I.; Ralph, S.J. Mitocans as anti-cancer agents targeting mitochondria: Lessons from studies with vitamin E analogues, inhibitors of complex II. J. Bioenerg. Biomembr. 2007, 39, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Wang, X.-F.; Dong, L.-F.; Low, P.; Ralph, S.J. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006, 580, 5125–5129. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Andera, L.; Reynolds, B.A.; Ralph, S.J.; Neuzil, J. Future use of mitocans against tumour-initiating cells? Mol. Nutr. Food Res. 2009, 53, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.; Polanczyk, M.J.; Borodovsky, A.; Ramanathapuram, L.V.; Akporiaye, E.T.; Ralph, S.J. Use of anti-cancer drugs, mitocans, to enhance the immune responses against tumors. Curr. Pharm. Biotechnol. 2013, 14, 357–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, L.; Spengler, G.; Molnar, J. Identification of Important Compounds Isolated from Natural Sources that Have Activity Against Multidrug-resistant Cancer Cell Lines: Effects on Proliferation, Apoptotic Mechanism and the Efflux Pump Responsible for Multi-resistance Phenotype. Anticancer. Res. 2016, 36, 5665–5672. [Google Scholar] [CrossRef] [Green Version]
- Abdelmageed, N.; Morad, S.A.; Elghoneimy, A.A.; Syrovets, T.; Simmet, T.; El-Zorba, H.; El-Banna, H.A.; Cabot, M.; Abdel-Aziz, M.I. Oleanolic acid methyl ester, a novel cytotoxic mitocan, induces cell cycle arrest and ROS-Mediated cell death in castration-resistant prostate cancer PC-3 cells. Biomed. Pharmacother. 2017, 96, 417–425. [Google Scholar] [CrossRef]
- Bhola, P.D.; Letai, A. Mitochondria—Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Jacotot, E.E.; Ferri, K.K.; El Hamel, C.C.; Brenner, C.C.; Druillennec, S.S.; Hoebeke, J.; Rustin, P.P.; Métivier, D.D.; Lenoir, C.C.; Geuskens, M.; et al. Control of Mitochondrial Membrane Permeabilization by Adenine Nucleotide Translocator Interacting with HIV-1 Viral Protein R and Bcl-2. J. Exp. Med. 2001, 193, 509–520. [Google Scholar] [CrossRef]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef]
