
Supramolecular Solvent-Based Microextraction of Selected Anticonvulsant and Nonsteroidal Anti-Inflammatory Drugs from Sediment Samples

 , and
, and
Abstract
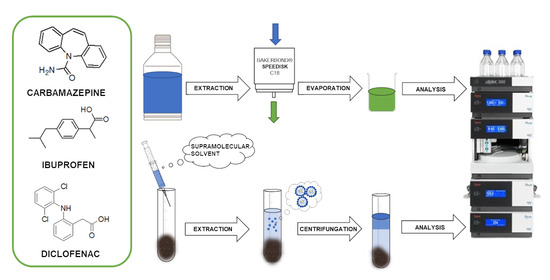
1. Introduction
2. Results and Discussion
2.1. Development of Chromatographic Conditions
2.2. SUPRAS Microextraction
2.2.1. Composition of Supramolecular Solvent
2.2.2. Volume of the Supramolecular Solvent
2.2.3. Sample Weight
2.2.4. Extraction Time
2.2.5. Centrifugation Time and Speed
2.2.6. SUPRAS Microextraction Procedure
2.3. Speedisk Solid-Phase Extraction
2.4. Method Validation
2.4.1. Linearity and Sensitivity
2.4.2. Accuracy and Precision
2.5. Comparison of Proposed SUPRAS-UHPLC-UV and Speedisk-UHPLC-UV Methods with Other Reported Methods
2.6. Study of the Sorption Efficiency of Selected Drugs on Soil
3. Materials and Methods
3.1. Standards, Chemicals, and Materials
3.2. Preparation of Standard Solutions
3.3. UHPLC-UV Conditions
3.4. Preparation of Supramolecular Solvent and Determination of Its Volume
3.5. Preparation of Bottom Sediment Samples–SUPRAS Microextraction
3.6. Preparation of Water Samples–Speedisk Technique
3.7. Preparation of Blank Samples
3.8. Method Validation
3.9. Study of Sorption Efficiency of Selected Drugs on Sediment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mompelat, S.; Le Bot, B.; Thomas, O. Occurrence and fate of pharmaceutical products and by-products, from resource to drinking water. Environ. Int. 2009, 35, 803–814. [Google Scholar] [CrossRef]
- Sammartino, M.P.; Castrucci, M.; Ruiu, D.; Visco, G.; Campanella, L. Photostability and toxicity of finasteride, diclofenac and naproxen under simulating sunlight exposure: Evaluation of the toxicity trend and of the packaging photoprotection. Chem. Cent. J. 2013, 7, 1–10. [Google Scholar] [CrossRef]
- Khetan, S.K.; Collins, T.J. Human pharmaceuticals in the aquatic environment: A challenge to green chemisty. Chem. Rev. 2007, 107, 2319–2364. [Google Scholar] [CrossRef]
- Seifrtová, M.; Nováková, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Gazal, G.; Al-Samadani, K.H. Comparison of paracetamol, ibuprofen, and diclofenac potassium for pain relief following dental extractions and deep cavity preparations. Saudi Med. J. 2017, 38, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Tayel, S.A.; Soliman, I.I.; Louis, D. Improvement of dissolution properties of Carbamazepine through application of the liquisolid tablet technique. Eur. J. Pharm. Biopharm. 2008, 69, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Osenbrück, K.; Gläser, H.R.; Knöller, K.; Weise, S.M.; Möder, M.; Wennrich, R.; Schirmer, M.; Reinstorf, F.; Busch, W.; Strauch, G. Sources and transport of selected organic micropollutants in urban groundwater underlying the city of Halle (Saale), Germany. Water Res. 2007, 41, 3259–3270. [Google Scholar] [CrossRef]
- Farré, M.; Petrovic, M.; Barceló, D. Recently developed GC/MS and LC/MS methods for determining NSAIDs in water samples. Anal. Bioanal. Chem. 2007, 387, 1203–1214. [Google Scholar] [CrossRef]
