
Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery


Abstract
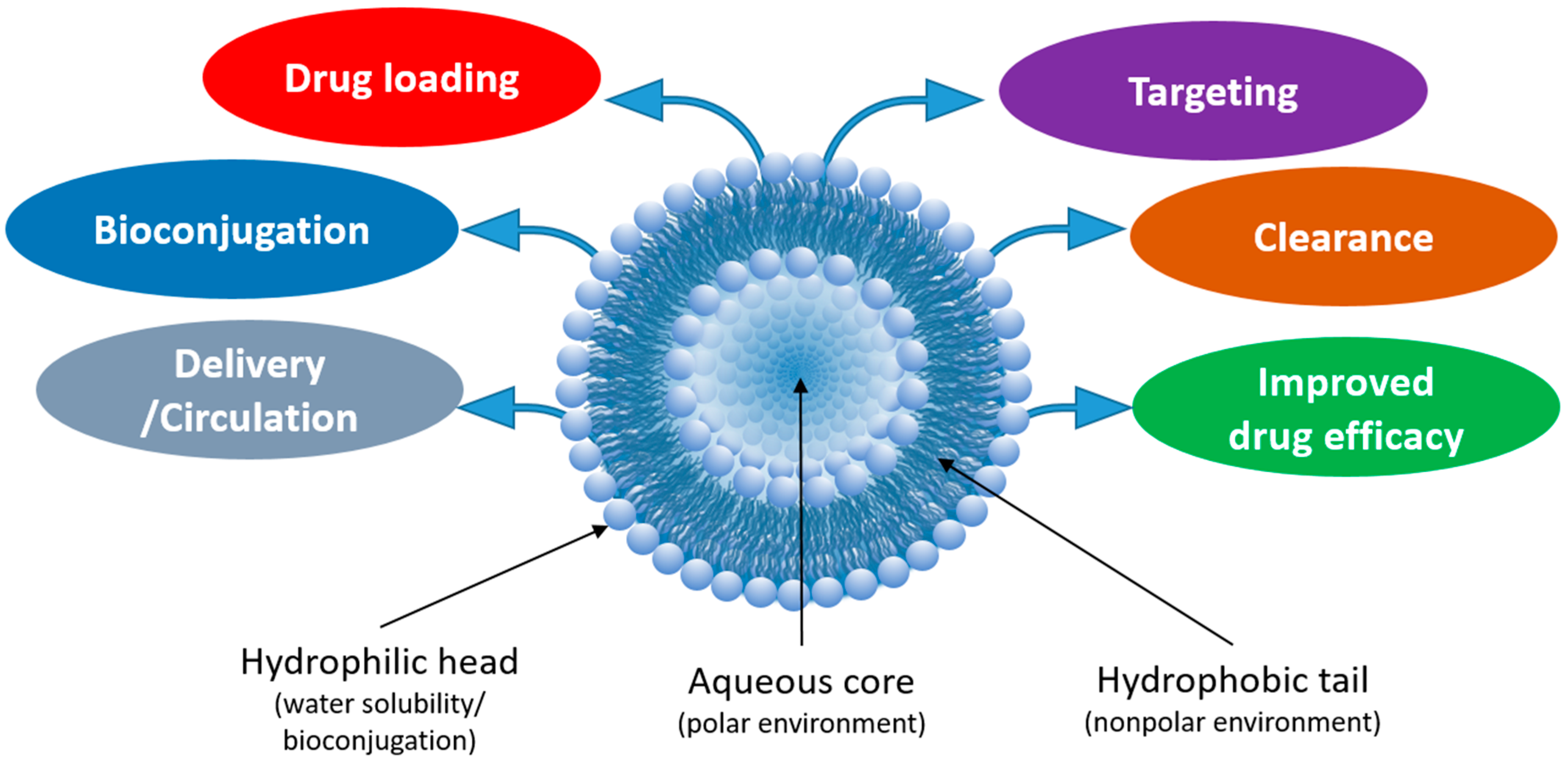
1. Introduction
2. Strategies for Incorporation of Drugs into Liposomal Carriers
3. Strategies for Drug Incorporation and Liposome Bioconjugation/Targeting
3.1. Drug Incorporation during Liposome Synthesis
3.2. Drug Incorporation after Liposome Synthesis

3.3. Drug Incorporation Using Covalent Conjugation
3.4. Drug Incorporation Using a Combination of Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Loading Method | Drug | Targeting Moiety | Liposome Composition | Application | Ref. |
|---|---|---|---|---|---|
| Incorporation during synthesis | Porphyrins (p-NH2, pOH, p-py) | HA (polymer) | DPPC, DOPG, chol-hy, HA | Delivery of photosensitizer porphyrins to CD44+ cells | [68] |
| ND | FA (vitamin) | FA-PEG-DSPE, ND and DSPC, chol, mPEG-DSPE | Treatment of P-glycoprotein+ and FA receptor+ tumors | [69] | |
| Anidulafungin | α-tocopherol (vitamin) | HSPC, PG, chol | Treatment of fungal infection | [70] | |
| DOX | FA (vitamin) | SPC, chol, DSPE-PEG or FA-PEG-DSPE | Comparison of two liposome fabrication methods for DOX | [71] | |
| Goniodiol | N/A | DSPC, PEG-A-DSPE or PEG-P-DSPE | Improved stability and activity of goniodiol for cancer treatment | [72] | |
| 7,8-DHF | N/A | SPC, chol, LF | Effects of crosslinking on drug release | [73] | |
| Calcein, TPS | N/A | DSPC, DOPE, chol, DSPE-PEG2000 | Light-triggered drug release for cancer | [74] | |
| 17β-estradiol | N/A | DPPC, DMPC or POPC, DDAB, chol, DSPE-PEG2000 | Preventing activation of undesired pathways while retaining drug activity | [75] | |
| Essential oils (e.g., estragole, isoeugenol, eucalyptol, pulegone, terpineol, thymol) | N/A | Lipoid S100, chol | Improved shelf life and activity | [76] | |
| KSP siRNA, PTX | N/A | DC-chol, DOPE, mPEG2000-DSPE | Dual-drug delivery for multi-drug resistant ovarian cancer tumors | [77] | |
| DOX | N/A | HSPC, corosolic acid, DSPE-PEG2000 | Increased cancer cell drug uptake and treatment | [78] | |
| DOX | N/A | di-LA-GPC prodrug | Improved liposome stability for cancer | [46] | |
| PTX-BSA | N/A | HEPC, DSPE-PEG2000 | Improved drug encapsulation and antitumor effect | [79] | |
| Curcumin | N/A | Soybean lecithin, chol, chitosan | Increased liposome stability | [80] | |
| Gemcitabine-copper(II) gluconate complex | N/A | DPPC, DSPC, DSPE-PEG2000 | Heat-triggered drug delivery | [81] | |
| RFP, CaO2 | N/A | DSPE-PEG3400, lecithin, lactic acid, stearic acid, PCM | Bacteria toxin-triggered antibiotic release | [82] | |
| Iridium(III) polypyridyl complexes | N/A | PC-98T:CHO-HP, PC-98T:DSPE-mPEG2000 | Improving anticancer efficacy of iridium(III) polypyridyl complexes | [83] | |
| Incorporation after synthesis | DOX | Porphyrins | DSPC, chol, DSPE-PEG, DOPC | Ultrasound-triggered, localized DOX release | [84] |
| Usnic Acid | N/A | Cationic or N-oxide surfactants, DMPC, chol | Improved antioxidant delivery | [48] | |
| Ciprofloxacin | N/A | DSPC, chol, DOPC, porphyrin-phospholipid, mPEG-2000-DSPE | Light-triggered antibiotic treatment | [50] | |
| DOX | N/A | DSPE-PEG2000, nitroimidazole, DPPC, chol, DMPC | Hypoxia-triggered DOX release | [54] | |
| DOX and Irinotecan | N/A | DSPC, chol, mPEG2000-DSPE | Combination treatment for treating cancer | [51] | |
| Sinomenine hydrochloride | N/A | DPPC, SPC, chol | Heat-triggered drug release for rheumatoid arthritis | [53] | |
| Covalent attachment to liposome surface | APL9 peptide | F4/80 (antibody) | PAM3CysSK4-peptide | Modified immune response in type 1 diabetes | [59] |
| GALA peptide | tbFGF lipopeptide | POPC, DPTE-lipopeptides | Endosomal escape and cell targeting | [57] | |
| Camptothecin | N/A | Di-CPT-GPC prodrug | CPT prodrug for anticancer treatment | [55] | |
| PTX | N/A | PTX-ss-PC prodrug, mPEG2000-DSPE, EPC, chol | Reduction-triggered, intracellular delivery | [56] | |
| Artesunate | N/A | Di-ART-GPC | Anti-inflammatory treatment of rheumatoid arthritis | [60] | |
| Combination of drug loading strategies | DOX | HER2 (antibody) | ICG-ODA, DSPE-PEG2000, PLsPC, S100 | Light-triggered drug release and ROS generation for chemotherapy | [52] |
