
Macrooxazoles A–D, New 2,5-Disubstituted Oxazole-4-Carboxylic Acid Derivatives from the Plant Pathogenic Fungus Phoma macrostoma

 and
and
Abstract
:1. Introduction
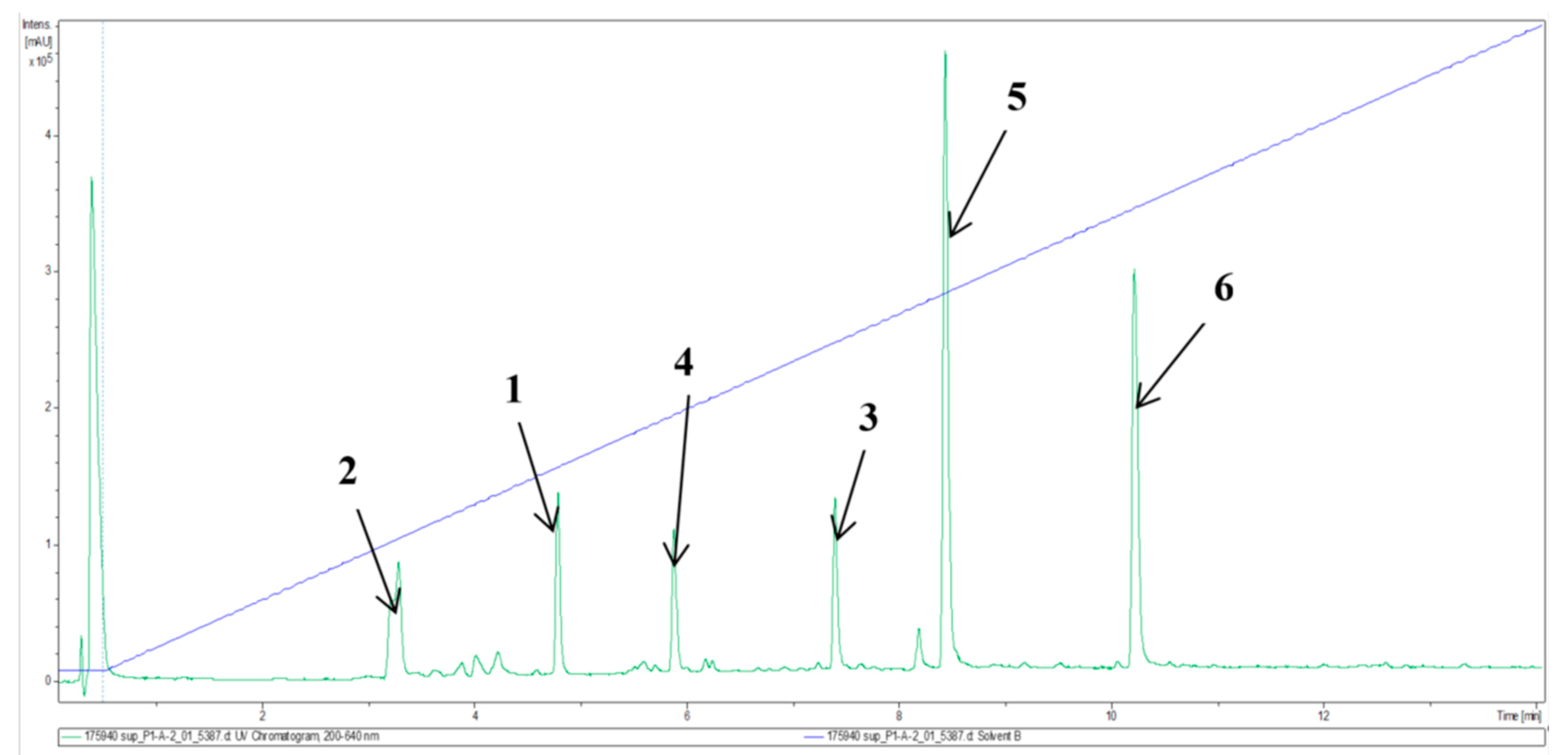
2. Results and Discussion
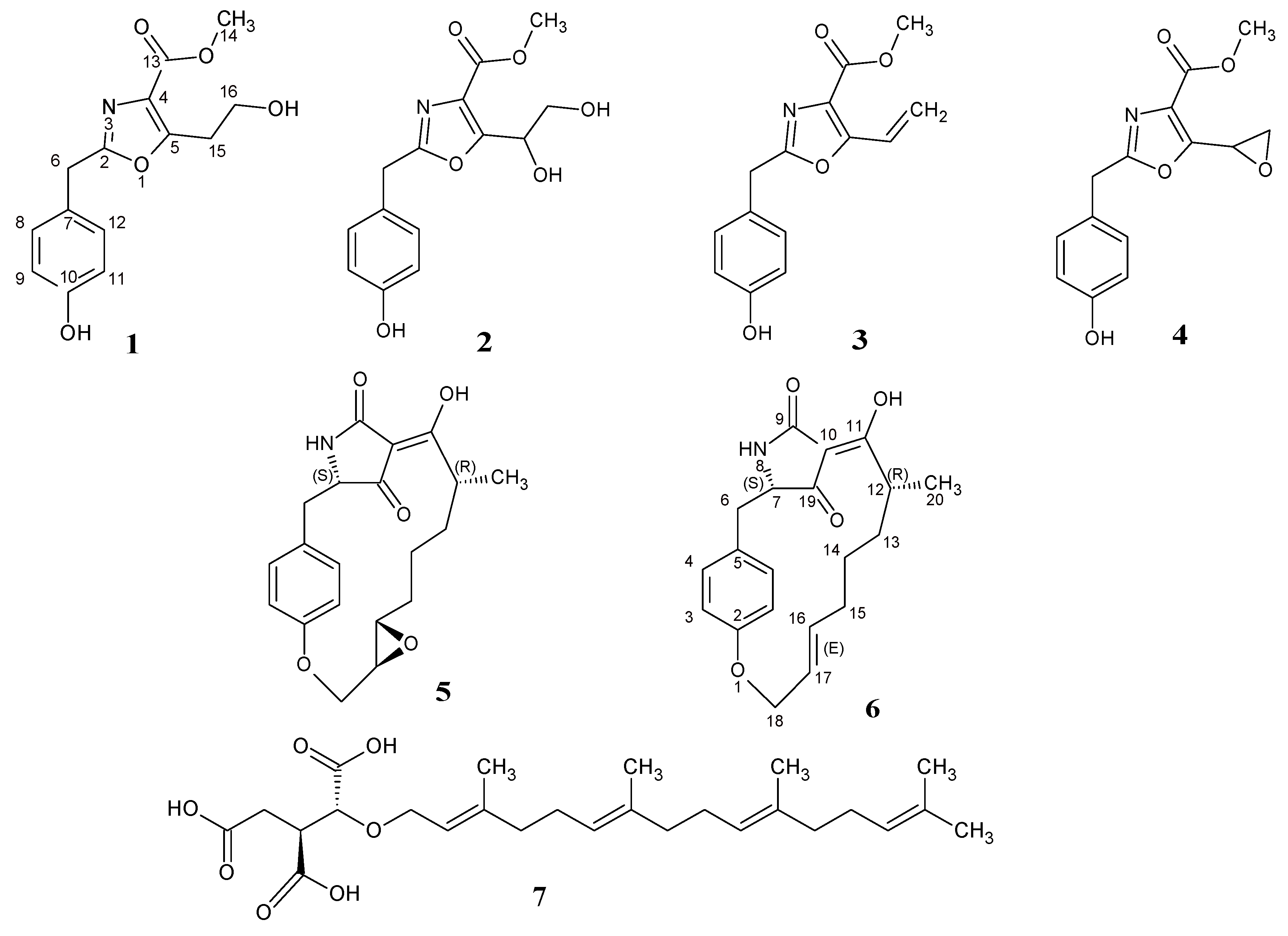
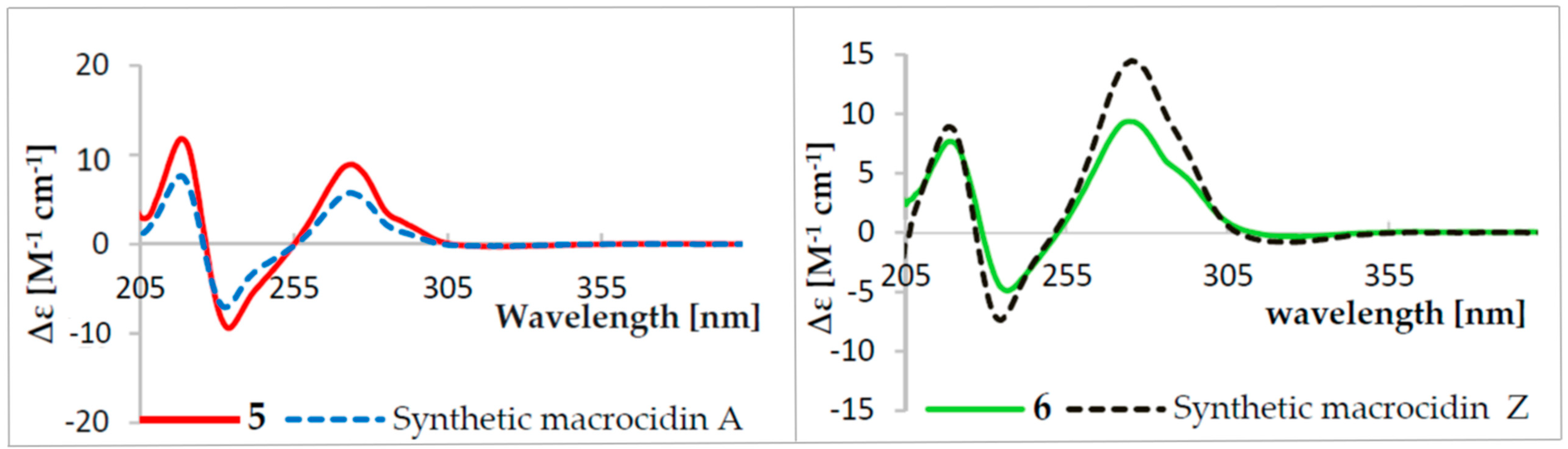
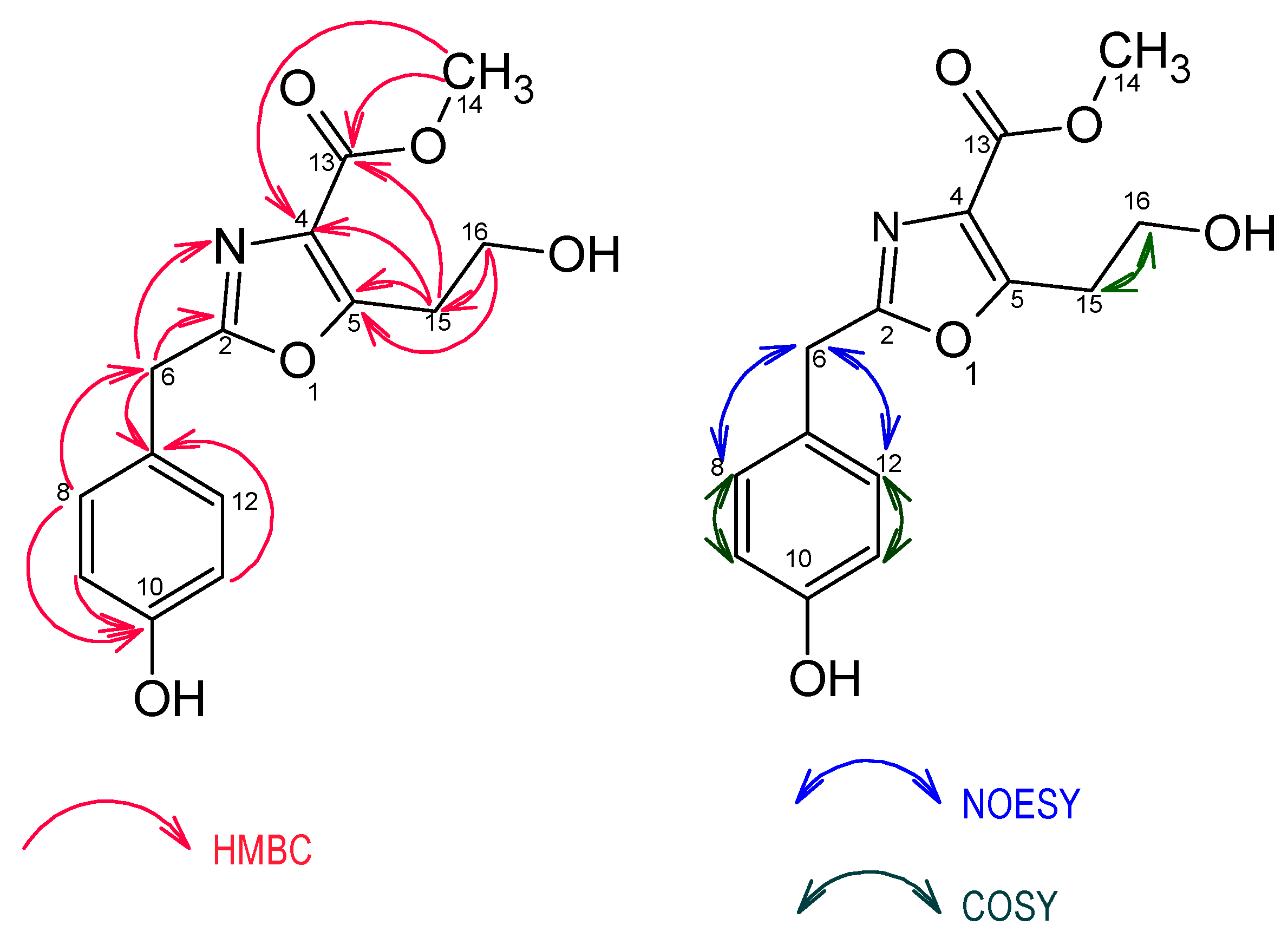
2.1. Structure Elucidation of Compounds 1–6
2.2. Biological Activities
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Fungal Material
3.3. Small-Scale Fermentation and Extraction
3.4. Scale Up of Production in Shake Flask Batches and Extraction
3.5. Isolation of Compounds 1–6
3.6. Physico-Chemical Characteristics of Compounds 1–6
3.7. Synthesis of Macrocidin Z (6)
3.8. Antimicrobial Assay
3.9. Cytotoxicity Assay
3.10. Biofilm Inhibition Assay
3.11. Dispersion of Preformed Biofilm
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kakkar, S.; Balasubramanian, N. A comprehensive review on biological activities of oxazole derivatives. BMC Chem. 2019, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Palta, K.; Kumar, M.; Bhargava, M.; Dahiya, L. Therapeutic potential of oxazole scaffold: A patent review (2006–2017). Expert Opin. Therap. Pat. 2018, 28, 783–812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, W.; Jiang, X.; Jiang, F.; Zhuang, H.; Fu, L. Design, synthesis and antimicrobial activity of chiral 2-(substituted-hydroxyl)-3-(benzo [d] oxazol-5-yl) propanoic acid derivatives. Eur. J. Med. Chem. 2011, 46, 3639–3650. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumar, N.M.; Sundaree, S.; Johnson, E.O.; Shah, K. An expeditious synthesis and anticancer activity of novel 4-(3′-indolyl) oxazoles. Eur. J. Med. Chem. 2010, 45, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Chang, M.; Villegas-Estrada, A.; Franzblau, S.G.; Möllmann, U.; Miller, M.J. Structure–activity relationship of new anti-tuberculosis agents derived from oxazoline and oxazole benzyl esters. Eur. J. Med. Chem. 2010, 45, 1703–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eren, G.; Ünlü, S.; Nuñez, M.T.; Labeaga, L.; Ledo, F.; Entrena, A.; Şahin, M.F. Synthesis, biological evaluation, and docking studies of novel heterocyclic diaryl compounds as selective COX-2 inhibitors. Bioorg. Med. Chem. 2010, 18, 6367–6376. [Google Scholar] [CrossRef]
- Ashton, W.T.; Sisco, R.M.; Dong, H.; Lyons, K.A.; He, H.; Doss, G.A.; Thornberry, N.A. Dipeptidyl peptidase IV inhibitors derived from β-aminoacylpiperidines bearing a fused thiazole, oxazole, isoxazole, or pyrazole. Bioorg. Med. Chem. Lett. 2005, 15, 2253–2258. [Google Scholar] [CrossRef]
- Jadhav, R.D.; Kadam, K.S.; Kandre, S.; Guha, T.; Reddy, M.M.K.; Brahma, M.K.; Enose, A.A. Synthesis and biological evaluation of isoxazole, oxazole, and oxadiazole containing heteroaryl analogs of biaryl ureas as DGAT1 inhibitors. Europ. J. Med. Chem. 2012, 54, 324–342. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Sudhakar, A. Total synthesis of bengazole A. Org. Lett. 2010, 12, 236–238. [Google Scholar] [CrossRef]
- Davyt, D.; Serra, G. Thiazole and oxazole alkaloids: Isolation and synthesis. Mar. Drugs 2010, 8, 2755–2780. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Bisht, A.S.; Juyal, D. Systematic scientific study of 1, 3-oxazole derivatives as a useful lead for pharmaceuticals: A review. Pharma Innov. 2017, 6, 109. [Google Scholar]