- Zamzami, N.; Maisse, C.; Métivier, D.; Kroemer, G. Chapter 8 Measurement of membrane permeability and permeability transition of mitochondria. Echinoderms Part B 2001, 65, 147–158. [Google Scholar] [CrossRef]
- Yang, H.; Dou, Q.P. Targeting apoptosis pathway with natural terpenoids: Implications for treatment of breast and prostate cancer. Curr. Drug Targets 2010, 11, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Kroemer, G. Mitochondria as Therapeutic Targets for the Treatment of Malignant Disease. Antioxidants Redox Signal. 2011, 15, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Serafim, T.L.; Carvalho, F.S.; Pereira, G.; Greene, A.; Perkins, E.; Holy, J.; Krasutsky, D.A.; Kolomitsyna, O.N.; Krasutsky, P.A.; Oliveira, P.J. Lupane triterpenoids as mitocans against breast cancer cells. Eur. J. Clin. Invest. 2013, 43, 48–49. [Google Scholar]
- Serafim, T.L.; Carvalho, F.S.; Bernardo, T.C.; Pereira, G.C.; Perkins, E.; Holy, J.; Krasutsky, D.A.; Kolomitsyna, O.N.; Krasutsky, P.A.; Oliveira, P.J. New derivatives of lupine triterpenoids disturb breast cancer mitochondria and induce cell death. Bioorg. Med. Chem. 2014, 22, 6270–6287. [Google Scholar] [CrossRef] [PubMed]
- Grymel, M.; Zawojak, M.; Adamek, J. Triphenylphosphonium Analogues of Betulin and Betulinic Acid with Biological Activity: A Comprehensive Review. J. Nat. Prod. 2019, 82, 1719–1730. [Google Scholar] [CrossRef]
- Kolevzon, N.; Kuflik, U.; Shmuel, M.; Benhamron, S.; Ringel, I.; Yavin, E. Multiple Triphenylphosphonium Cations as a Platform for the Delivery of a Pro-Apoptotic Peptide. Pharm. Res. 2011, 28, 2780–2789. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.; Odinokov, V.N.; Bel’Skii, Y.P.; Bel’Skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Akhmetova, V.R.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’Skii, Y.P.; Bel’Skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Huang, H.; Wu, H.; Huang, Y.; Zhang, S.; Lam, Y.-W.; Ao, N. Antitumor activity and antitumor mechanism of triphenylphosphonium chitosan against liver carcinoma. J. Mater. Res. 2018, 33, 2586–2597. [Google Scholar] [CrossRef]