| Calcein | FA (vitamin) | DOTAP, DOPC, AuNPs, VP | X-ray-triggered drug release for radiotherapy and chemotherapy | [64] | |
| DOX, Bcl-2 siRNA | N/A | TPGS or PEG-DSPE, DOTAP, DPPC, chol | Chemotherapy with dual suppression of drug resistance | [62] | |
| Gd-DTPA, DOX | N/A | Gd-DTPA-ONB | MRI-guided liposome drug delivery | [67] | |
| Disulfiram and DOX | N/A | DSPC, chol, mPEG2000-DSPE | Inhibit/reverse multidrug resistance in cancer cells | [63] | |
| N/A | N/A | HER2 (antibody) | DSPE-PEG2000, DPPC, chol | Improved targeting of HER2 cancer cells | [85] |
| N/A | HER2 (antibody) | FcBP, PEG-DSPE | Antibodies to increase targeting affinity | [86] | |
| N/A | CD11c (antibody) | DOPE, EPC, chol, DBCO-PEG | SPAAC modification | [87] |
4. Bioconjugation Strategies for Targeting and Delivery of Liposomes to Cells
4.1. Antibody–Liposome Bioconjugates (Immunoliposomes)
4.2. Aptamer–Liposome Bioconjugates

4.3. Peptide–Liposome Bioconjugates
4.4. Other Small Molecule–Liposome Bioconjugates

5. Clinical Use of Liposomes: Current State of the Art
| Drug Name | Year | Drug Cargo | Application | Trial Phase | ClinicalTrial.Gov ID or [ref] |
|---|---|---|---|---|---|
| Alprostadil | 2019 (2021) | Alprostadil | Peripheral artery disease | Phase II | NCT04197323 |
| Amikacin | 2019 | Amikacin (antibiotic) | Mycobacterium abscesses lung disease | Phase II | NCT03038178 |
| Annamycin | 2018 (2021) | Annamycin | Acute myeloid leukemia | Phase I | NCT03315039 |
| ARB-001467 TKM-HPV | 2018 | Three siRNA targeting HBV RNA | Hepatitis B Virus | Phase II | NCT02631096, [128] |
| Atu027 | 2016 | Atu027 (siRNA) targeting PKN3 (in conjunction with Gemcitabine) | Advanced pancreatic carcinoma | Phase I/II | NCT01808638, [130] |
| Bupivacaine | 2018 (2021) | Bupivacaine | Pain control during colorectal surgery | Phase III | NCT03702621 |
| Cyclosporine A | 2019 (2022) | Cyclosporine A | Bronchiolitis Obliterans, Lung Transplant Rejection | Phase III | NCT03657342 NCT03656926 |
| E7389 | 2017 (2021) | E7389 | Solid tumor therapy (breast cancer, adenoid cystic carcinoma, gastric cancer, esophageal cancer, and small cell lung cancer) | Phase I | NCT03207672, [131] |
| FF-10832 Gemcitabine | 2018 (2021) | Gemcitabine (in conjunction with free Paclitaxel) | Advanced solid tumors | Phase I | NCT03440450 |
| HIV-1 gp41 MPER-656 | 2019 (2021) | HIV-1 gp41 | HIV-1 vaccine | Phase I | NCT03934541 |
| LipocurcTM | 2017 | Curcumin | Advanced cancer (solid tumors) who have failed standard of care therapy | Phase I/II | NCT02138955, [132] |
| ND-L02-s0201 | 2016 | Heat shock protein 47 siRNA | Hepatic fibrosis | Phase I | NCT02227459, [128] |
| Onivyde® | 2015 | Irinotecan, Fluorouracil | Metastatic pancreatic cancer | FDA approved | [121] |
| Onpattro® | 2018 | siRNA (antitransthyretin)/Patrisiran | Transthyretin-mediated amyloidosis | FDA approved | [118,125] |
| ThermoDox® (Tardox) | 2019 (Ph I), 2018 (Ph III) | DOX | Temperature-triggered DOX release; liver cancer (Ph I), hepatocellular carcinoma (Ph III) | Phase I & Phase III | [120,133], NCT02181075 (Ph I) NCT02112656 (Ph III) |
| TLC599 | 2019 (2021) | Dexamethasone | Knee osteoarthritis | Phase III | NCT04123561 |
| Vyxeos® | 2017 | Daunorubicin and Cytarabine | Acute myeloid leukemia | FDA approved | [118,119,121] |
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent advancements in liposome technology. Adv. Drug Deliver. Rev. 2020. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-based medicines: A review of FDA-approved materials and clinical trials to date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Mufamadi, M.S.; Pillay, V.; Choonara, Y.E.; Du Toit, L.C.; Modi, G.; Naidoo, D.; Ndesendo, V.M.K. A review on composite liposomal technologies for specialized drug delivery. J. Drug Deliv. 2011, 2011, 939851. [Google Scholar] [CrossRef] [PubMed]
- Nag, O.K.; Awasthi, V. Surface engineering of liposomes for stealth behavior. Pharmaceutics 2013, 5, 542–569. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, P.; Pal, P.; Das, A.K.; Ray, S.; Bhattacharjee, A.; Mazumder, B. Nano lipid-drug conjugate: An integrated review. Int. J. Pharm. 2017, 529, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Abri Aghdam, M.; Bagheri, R.; Mosafer, J.; Baradaran, B.; Hashemzaei, M.; Baghbanzadeh, A.; de la Guardia, M.; Mokhtarzadeh, A. Recent advances on thermosensitive and pH-sensitive liposomes employed in controlled release. J. Control. Release 2019, 315, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal drug delivery systems: An update review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef]