- Ichiba, T.; Yoshida, W.Y.; Scheuer, P.J.; Higa, T.; Gravalos, D.G. Hennoxazoles, bioactive bisoxazoles from a marine sponge. J. Am. Chem. Soc. 1991, 113, 3173–3174. [Google Scholar] [CrossRef]
- Yokokawa, F.; Asano, T.; Shioiri, T. Total synthesis of the antiviral marine natural product (−)-hennoxazole A. Org. Lett. 2000, 2, 4169–4172. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, K.; Arai, N.; Shinose, M.; Takahashi, Y.; Yoshida, H.; Iwabuchi, J.; OMURA, S. New antibiotics phthoxazolins B, C and D produced by Streptomyces sp. KO-7888. J. Antibio. 1995, 48, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graupner, P.R.; Carr, A.; Clancy, E.; Gilbert, J.; Bailey, K.L.; Derby, J.A.; Gerwick, B.C. The macrocidins: Novel cyclic tetramic acids with herbicidal activity produced by Phoma macrostoma. J. Nat. Prod. 2003, 66, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- Graupner, P.R.; Gerwick, B.C.; Siddall, T.L.; Carr, A.W.; Clancy, E.; Gilbert, J.R.; Derby, J.A. Chlorosis inducing phytotoxic metabolites: New herbicides from Phoma macrostoma. ACS Symp. Ser. 2006, 927, 37–47. [Google Scholar]
- Haase, R.G.; Schobert, R. Synthesis of the bioherbicidal fungus metabolite macrocidin A. Org. Lett. 2016, 18, 6352–6355. [Google Scholar] [CrossRef]
- Andrade, C.K.; Rocha, R.O.; Vercillo, O.E.; Silva, W.A.; Matos, R.A. DCC/DMAP-mediated coupling of carboxylic acids with oxazolidinones and thiazolidinethiones. Synlett 2003, 35, 2351–2352. [Google Scholar] [CrossRef]
- Barnickel, B.; Schobert, R. Toward the macrocidins: Macrocyclization via Williamson etherification of a phenolate. J. Org. Chem. 2010, 75, 6716–6719. [Google Scholar] [CrossRef]