- Strobykina, I.Y.; Andreeva, O.V.; Belenok, M.G.; Semenova, M.N.; Semenov, V.V.; Chuprov-Netochin, R.N.; Sapunova, A.S.; Voloshina, A.D.; Dobrynin, A.B.; Semenov, V.E.; et al. Triphenylphosphonium conjugates of 1,2,3-triazolyl nucleoside analogues. Synthesis and cytotoxicity evaluation. Med. Chem. Res. 2020, 29, 1–15. [Google Scholar] [CrossRef]
- Jara, J.A.; Castro-Castillo, V.; Saavedra-Olavarría, J.; Peredo, L.; Pavanni, M.; Jaña, F.; Letelier, M.E.; Parra, E.; Becker, M.I.; Morello, A.; et al. Antiproliferative and Uncoupling Effects of Delocalized, Lipophilic, Cationic Gallic Acid Derivatives on Cancer Cell Lines. Validation in Vivo in Singenic Mice. J. Med. Chem. 2014, 57, 2440–2454. [Google Scholar] [CrossRef] [PubMed]
- Kafkova, A.; Trnka, J. Mitochondria-targeted compounds in the treatment of cancer. Neoplasma 2020, 67, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef]
- Sarek, J.; Biedermann, D.; Eignerova, B.; Hajduch, M. Synthesis and Evaluation of Biological Activity of the Quaternary Ammonium Salts of Lupane-, Oleanane-, and Ursane-Type Acids. Synthesis 2010, 22, 3839–3848. [Google Scholar] [CrossRef]
- Kataev, V.E.; Strobykina, I.Y.; Zakharova, L.Y. Quaternary ammonium derivatives of natural terpenoids. Synthesis and properties. Russ. Chem. Bull. 2014, 63, 1884–1900. [Google Scholar] [CrossRef]
- Brandes, B.; Koch, L.; Hoenke, S.; Deigner, H.-P.; Csuk, R. The presence of a cationic center is not alone decisive for the cytotoxicity of triterpene carboxylic acid amides. Steroids 2020, 163, 108713. [Google Scholar] [CrossRef]
- Perreault, M.; Maltais, R.; Dutour, R.; Poirier, D. Explorative study on the anticancer activity, selectivity and metabolic stability of related analogs of aminosteroid RM-133. Steroids 2016, 115, 105–113. [Google Scholar] [CrossRef]