- Lagner, M.; Kral, T.E. Liposome-based drug delivery systems. Pol. J. Pharmacol. 1999, 51, 211–222. [Google Scholar]
- Zununi Vahed, S.; Salehi, R.; Davaran, S.; Sharifi, S. Liposome-based drug co-delivery systems in cancer cells. Mater. Sci. Eng. C 2017, 71, 1327–1341. [Google Scholar] [CrossRef]
- Mohammed, A.R.; Weston, N.; Coombes, A.G.A.; Fitzgerald, M.; Perrie, Y. Liposome formulation of poorly water soluble drugs: Optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 2004, 285, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Adv. Pharm. Bull. 2017, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Cagdas, M.; Sezer, A.D.; Bucak, S. Liposomes as potential drug carrier systems for drug delivery. In Application of Nanotechnology in Drug Delivery; Sezer, A.D., Ed.; IntechOpen Limited: London, UK, 2014. [Google Scholar] [CrossRef]
- Maurer, N.; Fenske, D.B.; Cullis, P.R. Developments in liposomal drug delivery systems. Expert Opin. Biol. Ther. 2001, 1, 923–947. [Google Scholar] [CrossRef]
- Kim, J.S. Liposomal drug delivery system. J. Pharm. Investig. 2016, 46, 387–392. [Google Scholar] [CrossRef]
- Minko, T.; Pakunlu, R.I.; Wang, Y.; Khandare, J.J.; Saad, M. New generation of liposomal drugs for cancer. Anticancer Agents Med. Chem. 2006, 6, 537–552. [Google Scholar] [CrossRef]
- Allen, T.M.; Martin, F.J. Advantages of liposomal delivery systems for anthracyclines. Semin. Oncol. 2004, 31, 5–15. [Google Scholar] [CrossRef]
- Vemuri, S.; Rhodes, C.T. Preparation and characterization of liposomes as therapeutic delivery systems: A review. Pharm. Acta Helv. 1995, 70, 95–111. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Ishida, T. Liposomal Delivery Systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Nag, O.K.; Yadav, V.R.; Croft, B.; Hedrick, A.; Awasthi, V. Liposomes modified with superhydrophilic polymer linked to a nonphospholipid anchor exhibit reduced complement activation and enhanced circulation. J. Pharm. Sci. 2015, 104, 114–123. [Google Scholar] [CrossRef]
- Alyane, M.; Barratt, G.; Lahouel, M. Remote loading of doxorubicin into liposomes by transmembrane pH gradient to reduce toxicity toward H9c2 cells. Saudi Pharm. J. 2016, 24, 165–175. [Google Scholar] [CrossRef]
- Bolotin, E.M.; Cohen, R.; Bar, L.K.; Emanuel, N.; Ninio, S.; Barenholz, Y.; Lasic, D.D. Ammonium sulfate gradients for efficient and stable remote loading of amphipathic weak bases into liposomes and ligandoliposomes. J. Liposome Res. 1994, 4, 455–479. [Google Scholar] [CrossRef]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef]
- Yang, W.; Yang, Z.; Fu, J.; Guo, M.; Sun, B.; Wei, W.; Liu, D.; Liu, H. The influence of trapping agents on the antitumor efficacy of irinotecan liposomes: Head-to-head comparison of ammonium sulfate, sulfobutylether-β-cyclodextrin and sucrose octasulfate. Biomater. Sci. 2019, 7, 419–428. [Google Scholar] [CrossRef]
- Sur, S.; Fries, A.C.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Remote loading of preencapsulated drugs into stealth liposomes. Proc. Natl. Acad. Sci. USA 2014, 111, 2283–2288. [Google Scholar] [CrossRef]
- Pauli, G.; Tang, W.L.; Li, S.D. Development and characterization of the solvent-assisted active loading technology (SALT) for liposomal loading of poorly water-soluble compounds. Pharmaceutics 2019, 11, 465. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, X.; Liu, Y.; Lin, Z.Y.; Liu, B.; Liu, J. Profiling metal oxides with lipids: Magnetic liposomal nanoparticles displaying DNA and proteins. Angew. Chem. Int. Ed. 2016, 55, 12063–12067. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J. Leakage and rupture of lipid membranes by charged polymers and nanoparticles. Langmuir 2020, 36, 810–818. [Google Scholar] [CrossRef]