- Jouin, P.; Castro, B.; Nisato, D. Stereospecific synthesis of N-protected statine and its analogues via chiral tetramic acid. J. Chem. Soc. Perkin Trans. I 1987, 1177–1182. [Google Scholar] [CrossRef]
- Hori, K.; Arai, M.; Nomura, K.; Yoshii, E. An efficient 3(C)-acylation of tetramic acids involving acyl migration of 4(O)-acylates. Chem. Pharm. Bull. 1987, 35, 4368–4371. [Google Scholar] [CrossRef] [Green Version]
- Sengoku, T.; Nagae, Y.; Ujihara, Y.; Takahashi, M.; Yoda, H. A synthetic approach to diverse 3-acyltetramic acids via O- to C-acyl rearrangement and application to the total synthesis of penicillenol series. J. Org. Chem. 2012, 77, 4391–4401. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Derby, J. Fungal Isolates and Biological Control Compositions for the Control of Weeds. U.S. Patent No. 7772,155, 10 August 2010. [Google Scholar]
- Chepkirui, C.; Richter, C.; Matasyoh, J.C.; Stadler, M. Monochlorinated calocerins A-D and 9-oxostrobilurin derivatives from the basidiomycete Favolaschia calocera. Phytochemistry 2016, 132, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Post, S.J.; Keohane, C.E.; Rossiter, L.M.; Kaplan, A.R.; Khowsathit, J.; Matuska, K.; Karanicolas, J.; Wuest, W.M. Target-based design of promysalin analogues identifies a new putative binding cleft in succinate dehydrogenase. ACS Infect. Dis. 2020, 6, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Wessel, A.C.; Luangsa-ard, J.J.; Stadler, M. Viridistratins A-C, antimicrobial and cytotoxic benzo[j]fluoranthenes from stromata of Annulohypoxylon viridistratum (Hypoxylaceae, Ascomycota). Biomolecules 2020, 10, 805. [Google Scholar] [CrossRef]