- Barron, L.; Tobin, J.; Paull, B. Multi-residue determination of pharmaceuticals in sludge and sludge enriched soils using pressurized liquid extraction, solid phase extraction and liquid chromatography with tandem mass spectrometry. J. Environ. Monit. 2008, 10, 353–361. [Google Scholar] [CrossRef]
- Lin, K.; Gan, J. Sorption and degradation of wastewater-associated non-steroidal anti-inflammatory drugs and antibiotics in soils. Chemosphere 2011, 83, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Jelic, A.; Gros, M.; Ginebreda, A.; Cespedes-Sánchez, R.; Ventura, F.; Petrovic, M.; Barcelo, D. Occurrence, partition and removal of pharmaceuticals in sewage water and sludge during wastewater treatment. Water Res. 2011, 45, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Nieto, A.; Borrull, F.; Pocurull, E.; Marcé, R.M. Pressurized liquid extraction of pharmaceuticals from sewage-sludge. J. Sep. Sci. 2007, 30, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.; Santos, J.L.; Aparicio, I.; Alonso, E. Multi-residue method for the analysis of pharmaceutical compounds in sewage sludge, compost and sediments by sonication-assisted extraction and LC determination. J. Sep. Sci. 2010, 33, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Yamashita, N.; Tanaka, H.; Matsukawa, H.; Tanabe, K. Development of extraction method of pharmaceuticals and their occurrences found in Japanese wastewater treatment plants. Environ. Int. 2009, 35, 815–820. [Google Scholar] [CrossRef]
- Ternes, T.A.; Bonerz, M.; Herrmann, N.; Löffler, D.; Keller, E.; Lacida, B.B.; Alder, A.C. Determination of pharmaceuticals, iodinated contrast media and musk fragrances in sludge by LC tandem MS and GC/MS. J. Chromatogr. A 2005, 1067, 213–223. [Google Scholar] [CrossRef]
- Radjenović, J.; Jelić, A.; Petrović, M.; Barceló, D. Determination of pharmaceuticals in sewage sludge by pressurized liquid extraction (PLE) coupled to liquid chromatography-tandem mass spectrometry (LC-MS/MS). Anal. Bioanal. Chem. 2009, 393, 1685–1695. [Google Scholar] [CrossRef]
- Sagristà, E.; Larsson, E.; Ezoddin, M.; Hidalgo, M.; Salvadó, V.; Jönsson, J.Å. Determination of non-steroidal anti-inflammatory drugs in sewage sludge by direct hollow fiber supported liquid membrane extraction and liquid chromatography-mass spectrometry. J. Chromatogr. A 2010, 1217, 6153–6158. [Google Scholar] [CrossRef]
- Saleh, A.; Larsson, E.; Yamini, Y.; Jönsson, J.Å. Hollow fiber liquid phase microextraction as a preconcentration and clean-up step after pressurized hot water extraction for the determination of non-steroidal anti-inflammatory drugs in sewage sludge. J. Chromatogr. A 2011, 1218, 1331–1339. [Google Scholar] [CrossRef]
- Ballesteros-Gómez, A.; Rubio, S.; Pérez-Bendito, D. Potential of supramolecular solvents for the extraction of contaminants in liquid foods. J. Chromatogr. A 2009, 1216, 530–539. [Google Scholar] [CrossRef]
- Styszko, K. Sorption of emerging organic micropollutants onto fine sediments in a water supply dam reservoir, Poland. J. Soils Sediments 2016, 16, 677–686. [Google Scholar] [CrossRef]
- Sosa Ferrera, Z.; Padrón Sanz, C.; Mahugo Santana, C.; Santana Rodríguez, J.J. The use of micellar systems in the extraction and pre-concentration of organic pollutants in environmental samples. TrAC Trends Anal. Chem. 2004, 23, 469–479. [Google Scholar] [CrossRef]