- Al-Ahmady, Z.; Lozano, N.; Mei, K.C.; Al-Jamal, W.T.; Kostarelos, K. Engineering thermosensitive liposome-nanoparticle hybrids loaded with doxorubicin for heat-triggered drug release. Int. J. Pharm. 2016, 514, 133–141. [Google Scholar] [CrossRef]
- Kwon, H.J.; Byeon, Y.; Jeon, H.N.; Cho, S.H.; Han, H.D.; Shin, B.C. Gold cluster-labeled thermosensitive liposmes enhance triggered drug release in the tumor microenvironment by a photothermal effect. J. Control. Release 2015, 216, 132–139. [Google Scholar] [CrossRef]
- Singh, S.P.; Alvi, S.B.; Pemmaraju, D.B.; Singh, A.D.; Manda, S.V.; Srivastava, R.; Rengan, A.K. NIR triggered liposome gold nanoparticles entrapping curcumin as in situ adjuvant for photothermal treatment of skin cancer. Int. J. Biol. Macromol. 2018, 110, 375–382. [Google Scholar] [CrossRef]
- Zhan, C.; Wang, W.; McAlvin, J.B.; Guo, S.; Timko, B.P.; Santamaria, C.; Kohane, D.S. Phototriggered local anesthesia. Nano Lett. 2016, 16, 177–181. [Google Scholar] [CrossRef]
- Mathiyazhakan, M.; Yang, Y.; Liu, Y.; Zhu, C.; Liu, Q.; Ohl, C.D.; Tam, K.C.; Gao, Y.; Xu, C. Non-invasive controlled release from gold nanoparticle integrated photo-responsive liposomes through pulse laser induced microbubble cavitation. Colloid. Surface. B 2015, 126, 569–574. [Google Scholar] [CrossRef]
- Preiss, M.R.; Hart, A.; Kitchens, C.; Bothun, G.D. Hydrophobic nanoparticles modify the thermal release behavior of liposomes. J. Phys. Chem. B 2017, 121, 5040–5047. [Google Scholar] [CrossRef]
- Guo, H.; Kim, J.C. Photothermally induced release from liposome suspended in mixture solution of gold nanoparticle and thermo-sensitive polymer. Colloid. Surface. A 2015, 469, 73–82. [Google Scholar] [CrossRef]
- Sahu, A.; Kim, M.; Ryu, J.; Son, J.G.; Lee, E.; Noh, D.Y.; Tae, G. Nanographene oxide as a switch for CW/pulsed NIR laser triggered drug release from liposomes. Mater. Sci. Eng. C 2018, 82, 19–24. [Google Scholar] [CrossRef]
- Tripathy, N.; Ahmad, R.; Ko, H.A.; Khang, G.; Hahn, Y.B. Enhanced anticancer potency using an acid-responsive ZnO-incorporated liposomal drug-delivery system. Nanoscale 2015, 7, 4088–4096. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, F.; Steffen, K.J.; Mallik, S. Enzyme-responsive liposomes for the delivery of anticancer drugs. Bioconj. Chem. 2017, 28, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Parham Sahandi, Z.; Soroush, M.; Shayan, S.; Behrad, M.; Maryam, M.; Hamid, H.; Zahra, J.; Yasamin Davatgaran, T.; Hura, H.; Mahdi, K.; et al. Stimulus-responsive liposomes as smart nanoplatforms for drug delivery applications. Nanotech. Rev. 2018, 7, 95–122. [Google Scholar]
- Jiang, L.; Li, L.; He, X.; Yi, Q.; He, B.; Cao, J.; Pan, W.; Gu, Z. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials 2015, 52, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Ling, L.; Ismail, M.; Du, Y.; Yao, C.; Li, X. Lipoic acid-derived cross-linked liposomes for reduction-responsive delivery of anticancer drug. Int. J. Pharm. 2019, 560, 246–260. [Google Scholar] [CrossRef]
- Plourde, K.; Derbali, R.M.; Desrosiers, A.; Dubath, C.; Vallée-Bélisle, A.; Leblond, J. Aptamer-based liposomes improve specific drug loading and release. J. Control. Release 2017, 251, 82–91. [Google Scholar] [CrossRef]
- Battista, S.; Campitelli, P.; Galantini, L.; Köber, M.; Vargas-Nadal, G.; Ventosa, N.; Giansanti, L. Use of N-oxide and cationic surfactants to enhance antioxidant properties of (+)-usnic acid loaded liposomes. Colloid. Surface. A 2020, 585, 124154. [Google Scholar] [CrossRef]
- Song, S.J.; Lee, S.; Lee, Y.; Choi, J.S. Enzyme-responsive destabilization of stabilized plasmid-lipid nanoparticles as an efficient gene delivery. Eur. J. Pharm. Sci. 2016, 91, 20–30. [Google Scholar] [CrossRef]