- Sandargo, B.; Michehl, M.; Praditya, D.; Steinmann, E.; Stadler, M.; Surup, F. Antiviral meroterpenoid rhodatin and sesquiterpenoids rhodocoranes A–E from the Wrinkled Peach Mushroom, Rhodotus palmatus. Organic Lett. 2019, 21, 3286–3289. [Google Scholar] [CrossRef]
- Chepkirui, C.; Yuyama, K.; Wanga, L.; Decock, C.; Matasyoh, J.; Abraham, W.R.; Stadler, M. Microporenic acids A-G, biofilm inhibitors and antimicrobial agents from the basidiomycete Microporus sp. J. Nat. Prod. 2018, 81, 778–784. [Google Scholar] [CrossRef]
- Yuyama, K.T.; Chepkirui, C.; Wendt, L.; Fortkamp, D.; Stadler, M.; Abraham, W.R. Bioactive compounds produced by Hypoxylon fragiforme against Staphylococcus aureus biofilms. Microorganisms 2017, 5, 80. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 6 | Synthetic 6 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 157.4, C | 157.3, C | ||
| 3/21 | 115.9, CH | 6.69, m | 115.7, CH | 6.71, m |
| 4/22 | 132.6, CH | 6.98, m | 132.5, CH | 6.97, m |
| 5 | 127.6, C | 127.3, C | ||
| 6 | 36.6, CH2 | 3.07, dd (14.1, 3.9) | 36.5, CH2 | 3.07, dd (14.1, 3.9) |
| 2.90, dd (14.1, 3.3) | 2.89, dd (14.1, 3.1) | |||
| 7 | 63.8, CH | 4.10, t (3.6) | 63.8, CH | 4.10, m |
| 9 | 177.3, C | 175.5, C | ||
| 10 | 102.3, C | 102.1, C | ||
| 11 | 194.0, C | 191.8, C | ||
| 12 | 37.2, CH | 3.40, sxt (6.8) | 36.8, C | 3.39, sxt (6.8) |
| 13 | 35.3, CH2 | 1.16, tdd (12.9, 6.4, 4.3) | 35.1, CH2 | 1.13, m |
| 1.09, m | ||||
| 14 | 28.3, CH2 | 0.83, tddd (12.9, 8.5, 6.5, 4.4) | 28.1, CH2 | 0.83, m |
| 1.32, m | 1.32, m | |||
| 15 | 33.6, CH2 | 2.06, dq (12.8, 6.2) | 33.4, CH2 | 2.06, m |
| 1.79, m | 1.79, m | |||
| 16 | 139.1, CH | 5.67, ddd (15.5, 8.8, 5.9) | 139.0, CH | 5.68, m |
| 17 | 126.6, CH | 5.26, ddd (15.5, 8.9, 3.8) | 126.7, CH | 5.26, m |