- Gu, T.; Galera-Gómez, P.A. The effect of different alcohols and other polar organic additives on the cloud point of Triton X-100 in water. Colloids Surf. A Physicochem. Eng. Asp. 1999, 147, 365–370. [Google Scholar] [CrossRef]
- López-Jiménez, F.J.; Rosales-Marcano, M.; Rubio, S. Restricted access property supramolecular solvents for combined microextraction of endocrine disruptors in sediment and sample cleanup prior to their quantification by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2013, 1303, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Caballo, C.; Sicilia, M.D.; Rubio, S. Fast, simple and efficient supramolecular solvent-based microextraction of mecoprop and dichlorprop in soils prior to their enantioselective determination by liquid chromatography-tandem mass spectrometry. Talanta 2014, 119, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Moral, A.; Caballo, C.; Sicilia, M.D.; Rubio, S. Highly efficient microextraction of chlorophenoxy acid herbicides in natural waters using a decanoic acid-based nanostructured solvent prior to their quantitation by liquid chromatography-mass spectrometry. Anal. Chim. Acta 2012, 709, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tayyebi, M.; Yamini, Y.; Moradi, M. Reverse micelle-mediated dispersive liquid-liquid microextraction of 2,4-dichlorophenoxyacetic acid and 4-chloro-2-methylphenoxyacetic acid. J. Sep. Sci. 2012, 35, 2491–2498. [Google Scholar] [CrossRef]
- Caballo, C.; Sicilia, M.D.; Rubio, S. Stereoselective quantitation of mecoprop and dichlorprop in natural waters by supramolecular solvent-based microextraction, chiral liquid chromatography and tandem mass spectrometry. Anal. Chim. Acta 2013, 761, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.; Yamini, Y.; Tayyebi, M.; Asiabi, H. Ultrasound-assisted liquid-phase microextraction based on a nanostructured supramolecular solvent. Anal. Bioanal. Chem. 2013, 405, 4235–4243. [Google Scholar] [CrossRef]
- Ebrahimpour, B.; Yamini, Y.; Seidi, S.; Rezaei, F. Nanostructured solvent based microextraction followed by a novel strategy for online phase separation coupled with HPLC for determination of ethinyl estradiol. Anal. Methods 2014, 6, 2936–2942. [Google Scholar] [CrossRef]
- Radjenović, J.; Petrović, M.; Barceló, D. Fate and distribution of pharmaceuticals in wastewater and sewage sludge of the conventional activated sludge (CAS) and advanced membrane bioreactor (MBR) treatment. Water Res. 2009, 43, 831–841. [Google Scholar] [CrossRef]
- Stepnowski, P.; Synak, E.; Szafranek, B.; Kaczyński, Z. Techniki Separacyjne, 1st ed.; Wydawnictwo Uniwersytetu Gdańskiego: Gdańsk, Poland, 2010; ISBN 978-83-7326-712-1. [Google Scholar]
- Ruiz, F.J.; Rubio, S.; Pérez-Bendito, D. Water-induced coacervation of alkyl carboxylic acid reverse micelles: Phenomenon description and potential for the extraction of organic compounds. Anal. Chem. 2007, 79, 7473–7484. [Google Scholar] [CrossRef] [PubMed]
- López-Jiménez, F.J.; Rubio, S.; Pérez-Bendito, D. Supramolecular solvent-based microextraction of Sudan dyes in chilli-containing foodstuffs prior to their liquid chromatography-photodiode array determination. Food Chem. 2010, 121, 763–769. [Google Scholar] [CrossRef]