- Brandes, B.; Hoenke, S.; Fischer, L.; Csuk, R. Design, synthesis and cytotoxicity of BODIPY FL labelled triterpenoids. Eur. J. Med. Chem. 2020, 185, 111858. [Google Scholar] [CrossRef]
- Barut, B.; Çoban, Ö.; Özgür, Y.C.; Baş, H.; Sari, S.; Biyiklioglu, Z.; Demirbaş, Ü.; Özel, A. Synthesis, DNA interaction, in vitro/in silico topoisomerase II inhibition and photodynamic therapy activities of two cationic BODIPY derivatives. Dye. Pigment. 2020, 174, 108072. [Google Scholar] [CrossRef]
- Barut, B.; Özgür, Y.C.; Sari, S.; Çoban, Ö.; Keleş, T.; Biyiklioglu, Z.; Abudayyak, M.; Demirbaş, Ü.; Özel, A. Novel water soluble BODIPY compounds: Synthesis, photochemical, DNA interaction, topoisomerases inhibition and photodynamic activity properties. Eur. J. Med. Chem. 2019, 183, 11685. [Google Scholar] [CrossRef] [PubMed]
- Taki, S.; Ardestani, M.S. Novel nanosized AS1411-chitosan-BODIPY conjugate for molecular fluorescent imaging. Int. J. Nanomed. 2019, 14, 3543–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, X.; Cao, Z.; Wu, Y.; Chen, J.-A.; Gao, J.; Wang, Z.; Guo, W.; Gu, X. Mitochondria-targeting BODIPY-loaded micelles as novel class of photosensitizer for photodynamic therapy. Eur. J. Med. Chem. 2018, 157, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Kraft, O.; Kozubek, M.; Hoenke, S.; Serbian, I.; Major, D.; Csuk, R. Cytotoxic triterpenoid–safirinium conjugates target the endoplasmic reticulum. Eur. J. Med. Chem. 2020, 112920. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, C. Quirks of dye nomenclature. 5. Rhodamines. Biotech. Histochem. 2015, 91, 1–6. [Google Scholar] [CrossRef]
- Ceresole, M. Rhodamin Dye. U.S. Patent 516589 A 18940313, 13 March 1894. [Google Scholar]
- Ceresole, M. Red Dyestuff. U.S. Patent 456081 18910714, 14 July 1891. [Google Scholar]
- Kim, H.N.; Lee, M.H.; Kim, H.J.; Kim, J.S.; Yoon, J. A new trend in rhodamine-based chemosensors: Application of spirolactam ring-opening to sensing ions. Chem. Soc. Rev. 2008, 37, 1465–1472. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.; Li, Y.; Song, Y.; Song, Y.; Zhang, M.; Yu, H. A single rhodamine spirolactam probe for localization and pH monitoring of mitochondrion/lysosome in living cells. New J. Chem. 2018, 42, 11102–11108. [Google Scholar] [CrossRef]