| 18 | 68.1, CH2 | 4.64, dd (13.4, 8.9) | 67.9, CH2 | 4.64, dd (13.4, 9.5) |
| 4.53, dd (13.4, 3.8) | 4.53, m | |||
| 19 | 197.3, C | 197.1, C | ||
| 20 | 15.4, CH3 | 1.05, d (6.8) | 15.2, CH3 | |
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 164.4, C | - | 165.1, C | - |
| 4 | 129.0, C | - | 129.6, C | - |
| 5 | 159.1, C | - | 159.3, C | - |
| 6 | 34.3, CH2 | 4.00, s | 34.3, CH2 | 4.04, s |
| 7 | 127.0, C | - | 126.9, C | - |
| 8 | 131.0, CH | 7.10, d (9.0) | 131.0, CH | 7.12, d (9.0) |
| 9 | 116.7, CH | 6.73, d (9.0) | 116.7, CH | 6.73, d (9.0) |
| 10 | 157.9, C | - | 158.0, C | - |
| 11 | 116.7, CH | 6.73, d (9.0) | 116.7, CH | 6.73, d (9.0) |
| 12 | 131.0, CH | 7.10, d (9.0) | 131.0, CH | 7.12, d (9.0) |
| 13 | 163.9, C | - | 163.6, C | - |
| 14 | 52.4, CH3 | 3.87, s | 52.6, CH3 | 3.88, s |
| 15 | 30.8, CH2 | 3.19, t (6.5) | 67.3, CH | 5.33, t (6.5) |
| 16 | 60.6, CH2 | 3.80, t (6.5) | 65.1, CH2 | 3.73, dd (11, 6.5) 3.78, dd (11, 6.5) |
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 2 | 164.8, C | - | 165.3, C | - |
| 4 | 128.1, C | - | 133.0, C | - |
| 5 | 155.6, C | - | 155.1, C | - |
| 6 | 34.3, CH2 | 4.04, s | 34.2, CH2 | 4.00, s |
| 7 | 126.8, C | - | 126.6, C | |
| 8 | 131.0, CH | 7.12, d (9.0) | 131.0, CH | 7.08, d (9.0) |
| 9 | 116.8, CH | 6.75, d (9.0) | 116.7, CH | 6.73, d (9.0) |
| 10 | 158.0, C | - | 158.0, C | - |
| 11 | 116.8, CH | 6.75, d (9.0) | 116.7, CH | 6.73, d (9.0) |
| 12 | 131.0, CH | 7.12, d (9.0) | 131.0, CH | 7.08, d (9.0) |
| 13 | 163.4, C | - | 163.2, C | - |
| 14 | 52.6, CH3 | 3.89, s | 52.8, CH3 | 3.92, s |
| 15 | 123.2, CH | 7.14, dd (17.5, 11.5) | 44.8, CH | 4.51, t (3.5) |