- Ferdowsi, M.; Taghian, A.; Najafi, A.; Moradi, M. Application of a nanostructured supramolecular solvent for the microextraction of diphenylamine and its mono-Nitrated derivatives from unburned single-Base propellants. J. Sep. Sci. 2015, 38, 276–282. [Google Scholar] [CrossRef]
- Sacher, F.; Lange, F.T.; Brauch, H.-J.; Blankenhorn, I. Pharmaceuticals in groundwaters Analytical methods and results of a monitoring program in Baden-Wurttemberg, Germany. J. Chromatogr. A 2001, 938, 199–210. [Google Scholar] [CrossRef]
- Weigel, S.; Berger, U.; Jensen, E.; Kallenborn, R.; Thoresen, H.; Hühnerfuss, H. Determination of selected pharmaceuticals and caffeine in sewage and seawater from Tromsø/Norway with emphasis on ibuprofen and its metabolites. Chemosphere 2004, 56, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, R.; Marotta, R.; Paxéus, N. Pharmaceuticals in STP effluents and their solar photodegradation in aquatic environment. Chemosphere 2003, 50, 1319–1330. [Google Scholar] [CrossRef]
- Ashton, D.; Hilton, M.; Thomas, K.V. Investigating the environmental transport of human pharmaceuticals to streams in the United Kingdom. Sci. Total Environ. 2004, 333, 167–184. [Google Scholar] [CrossRef]
- Lin, W.C.; Chen, H.C.; Ding, W.H. Determination of pharmaceutical residues in waters by solid-phase extraction and large-volume on-line derivatization with gas chromatography-mass spectrometry. J. Chromatogr. A 2005, 1065, 279–285. [Google Scholar] [CrossRef]
- Koutsouba, V.; Heberer, T.; Fuhrmann, B.; Schmidt-Baumler, K.; Tsipi, D.; Hiskia, A. Determination of polar pharmaceuticals in sewage water of Greece by gas chromatography-mass spectrometry. Chemosphere 2003, 51, 69–75. [Google Scholar] [CrossRef]
- Nebot, C.; Gibb, S.W.; Boyd, K.G. Quantification of human pharmaceuticals in water samples by high performance liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2007, 598, 87–94. [Google Scholar] [CrossRef]
- Ternes, T.A. Analytical methods for the determination of pharmaceuticals in aqueous environmental samples. TrAC Trends Anal. Chem. 2001, 20, 419–434. [Google Scholar] [CrossRef]
- Kosjek, T.; Heath, E.; Kompare, B. Removal of pharmaceutical residues in a pilot wastewater treatment plant. Anal. Bioanal. Chem. 2007, 387, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, A.; Souhail, B.; Ballesteros, E. Continuous solid-phase extraction and gas chromatography-mass spectrometry determination of pharmaceuticals and hormones in water samples. J. Chromatogr. A 2010, 1217, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Kowal, A.; Świderska-Bróż, M. Oczyszczanie Wody. Podstawy Teoretyczne i Technologiczne, Procesy i Urządzenia, 6th ed.; PWN: Warsaw, Poland, 2009; ISBN 978-83-011-5871-2. [Google Scholar]
- Mitra, S.; Benfield, B. Ibuprofen Sorption to Coastal Plain Soils. Water. Air. Soil Pollut. 2018, 229, 229–295. [Google Scholar] [CrossRef]
- Hiller, E.; Šebesta, M. Effect of temperature and Soil pH on the sorption of ibuprofen in agricultural soil. Soil Water Res. 2017, 12, 78–85. [Google Scholar] [CrossRef]