- Adamovich, L.P.; Mel’nik, V.V.; McHedlov-Petrosyan, N.O. Rhodamine B equilibriums in water-salt solutions. Zh. Fiz. Khim. 1979, 53, 356–359. [Google Scholar]
- Hinckley, D.A.; Seybold, P.G. Thermodynamics of the rhodamine B lactone zwitterion equilibrium: An undergraduate laboratory experiment. J. Chem. Educ. 1987, 64, 362. [Google Scholar] [CrossRef]
- Ramart-Lucas, P. Structure of rhodamine from its absorption spectrum. Compt. Rend. 1938, 207, 1416–1418. [Google Scholar]
- Ramette, R.W.; Blackburn, T.R. Specific effects of cations on rhodamine B equilibriums. J. Phys. Chem. 1958, 62, 1601–1603. [Google Scholar] [CrossRef]
- Ramette, R.W.; Sandell, E.B. Rhodamine B equilibriums. J. Am. Chem. Soc. 1956, 78, 4872–4878. [Google Scholar] [CrossRef]
- Hinckley, D.A.; Seybold, P.G.; Borris, D.P. Solvatochromism and thermochromism of rhodamine solutions. Spectrochim. Acta Part A Mol. Spectrosc. 1986, 42, 747–754. [Google Scholar] [CrossRef]
- Beija, M.; Afonso, C.A.M.; Martinho, J.M.G. Synthesis and applications of Rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. [Google Scholar] [CrossRef] [Green Version]
- Arbeloa, F.L.; Estevez, M.J.T. Photophysics of rhodamines: Molecular structure and solvent effects. J. Phys. Chem. 1991, 95, 2203–2208. [Google Scholar] [CrossRef]
- Arbeloa, T.L.; Estévez, M.T.; López-Arbeloa, F.; Aguirresacona, I.U.; Arbeloa, I.L. Luminescence properties of rhodamines in water/ethanol mixtures. J. Lumin 1991, 48, 400–404. [Google Scholar] [CrossRef]
- Milvy, P.; Kay, K. Mutagenicity of 19 major graphic arts and printing dyes. J. Toxicol. Environ. Heal. Part A 1978, 4, 31–36. [Google Scholar] [CrossRef]
- Smart, P.L. A review of the toxicity of twelve fluorescent dyes used for water tracing. NSS Bull. 1984, 46, 21–33. [Google Scholar]