| 16 | 121.0, CH2 | 5.58, dd (11.5, 1.1) 5.96, dd (17.5, 1.1) | 48.9 *, CH2 | 3.22, dd (3.5, 1.6) |
| Compounds | Inhibition of Biofilm Formation (%) | Destruction of Preformed Biofilm (%) |
|---|---|---|
| 1 | - | - |
| 2 | 65 (250 µg/mL) 43 (125 µg/mL) | 36 (250 µg/mL) 31 (125 µg/mL) |
| 3 | 75 (250 µg/mL) 59 (125 µg/mL) | 57 (250 µg/mL) 48 (125 µg/mL) |
| 4 | n.t | n.t |
| 5 | 79 (250 µg/mL) 77 (62.5 µg/mL) 61 (15.6 µg/mL) | 75 (250 µg/mL) 65 (62.5 µg/mL) 31 (15.6 µg/mL) |
| 6 | 76 (250 µg/mL) 70 (62.5 µg/mL) 19 (15.6 µg/mL) | 73 (250 µg/mL) 59 (62.5 µg/mL) 40 (15.6 µg/mL) |
| Microporenic acid A | 83 (250 µg/mL) 81 (62.5 µg/mL) 48 (15.6 µg/mL) | 71 (250 µg/mL) 70 (62.5 µg/mL) 39 (15.6 µg/mL) |
Sample Availability: Samples of the compounds are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matio Kemkuignou, B.; Treiber, L.; Zeng, H.; Schrey, H.; Schobert, R.; Stadler, M. Macrooxazoles A–D, New 2,5-Disubstituted Oxazole-4-Carboxylic Acid Derivatives from the Plant Pathogenic Fungus Phoma macrostoma. Molecules 2020, 25, 5497. https://doi.org/10.3390/molecules25235497
Matio Kemkuignou B, Treiber L, Zeng H, Schrey H, Schobert R, Stadler M. Macrooxazoles A–D, New 2,5-Disubstituted Oxazole-4-Carboxylic Acid Derivatives from the Plant Pathogenic Fungus Phoma macrostoma. Molecules. 2020; 25(23):5497. https://doi.org/10.3390/molecules25235497
Chicago/Turabian StyleMatio Kemkuignou, Blondelle, Laura Treiber, Haoxuan Zeng, Hedda Schrey, Rainer Schobert, and Marc Stadler. 2020. "Macrooxazoles A–D, New 2,5-Disubstituted Oxazole-4-Carboxylic Acid Derivatives from the Plant Pathogenic Fungus Phoma macrostoma" Molecules 25, no. 23: 5497. https://doi.org/10.3390/molecules25235497