- Babić, S.; Horvat, A.J.M.; Mutavdžić Pavlović, D.; Kaštelan-Macan, M. Determination of pKa values of active pharmaceutical ingredients. TrAC - Trends Anal. Chem. 2007, 26, 1043–1061. [Google Scholar] [CrossRef]
- Latawiec, A.E.; Peake, L.; Baxter, H.; Cornelissen, G.; Grotkiewicz, K.; Hale, S.; Królczyk, J.B.; Kubon, M.; Łopatka, A.; Medynska-Juraszek, A.; et al. A reconnaissance-scale GIS-based multicriteria decision analysis to support sustainable biochar use: Poland as a case study. J. Environ. Eng. Landsc. Manag. 2017, 25, 208–222. [Google Scholar] [CrossRef]
- Behera, S.K.; Oh, S.Y.; Park, H.S. Sorptive removal of ibuprofen from water using selected soil minerals and activated carbon. Int. J. Environ. Sci. Technol. 2012, 9, 85–94. [Google Scholar] [CrossRef]
- Xu, Y.; Yu, X.; Xu, B.; Peng, D.; Guo, X. Sorption of pharmaceuticals and personal care products on soil and soil components: Influencing factors and mechanisms. Sci. Total Environ. 2021, 753, 141891. [Google Scholar] [CrossRef]
- Foolad, M.; Hu, J.; Tran, N.H.; Ong, S.L. Sorption and biodegradation characteristics of the selected pharmaceuticals and personal care products onto tropical soil. Water Sci. Technol. 2016, 73, 51–59. [Google Scholar] [CrossRef]
- Biel-Maeso, M.; González-González, C.; Lara-Martín, P.A.; Corada-Fernández, C. Sorption and degradation of contaminants of emerging concern in soils under aerobic and anaerobic conditions. Sci. Total Environ. 2019, 666, 662–671. [Google Scholar] [CrossRef]
- Conkle, J.L.; Gan, J.; Anderson, M.A. Degradation and sorption of commonly detected PPCPs in wetland sediments under aerobic and anaerobic conditions. J. Soils Sediments 2012, 12, 1164–1173. [Google Scholar] [CrossRef]
- Radović, T.T.; Grujić, S.D.; Kovačević, S.R.; Laušević, M.D.; Dimkić, M.A. Sorption of selected pharmaceuticals and pesticides on different river sediments. Environ. Sci. Pollut. Res. 2016, 23, 25232–25244. [Google Scholar] [CrossRef] [PubMed]
- Khazri, H.; Ghorbel-Abid, I.; Kalfat, R.; Trabelsi-Ayadi, M. Removal of ibuprofen, naproxen and carbamazepine in aqueous solution onto natural clay: Equilibrium, kinetics, and thermodynamic study. Appl. Water Sci. 2017, 7, 3031–3040. [Google Scholar] [CrossRef]
- Costi, E.M.; Sicilia, M.D.; Rubio, S. Supramolecular solvents in solid sample microextractions: Application to the determination of residues of oxolinic acid and flumequine in fish and shellfish. J. Chromatogr. A 2010, 1217, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Moral, A.; Sicilia, M.D.; Rubio, S. Supramolecular solvent-based extraction of benzimidazolic fungicides from natural waters prior to their liquid chromatographic/fluorimetric determination. J. Chromatogr. A 2009, 1216, 3740–3745. [Google Scholar] [CrossRef]
- García-Fonseca, S.; Ballesteros-Gómez, A.; Rubio, S.; Pérez-Bendito, D. Supramolecular solvent-based microextraction of ochratoxin A in raw wheat prior to liquid chromatography-fluorescence determination. J. Chromatogr. A 2010, 1217, 2376–2382. [Google Scholar] [CrossRef]