- Ghosh, S.; Qi, R.; Carter, K.A.; Zhang, G.; Pfeifer, B.A.; Lovell, J.F. Loading and releasing ciprofloxacin in photoactivatable liposomes. Biochem. Eng. J. 2019, 141, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chi, D.; Pan, S.; Zhao, L.; Wang, X.; Wang, D.; Wang, Y. Effective co-encapsulation of doxorubicin and irinotecan for synergistic therapy using liposomes prepared with triethylammonium sucrose octasulfate as drug trapping agent. Int. J. Pharm. 2019, 557, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, W.; Di, H.; Luo, L.; Zhu, C.; Yang, J.; Yin, X.; Yin, H.; Gao, J.; Du, Y.; et al. A photosensitive liposome with NIR light triggered doxorubicin release as a combined photodynamic-chemo therapy system. J. Control. Release 2018, 277, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Zhang, X.; Qi, J.; Shu, G.; Du, Y.; Ying, X. Sinomenine hydrochloride loaded thermosensitive liposomes combined with microwave hyperthermia for the treatment of rheumatoid arthritis. Int. J. Pharm. 2020, 576, 119001. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, A.; Long, M.; Cui, L.; Chen, Z.; Zhu, L. Nitroimidazole derivative incorporated liposomes for hypoxia-triggered drug delivery and enhanced therapeutic efficacy in patient-derived tumor xenografts. Acta Biomater. 2019, 83, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Hou, Y.; Ling, L.; Wang, D.; Ismail, M.; Du, Y.; Zhang, Y.; Yao, C.; Li, X. Dimeric camptothecin derived phospholipid assembled liposomes with high drug loading for cancer therapy. Colloid. Surface. B 2018, 166, 235–244. [Google Scholar] [CrossRef]
- Wang, Z.; Ling, L.; Du, Y.; Yao, C.; Li, X. Reduction responsive liposomes based on paclitaxel-ss-lysophospholipid with high drug loading for intracellular delivery. Int. J. Pharm. 2019, 564, 244–255. [Google Scholar] [CrossRef]
- de la Fuente-Herreruela, D.; Monnappa, A.K.; Muñoz-Úbeda, M.; Morallón-Piña, A.; Enciso, E.; Sánchez, L.; Giusti, F.; Natale, P.; López-Montero, I. Lipid–peptide bioconjugation through pyridyl disulfide reaction chemistry and its application in cell targeting and drug delivery. J. Nanobiotechnology 2019, 17, 77. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, W.; He, R.; Ismail, M.; Ling, L.; Yao, C.; Fu, Z.; Li, X. Dual 7-ethyl-10-hydroxycamptothecin conjugated phospholipid prodrug assembled liposomes with in vitro anticancer effects. Bioorg. Med. Chem. 2017, 25, 3247–3258. [Google Scholar] [CrossRef]
- Nandedkar-Kulkarni, N.; Vartak, A.R.; Sucheck, S.J.; Wall, K.A.; Quinn, A.; Morran, M.P.; McInerney, M.F. Development of a bioconjugate platform for modifying the immune response of autoreactive cytotoxic T lymphocytes involved in type 1 diabetes. Bioconjug. Chem. 2019, 30, 2049–2059. [Google Scholar] [CrossRef]
- Zhang, Y.; He, W.; Du, Y.; Du, Y.; Zhao, C.; Zhang, Y.; Zhang, H.; Yin, L.; Li, X. Dimeric artesunate phospholipid-conjugated liposomes as promising anti-inflammatory therapy for rheumatoid arthritis. Int. J. Pharm. 2020, 579, 119178. [Google Scholar] [CrossRef]
- Signorell, R.D.; Luciani, P.; Brambilla, D.; Leroux, J.C. Pharmacokinetics of lipid-drug conjugates loaded into liposomes. Eur. J. Pharm. Biopharm. 2018, 128, 188–199. [Google Scholar] [CrossRef]
- Li, Y.; Tan, X.; Liu, X.; Liu, L.; Fang, Y.; Rao, R.; Ren, Y.; Yang, X.; Liu, W. Enhanced anticancer effect of doxorubicin by TPGS-coated liposomes with Bcl-2 siRNA-corona for dual suppression of drug resistance. Asian J. Pharm. Sci. 2019. [Google Scholar] [CrossRef]
- Rolle, F.; Bincoletto, V.; Gazzano, E.; Rolando, B.; Lollo, G.; Stella, B.; Riganti, C.; Arpicco, S. Coencapsulation of disulfiram and doxorubicin in liposomes strongly reverses multidrug resistance in breast cancer cells. Int. J. Pharm. 2020, 580, 119191. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Chen, W.; Clement, S.; Guller, A.; Zhao, Z.; Engel, A.; Goldys, E.M. Controlled gene and drug release from a liposomal delivery platform triggered by X-ray radiation. Nat. Commun. 2018, 9, 2713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chen, H.; Liu, A.Y.; Shen, J.J.; Shah, V.; Zhang, C.; Hong, J.; Ding, Y. Gold conjugate-based liposomes with hybrid cluster bomb structure for liver cancer therapy. Biomaterials 2016, 74, 280–291. [Google Scholar] [CrossRef]
- Salvatore, A.; Montis, C.; Berti, D.; Baglioni, P. Multifunctional magnetoliposomes for sequential controlled release. ACS Nano 2016, 10, 7749–7760. [Google Scholar] [CrossRef]
- Liu, C.; Ewert, K.K.; Wang, N.; Li, Y.; Safinya, C.R.; Qiao, W. A multifunctional lipid that forms contrast-agent liposomes with dual-control release capabilities for precise MRI-guided drug delivery. Biomaterials 2019, 221, 119412. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wang, J.; Song, H.; Zhuo, L.G.; Wang, G.; Liao, W.; Feng, Y.; Wei, H.; Chen, Y.; Yang, Y. Uptake and light-induced cytotoxicity of hyaluronic acid-grafted liposomes containing porphyrin in tumor cells. J. Drug Deliv. Sci. Technol. 2018, 47, 137–143. [Google Scholar] [CrossRef]