- Umeda, M. Experimental production of sarcoma in rats by injection of rhodamine B. I. Gann 1952, 43, 120–122. [Google Scholar]
- Umeda, M. Experimental study of xanthene dyes as carcinogenic agents. Gann 1956, 47, 51–78. [Google Scholar]
- Nestmann, E.R.; Douglas, G.R.I.; Matula, T.E.; Grant, C.; Kowbel, D.J. Mutagenic activity of rhodamine dyes and their impurities as detected by mutation induction in Salmonella and DNA damage in Chinese hamster ovary cells. Cancer Res. 1979, 39, 4412–4417. [Google Scholar] [PubMed]
- Elliott, G.S.; Mason, R.W.; Edwards, I.R. Studies on the pharmacokinetics and mutagenic potential of rhodamine b. J. Toxicol. Clin. Toxicol. 1990, 28, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Macasoi, I.; Mioc, M.; Vaduva, D.B.; Ghiulai, R.; Mioc, A.; Soica, C.; Muntean, D.; Dumitrascu, V. In silico Evaluation of the Antiproliferative Mithocondrial Targeted Mechanism of Action of Some Pentacyclic Triterpene Derivatives. Rev. Chim. 2019, 69, 3361–3363. [Google Scholar] [CrossRef]
- Friedrich, S.; Serbian, I.; Hoenke, S.; Wolfram, R.K.; Csuk, R. Synthesis and cytotoxic evaluation of malachite green derived oleanolic and ursolic acid piperazineamides. Med. Chem. Res. 2020, 29, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Serbian, I.; Hoenke, S.; Kraft, O.; Csuk, R. Ester and amide derivatives of rhodamine B exert cytotoxic effects on different human tumor cell lines. Med. Chem. Res. 2020, 29, 1655–1661. [Google Scholar] [CrossRef]
- Kozubek, M.; Serbian, I.; Hoenke, S.; Kraft, O.; Csuk, R. Synthesis and cytotoxic evaluation of hydroxycinnamic acid rhodamine B conjugates. Results Chem. 2020, 2, 100057. [Google Scholar] [CrossRef]
- Serbian, I.; Hoenke, S.; Csuk, R. Synthesis of some steroidal mitocans of nanomolar cytotoxicity acting by apoptosis. Eur. J. Med. Chem. 2020, 199, 112425. [Google Scholar] [CrossRef]