- Costi, E.M.; Sicilia, M.D.; Rubio, S. Multiresidue analysis of sulfonamides in meat by supramolecular solvent microextraction, liquid chromatography and fluorescence detection and method validation according to the 2002/657/EC decision. J. Chromatogr. A 2010, 1217, 6250–6257. [Google Scholar] [CrossRef]
- Rezaee, M.; Assadi, Y.; Milani Hosseini, M.R.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef]
- Chen, H. Antibiotic Sorption and Transport in Porous Media; University of Florida: Gainesville, FL, USA, 2012. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration (µg g−1)/(µg L−1) | Recovery (%) | RSD (%) |
|---|---|---|---|
| SUPRAS procedure | |||
| CBZ | 1.25 | 115 | 7.2 |
| 10 | 96.3 | 4.1 | |
| 20 | 98.8 | 7.3 | |
| DIC | 1.25 | 97.0 | 8.2 |
| 10 | 88.5 | 3.5 | |
| 20 | 90.8 | 1.6 | |
| IBU | 1.25 | 112 | 2.8 |
| 10 | 103 | 0.3 | |
| 20 | 98.4 | 3.2 | |
| Speedisk solid phase extraction procedure | |||
| CBZ | 0.5 | 100 | 1.1 |
| 2 | 97.8 | 2.4 | |
| 4 | 97.3 | 0.6 | |
| DIC | 0.5 | 103 | 2.6 |
| 2 | 106 | 2.0 | |
| 4 | 103 | 1.8 | |
| IBU | 0.5 | 81.0 | 2.9 |
| 2 | 98.5 | 1.8 | |
| 4 | 102 | 5.6 | |
| Compound | Linear Range | Calibration Curve Equation | Sxy | Sa | Sb | R2 a | LOQ b | LOD c |
|---|---|---|---|---|---|---|---|---|
| Bottom sediment samples | ||||||||
| CBZ | 0.5–25.0 µg g−1 | y = 0.4853x + 0.1092 | 0.4635 | 0.0204 | 0.2619 | 0.9912 | 1.25 µg g−1 | 0.42 µg g−1 |
| DIC | y = 0.1196x + 0.0517 | 0.0605 | 0.0027 | 0.0342 | 0.9975 | |||
| IBU | y = 0.0786x + 0.0849 | 0.0413 | 0.0018 | 0.0233 | 0.9973 | |||
| Water samples | ||||||||
| CBZ | 0.05–5.0 µg L−1 | y = 0.6159x − 0.0264 | 0.0191 | 0.0042 | 0.0118 | 0.9998 | 0.05 µg L−1 | 0.017 µg L−1 |
| DIC | y = 0.1571x − 0.0004 | 0.0132 | 0.0029 | 0.0081 | 0.9983 | |||
| IBU | y = 0.2503x + 0.0217 | 0.0317 | 0.0070 | 0.0196 | 0.9961 | |||
| Method | Detection | Compound | LOD/LOQ | Linear Range | Recovery (%) | Volume of Extractant | Extraction/Analysis Time | Ref. |
|---|---|---|---|---|---|---|---|---|
| PLE/SPE-LC | MS/MS | ibuprofen diclofenac carbamazepine | LOD 3.0–30.0 ng g−1 | 0.05–10 µg g−1 | 35–135 | 1 mL | 11 min + SPE/45 min | [9] |
| SLE/SPE-UPLC | MS/MS | ibuprofen diclofenac | - | – | 85–106.5 | 1 mL | 210 min + SPE/– | [10] |
| PLE/SPE-HPLC | MS/MS | diclofenac carbamazepine | LOQ 0.2–2.0 ng g−1 | – | – | 1 mL | 15 min + SPE/– | [11] |
| PLE-HPLC | MS | ibuprofen diclofenac carbamazepine | LOQ 14.0–29.0 ng g−1 | 10–100 µg L−1 | 68–112 | 40 mL | 40 min/25–30 min | [12] |
| USE-SPE-HPLC | DAD | ibuprofen diclofenac carbamazepine | LOQ 1.1–187.0 ng g−1 | – | 85.8–102 | 150 µL | 35.5 min + SPE/– | [13] |
| PLE-SPE-LC USE-SPE-LC | MS/MS | ibuprofen diclofenac carbamazepine | - | – | 40–130 | 1 mL | 15–35 min + SPE/– | [14] |
| USE-SPE-LC | MS/MS | ibuprofen diclofenac carbamazepine | LOQ 20.0 ng g−1 | 10 ng g−1–20 µg g−1 | 44–81 | 200 µL | 10 min + SPE/28 min | [15] |
| PLE-SPE-LC | MS/MS | ibuprofen dikolfenak carbamazepine | MQL 0.78–163.7 ng g−1 | 10 ng g−1–2 µg g−1 | 60–82 | 1 mL | 31 min + SPE/– | [16] |
| SBSE-HF-LPME-LC | MS | ibuprofen diclofenac | - | 0.5–8 mg L−1 | 57–62 | 10 µL | 20–22 h/– | [17] |
| PLE-HF-LPME-LC | MS | ibuprofen diclofenac | MLD 0.4–1.4 ng g−1 | 3.9–4000 ng mL−1 | 101–109 | 25 µL | 176 min/26 min | [18] |