- Gazzano, E.; Rolando, B.; Chegaev, K.; Salaroglio, I.C.; Kopecka, J.; Pedrini, I.; Saponara, S.; Sorge, M.; Buondonno, I.; Stella, B. Folate-targeted liposomal nitrooxy-doxorubicin: An effective tool against P-glycoprotein-positive and folate receptor-positive tumors. J. Control. Release 2018, 270, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Vera-González, N.; Bailey-Hytholt, C.M.; Langlois, L.; de Camargo Ribeiro, F.; de Souza Santos, E.L.; Junqueira, J.C.; Shukla, A. Anidulafungin liposome nanoparticles exhibit antifungal activity against planktonic and biofilm Candida albicans. J. Biomed. Mat. Res. A 2020, 108, 2263–2276. [Google Scholar] [CrossRef]
- Bi, D.; Zhao, L.; Li, H.; Guo, Y.; Wang, X.; Han, M. A comparative study of polydopamine modified and conventional chemical synthesis method in doxorubicin liposomes form the aspect of tumor targeted therapy. Int. J. Pharm. 2019, 559, 76–85. [Google Scholar] [CrossRef]
- Charoensit, P.; Pompimon, W.; Khorana, N.; Sungthongjeen, S. Effect of amide linkage of PEG-lipid conjugates on the stability and cytotoxic activity of goniodiol loaded in PEGylated liposomes. J. Drug Deliv. Sci. Technol. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, G.; Zhao, Z.; Xue, F.; Gu, Y.; Chen, C.; Zhang, Y. 7,8-Dihydroxyflavone nano-liposomes decorated by crosslinked and glycosylated lactoferrin: Storage stability, antioxidant activity, in vitro release, gastrointestinal digestion and transport in Caco-2 cell monolayers. J. Funct. Foods 2020, 65, 103742. [Google Scholar] [CrossRef]
- Fuse, T.; Tagami, T.; Tane, M.; Ozeki, T. Effective light-triggered contents release from helper lipid-incorporated liposomes co-encapsulating gemcitabine and a water-soluble photosensitizer. Int. J. Pharm. 2018, 540, 50–56. [Google Scholar] [CrossRef]
- Gallez, A.; Palazzo, C.; Blacher, S.; Tskitishvili, E.; Noël, A.; Foidart, J.-M.; Evrard, B.; Pequeux, C.; Piel, G. Liposomes and drug-in-cyclodextrin-in-liposomes formulations encapsulating 17β-estradiol: An innovative drug delivery system that prevents the activation of the membrane-initiated steroid signaling (MISS) of estrogen receptor α. Int. J. Pharm. 2020, 573, 118861. [Google Scholar] [CrossRef]
- Hammoud, Z.; Gharib, R.; Fourmentin, S.; Elaissari, A.; Greige-Gerges, H. Drug-in-hydroxypropyl-β-cyclodextrin-in-lipoid S100/cholesterol liposomes: Effect of the characteristics of essential oil components on their encapsulation and release. Int. J. Pharm. 2020, 579, 119151. [Google Scholar] [CrossRef]
- Lee, J.; Cho, Y.J.; Lee, J.W.; Ahn, H.J. KSP siRNA/paclitaxel-loaded PEGylated cationic liposomes for overcoming resistance to KSP inhibitors: Synergistic antitumor effects in drug-resistant ovarian cancer. J. Control. Release 2020, 321, 184–197. [Google Scholar] [CrossRef]
- Li, X.; Widjaya, A.S.; Liu, J.; Liu, X.; Long, Z.; Jiang, Y. Cell-penetrating corosolic acid liposome as a functional carrier for delivering chemotherapeutic drugs. Acta Biomater. 2020, 106, 301–313. [Google Scholar] [CrossRef]
- Okamoto, Y.; Taguchi, K.; Imoto, S.; Giam Chuang, V.T.; Yamasaki, K.; Otagiri, M. Cell uptake and anti-tumor effect of liposomes containing encapsulated paclitaxel-bound albumin against breast cancer cells in 2D and 3D cultured models. J. Drug Deliv. Sci. Technol. 2020, 55, 101381. [Google Scholar] [CrossRef]
- Tai, K.; Rappolt, M.; Mao, L.; Gao, Y.; Li, X.; Yuan, F. The stabilization and release performances of curcumin-loaded liposomes coated by high and low molecular weight chitosan. Food Hydrocoll. 2020, 99, 105355. [Google Scholar] [CrossRef]
- Tucci, S.T.; Kheirolomoom, A.; Ingham, E.S.; Mahakian, L.M.; Tam, S.M.; Foiret, J.; Hubbard, N.E.; Borowsky, A.D.; Baikoghli, M.; Cheng, R.H.; et al. Tumor-specific delivery of gemcitabine with activatable liposomes. J. Control. Release 2019, 309, 277–288. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Z.; Wang, H.; Han, H. Endogenous stimulus-powered antibiotic release from nanoreactors for a combination therapy of bacterial infections. Nat. Commun. 2019, 10, 4464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Du, F.; He, M.; Bai, L.; Gu, Y.Y.; Yang, L.L.; Liu, Y.J. Studies of anticancer activity in vitro and in vivo of iridium(III) polypyridyl complexes-loaded liposomes as drug delivery system. Eur. J. Med. Chem. 2019, 178, 390–400. [Google Scholar] [CrossRef]