- Wiemann, J.; Fischer, L.; Kessler, J.; Ströhl, D.; Csuk, R. Ugi Multicomponent-reaction: Syntheses of cytotoxic dehydroabietylamine derivatives. Bioorganic Chem. 2018, 81, 567–576. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Heller, L.; Csuk, R. Targeting mitochondria: Esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018, 152, 21–30. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Fischer, L.; Kluge, R.; Ströhl, D.; Al-Harrasi, A.; Csuk, R. Homopiperazine-rhodamine B adducts of triterpenoic acids are strong mitocans. Eur. J. Med. Chem. 2018, 155, 869–879. [Google Scholar] [CrossRef]
- Siewert, B.; Pianowski, E.; Obernauer, A.; Csuk, R. Towards cytotoxic and selective derivatives of maslinic acid. Bioorg. Med. Chem. 2014, 22, 594–615. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kuhfs, J.; Csuk, R. Urea derivates of ursolic, oleanolic and maslinic acid induce apoptosis and are selective cytotoxic for several human tumor cell lines. Eur. J. Med. Chem. 2016, 119, 1–16. [Google Scholar] [CrossRef]
- Kahnt, M.; Wiemann, J.; Fischer, L.; Sommerwerk, S.; Csuk, R. Transformation of asiatic acid into a mitocanic, bimodal-acting rhodamine B conjugate of nanomolar cytotoxicity. Eur. J. Med. Chem. 2018, 159, 143–148. [Google Scholar] [CrossRef] [PubMed]
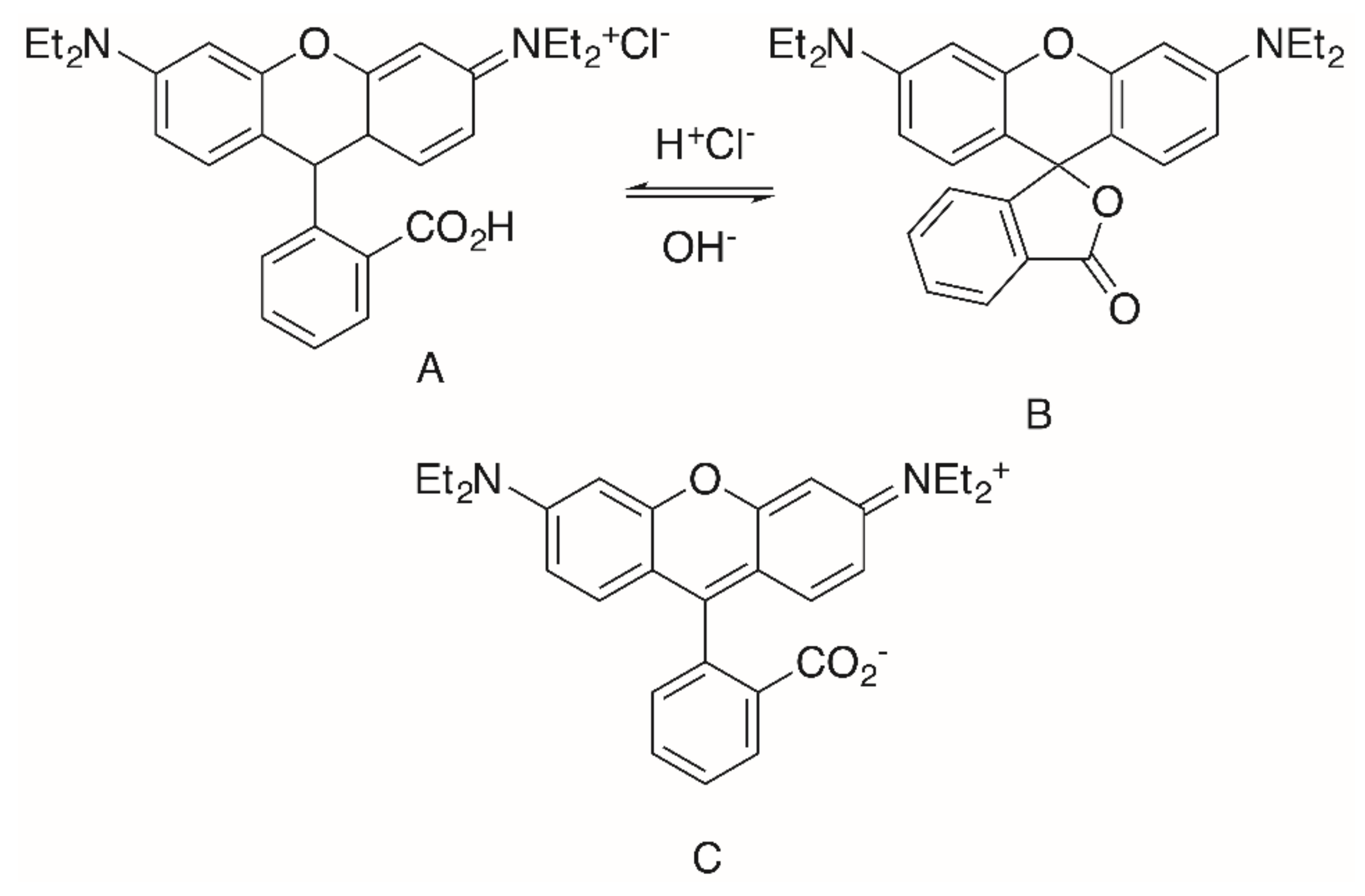

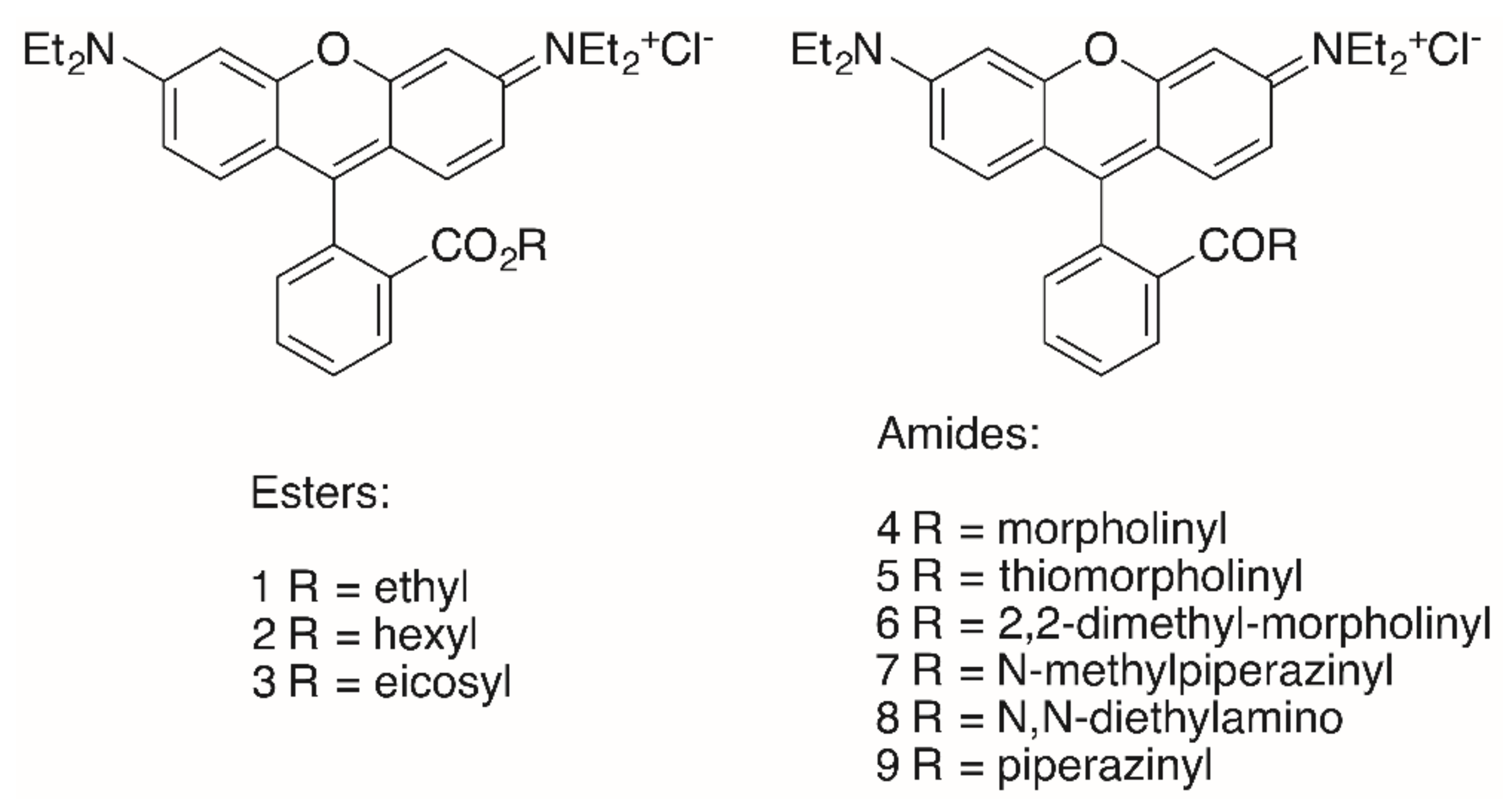



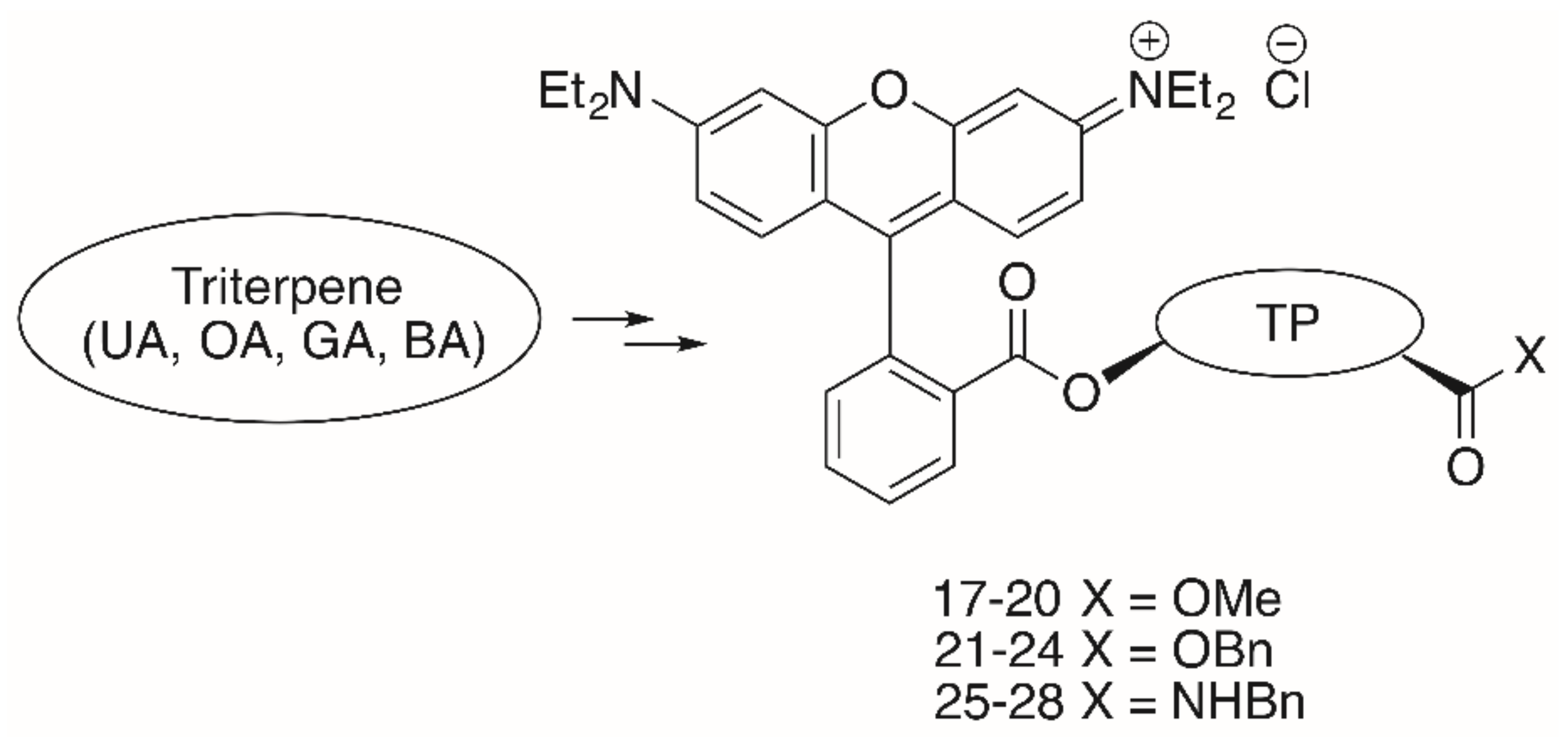

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| RhoB | >30 | >30 | >30 | >30 | >30 | >30 |
| 1 | 0.38 | 0.41 | 0.23 | 0.21 | 0.30 | 0.96 |
| 2 | 0.19 | 0.19 | 0.14 | 0.17 | 0.15 | 0.32 |
| 3 | >30 | >30 | >30 | >30 | >30 | >30 |
| 4 | 7.09 | 5.46 | 1.54 | 1.66 | 4.53 | >30 |
| 5 | 1.79 | 1.54 | 0.44 | 0.52 | 1.12 | 5.09 |
| 6 | 3.05 | 1.74 | 0.49 | 0.70 | 1.52 | 7.92 |
| 7 | 16.05 | 17.34 | 3.74 | 3.62 | 11.78 | >30 |
| 8 | 1.03 | 0.54 | 0.32 | 0.27 | 0.64 | 3.27 |
| 9 | >30 | >30 | 17.80 | 26.40 | >30 | >30 |
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| 10 | 0.16 | 0.12 | 0.06 | 0.08 | 0.26 | 0.25 |
| 11 | 0.11 | 0.64 | 0.21 | 0.31 | 0.40 | 1.81 |
| 12 | 0.22 | 0.21 | 0.23 | 0.13 | 0.24 | 0.37 |
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| 13 | 3.2 | 0.18 | 0.10 | 0.37 | 0.23 | 0.28 |