| SUPRAS-UHPLC | UV | ibuprofen diclofenac carbamazepine | LOD 0.42 µg g−1 | 0.5–25 µg g−1 | 88.5–115 | 700 µL | 30 min/7 min | this work |
| Method | Detection | Compound | LOD/LOQ | Linear Range | Recovery (%) | Volume of Extractant | Extraction/Analysis Time | Ref. |
|---|---|---|---|---|---|---|---|---|
| SPE-GC | MS | ibuprofen diclofenac carbamazepine | LOD: 12–32 ng L−1 | 5–50 ng L−1 | 67–80 | 200 µL | SPE/62 min | [35] |
| SPE-GC | MS | ibuprofen diclofenac carbamazepine | LOQ: 0.07–0.09 ng L−1 | – | 70–100 | 50 µL | SPE/38 min | [36] |
| SPE-LC | MS | ibuprofen diclofenac carbamazepine | – | – | 46–97 | 2 mL | SPE/40 min | [37] |
| SPE-LC | MS/MS | ibuprofen diclofenac | LOD: 20 ng L−1 | – | 62–117 | – | – | [38] |
| SPE-GC | MS | ibuprofen diclofenac carbamazepine | LOQ: 2–8 ng L−1 | 0.1–10 ng µL−1 | 77–93 | 100 µL | SPE/32 min | [39] |
| SPE-GC | MS | ibuprofen diclofenac | LOD: 36–38 ng L−1 | 10–2000 ng L−1 | 67–76 | 100 µL | SPE/50.5 min | [40] |
| SPE-HPLC | MS/MS | ibuprofen diclofenac | LOD: 0.14–0.52 µg L−1 | 1–250 µg L−1 | 56–77 | 300 µL | SPE/20–43 min | [41] |
| SPE-GC | MS | ibuprofen dikolfenak carbamazepine | LOQ: 5–10 ng L−1 | 10 ng g−1–2 µg g−1 | 50–99 | 0.5 mL | SPE/– | [42] |
| SPE-GC | MS | ibuprofen diclofenac | ILD: 20–32 µg L−1 | 0.02–25 mg mL−1 | 84–157 | 130 µL | SPE/56 min | [43] |
| SPE-GC | MS | ibuprofen diclofenac carbamazepine | LOD: 0.01–0.02 ng L−1 | 0.06–400 ng L−1 | 92–102 | 105 µL | SPE/– | [44] |
| Speedisk-UHPLC | UV | ibuprofen diclofenac carbamazepine | LOD: 0.17 µg L−1 | 0.05–10 µg L−1 | 81–106 | 500 µL | 60 min/7 min | this work |
| Time (min) | Solvent A (%) a | Solvent B (%) a | Flow Rate (mL min−1) |
|---|---|---|---|
| 0.0 | 40 | 60 | 0.2 |
| 1.0 | 45 | 55 | 0.7 |
| 2.0 | 45 | 55 | 0.6 |
| 3.0 | 45 | 55 | 0.5 |
| 3.5 | 80 | 20 | 0.7 |
| 4.0 | 80 | 20 | 0.7 |
| 4.5 | 85 | 15 | 0.4 |
| 4.9 | 85 | 15 | 0.3 |
| 5.0 | 100 | 0 | 0.7 |
| 7.0 | 100 | 0 | 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajkacz, S.; Adamczewska, P.; Kokoszka, K.; Kycia-Słocka, E.; Sochacki, A.; Felis, E. Supramolecular Solvent-Based Microextraction of Selected Anticonvulsant and Nonsteroidal Anti-Inflammatory Drugs from Sediment Samples. Molecules 2020, 25, 5671. https://doi.org/10.3390/molecules25235671
Bajkacz S, Adamczewska P, Kokoszka K, Kycia-Słocka E, Sochacki A, Felis E. Supramolecular Solvent-Based Microextraction of Selected Anticonvulsant and Nonsteroidal Anti-Inflammatory Drugs from Sediment Samples. Molecules. 2020; 25(23):5671. https://doi.org/10.3390/molecules25235671
Chicago/Turabian StyleBajkacz, Sylwia, Paulina Adamczewska, Klaudia Kokoszka, Elżbieta Kycia-Słocka, Adam Sochacki, and Ewa Felis. 2020. "Supramolecular Solvent-Based Microextraction of Selected Anticonvulsant and Nonsteroidal Anti-Inflammatory Drugs from Sediment Samples" Molecules 25, no. 23: 5671. https://doi.org/10.3390/molecules25235671
APA StyleBajkacz, S., Adamczewska, P., Kokoszka, K., Kycia-Słocka, E., Sochacki, A., & Felis, E. (2020). Supramolecular Solvent-Based Microextraction of Selected Anticonvulsant and Nonsteroidal Anti-Inflammatory Drugs from Sediment Samples. Molecules, 25(23), 5671. https://doi.org/10.3390/molecules25235671