- Wang, X.; Yan, F.; Liu, X.; Wang, P.; Shao, S.; Sun, Y.; Sheng, Z.; Liu, Q.; Lovell, J.F.; Zheng, H. Enhanced drug delivery using sonoactivatable liposomes with membrane-embedded porphyrins. J. Control. Release 2018, 286, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Nikkhoi, S.K.; Rahbarizadeh, F.; Ranjbar, S.; Khaleghi, S.; Farasat, A. Liposomal nanoparticle armed with bivalent bispecific single-domain antibodies, novel weapon in HER2 positive cancerous cell lines targeting. Mol. Immunol. 2018, 96, 98–109. [Google Scholar] [CrossRef]
- Shim, G.; Kim, D.; Lee, S.; Chang, R.S.; Byun, J.; Oh, Y.K. Staphylococcus aureus-mimetic control of antibody orientation on nanoparticles. Nanomed.-Nanotechnol. 2019, 16, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Gai, M.; Simon, J.; Lieberwirth, I.; Mailänder, V.; Morsbach, S.; Landfester, K. A bio-orthogonal functionalization strategy for site-specific coupling of antibodies on vesicle surfaces after self-assembly. Polym. Chem. 2020, 11, 527–540. [Google Scholar] [CrossRef]
- Maeda, H. Macromolecular therapeutics in cancer treatment: The EPR effect and beyond. J. Control. Release 2012, 164, 138–144. [Google Scholar] [CrossRef]
- Nogueira, E.; Gomes, A.C.; Preto, A.; Cavaco-Paulo, A. Design of liposomal formulations for cell targeting. Colloid. Surface. B 2015, 136, 514–526. [Google Scholar] [CrossRef]
- Khan, A.A.; Allemailem, K.S.; Almatroodi, S.A.; Almatroudi, A.; Rahmani, A.H. Recent strategies towards the surface modification of liposomes: An innovative approach for different clinical applications. 3 Biotech 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Eloy, J.O.; Petrilli, R.; Trevizan, L.N.F.; Chorilli, M. Immunoliposomes: A review on functionalization strategies and targets for drug delivery. Colloid. Surface. B 2017, 159, 454–467. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Van Vught, R.; Oliveira, S.; Roovers, R.; en Henegouwen, P.v.B.; Pieters, R.; Van Gulik, T.; Breukink, E.; Heger, M. Site-specific conjugation of single domain antibodies to liposomes enhances photosensitizer uptake and photodynamic therapy efficacy. Nanoscale 2016, 8, 6490–6494. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, M.; Kim, B.; Auguste, D.T. Incorporating gold nanoclusters and target-directed liposomes as a synergistic amplified colorimetric sensor for HER2-positive breast cancer cell detection. Theranostics 2017, 7, 899. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Thapa, R.K.; Shin, B.S.; Jeong, J.H.; Kim, J.R.; Yong, C.S.; Kim, J.O. CD9 monoclonal antibody-conjugated PEGylated liposomes for targeted delivery of rapamycin in the treatment of cellular senescence. Nanotechnology 2017, 28, 095101. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Contreras, O.A.; Brenner, J.S.; Kiseleva, R.Y.; Zuluaga-Ramirez, V.; Greineder, C.F.; Villa, C.H.; Hood, E.D.; Myerson, J.W.; Muro, S.; Persidsky, Y. Combining vascular targeting and the local first pass provides 100-fold higher uptake of ICAM-1-targeted vs. untargeted nanocarriers in the inflamed brain. J. Control. Release 2019, 301, 54–61. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Moosavian, S.A.; Sahebkar, A. Aptamer-functionalized liposomes for targeted cancer therapy. Cancer Lett. 2019, 448, 144–154. [Google Scholar] [CrossRef]
- Moosavian, S.A.; Abnous, K.; Badiee, A.; Jaafari, M.R. Improvement in the drug delivery and anti-tumor efficacy of PEGylated liposomal doxorubicin by targeting RNA aptamers in mice bearing breast tumor model. Colloid. Surface. B 2016, 139, 228–236. [Google Scholar] [CrossRef]
- Dou, X.Q.; Wang, H.; Zhang, J.; Wang, F.; Xu, G.L.; Xu, C.C.; Xu, H.H.; Xiang, S.S.; Fu, J.; Song, H.F. Aptamer–drug conjugate: Targeted delivery of doxorubicin in a HER3 aptamer-functionalized liposomal delivery system reduces cardiotoxicity. Int. J. Nanomed. 2018, 13, 763. [Google Scholar] [CrossRef]