| 14 | 0.23 | 0.32 | 0.16 | 0.57 | 0.35 | 0.41 |
| 15 | 0.20 | 0.28 | 0.12 | 0.66 | 0.32 | 0.44 |
| 16a/16b | >30 | >30 | >30 | >30 | >30 | >30 |
| Compound | TP | R | FaDu | A2780 | HT29 | MCF-7 | SW1736 | NIH 3T3 |
|---|---|---|---|---|---|---|---|---|
| 17 | UA | OMe | 1.96 | 1.75 | 1.85 | 1.83 | 1.72 | 1.84 |
| 18 | OA | OMe | 1.99 | 1.14 | 2.75 | 2.31 | 1.76 | 2.63 |
| 19 | GA | OMe | 0.19 | 0.08 | 0.15 | 0.18 | 0.15 | 0.20 |
| 20 | BA | OMe | 1.29 | 0.42 | 0.61 | 0.81 | 0.74 | 1.77 |
| 21 | UA | OBn | 15.79 | 10.10 | 11.41 | 13.75 | 12.66 | 15.42 |
| 22 | OA | OBn | 9.12 | 3.35 | 8.90 | 9.40 | 9.05 | 11.25 |
| 23 | GA | OBn | 1.54 | 0.90 | 1.42 | 1.47 | 1.13 | 1.28 |
| 24 | BA | OBn | 7.59 | 3.36 | 5.33 | 5.05 | 6.43 | 8.04 |
| 25 | UA | NBn | 0.44 | 0.34 | 0.45 | 0.30 | 0.24 | 0.37 |
| 26 | OA | NBn | 0.50 | 0.32 | 0.46 | 0.36 | 0.27 | 0.40 |
| 27 | GA | NBn | 0.06 | 0.02 | 0.06 | 0.04 | 0.04 | 0.08 |
| 28 | BA | NBn | 0.54 | 0.31 | 0.53 | 0.47 | 0.45 | 0.54 |
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| 29 | 0.19 | 0,19 | 0.094 | 0.066 | 0.074 | 0.21 |
| 30 | 0.012 | 0.012 | 0.022 | 0.004 | 0.004 | 0.164 |
| 31 | 0.14 | 0.16 | 0.0084 | 0.037 | 0.041 | 0.25 |
| 32 | 0.06 | 0.005 | 0.008 | 0.001 | 0.001 | 0.19 |
| 33 | n.d. | 0.017 | 0.012 | 0.008 | n.d. | 0.178 |
| EM2 | n.d. | 4.70 | 7.70 | 0.50 | n.d. | 33.8 |
Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules 2020, 25, 5443. https://doi.org/10.3390/molecules25225443
Hoenke S, Serbian I, Deigner H-P, Csuk R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules. 2020; 25(22):5443. https://doi.org/10.3390/molecules25225443
Chicago/Turabian StyleHoenke, Sophie, Immo Serbian, Hans-Peter Deigner, and René Csuk. 2020. "Mitocanic Di- and Triterpenoid Rhodamine B Conjugates" Molecules 25, no. 22: 5443. https://doi.org/10.3390/molecules25225443
APA StyleHoenke, S., Serbian, I., Deigner, H.-P., & Csuk, R. (2020). Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules, 25(22), 5443. https://doi.org/10.3390/molecules25225443