- Kim, D.M.; Kim, M.; Park, H.B.; Kim, K.S.; Kim, D.E. Anti-MUC1/CD44 Dual-aptamer-conjugated liposomes for cotargeting breast cancer cells and cancer stem cells. ACS Appl. Bio. Mater. 2019, 2, 4622–4633. [Google Scholar] [CrossRef]
- Li, F.; Mei, H.; Xie, X.; Zhang, H.; Liu, J.; Lv, T.; Nie, H.; Gao, Y.; Jia, L. Aptamer-conjugated chitosan-anchored liposomal complexes for targeted delivery of erlotinib to EGFR-mutated lung cancer cells. AAPS J. 2017, 19, 814–826. [Google Scholar] [CrossRef]
- Zhen, S.; Takahashi, Y.; Narita, S.; Yang, Y.C.; Li, X. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget 2017, 8, 9375. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Jeong, H.Y.; Kang, S.J.; Choi, M.J.; You, Y.M.; Im, C.S.; Lee, T.S.; Song, I.H.; Lee, C.G.; Rhee, K.J. Cancer-targeted nucleic acid delivery and quantum dot imaging using EGF receptor aptamer-conjugated lipid nanoparticles. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.X.; Chuang, E.Y.; Lin, C.C.; Ho, Y.C.; Lin, K.J.; Cheng, P.Y.; Chen, K.J.; Wei, H.J.; Sung, H.W. An AS1411 aptamer-conjugated liposomal system containing a bubble-generating agent for tumor-specific chemotherapy that overcomes multidrug resistance. J. Control. Release 2015, 208, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Xie, X.; Wang, Z.; Gong, W.; Zhang, H.; Li, Y.; Yu, F.; Li, Z.; Mei, X. Dual-modified liposomes with a two-photon-sensitive cell penetrating peptide and NGR ligand for siRNA targeting delivery. Biomaterials 2015, 48, 84–96. [Google Scholar] [CrossRef]
- Mousavizadeh, A.; Jabbari, A.; Akrami, M.; Bardania, H. Cell targeting peptides as smart ligands for targeting of therapeutic or diagnostic agents: A systematic review. Colloid. Surface. B 2017, 158, 507–517. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, L.; Zhang, L.; Shi, K.; Cun, X.; Yang, Y.; Liu, Y.; Gao, H.; He, Q. Dual-functionalized liposomal delivery system for solid tumors based on RGD and a pH-responsive antimicrobial peptide. Sci. Rep. 2016, 6, 19800. [Google Scholar] [CrossRef]
- Cheng, Y.; Ji, Y. RGD-modified polymer and liposome nanovehicles: Recent research progress for drug delivery in cancer therapeutics. Eur. J. Pharm. Sci. 2019, 128, 8–17. [Google Scholar] [CrossRef]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface functionalization and targeting strategies of liposomes in solid tumor therapy: A review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef]
- Shefrin, S.; Vijayan, V.; Nair, S.C. Enzymosomes: A rising effectual tool for targeted drug delivery system. Int. J. App. Pharm. 2017, 9, 1. [Google Scholar]
- Chatzikleanthous, D.; Schmidt, S.T.; Buffi, G.; Paciello, I.; Cunliffe, R.; Carboni, F.; Romano, M.R.; O’Hagan, D.T.; D’Oro, U.; Woods, S. Design of a novel vaccine nanotechnology-based delivery system comprising CpGODN-protein conjugate anchored to liposomes. J. Control. Release 2020, 323, 125–137. [Google Scholar] [CrossRef]
- Park, S.H.; Yoon, Y.I.; Moon, H.; Lee, G.H.; Lee, B.-H.; Yoon, T.J.; Lee, H.J. Development of a novel microbubble-liposome complex conjugated with peptide ligands targeting IL4R on brain tumor cells. Oncol. Rep. 2016, 36, 131–136. [Google Scholar] [CrossRef]
- Gouveia, V.M.; Lopes-de-Araújo, J.; Costa Lima, S.A.; Nunes, C.; Reis, S. Hyaluronic acid-conjugated pH-sensitive liposomes for targeted delivery of prednisolone on rheumatoid arthritis therapy. Nanomedicine 2018, 13, 1037–1049. [Google Scholar] [CrossRef]
- Chi, Y.; Yin, X.; Sun, K.; Feng, S.; Liu, J.; Chen, D.; Guo, C.; Wu, Z. Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J. Control. Release 2017, 261, 113–125. [Google Scholar] [CrossRef]
- Chen, J.; Son, H.N.; Hill, J.J.; Srinivasan, S.; Su, F.Y.; Stayton, P.S.; Convertine, A.J.; Ratner, D.M. Nanostructured glycopolymer augmented liposomes to elucidate carbohydrate-mediated targeting. Nanomed.-Nanotechnol. 2016, 12, 2031–2041. [Google Scholar] [CrossRef]
- Li, X.; Rao, X.; Cai, L.; Liu, X.; Wang, H.; Wu, W.; Zhu, C.; Chen, M.; Wang, P.G.; Yi, W. Targeting tumor cells by natural anti-carbohydrate antibodies using rhamnose-functionalized liposomes. ACS Chem. Biol. 2016, 11, 1205–1209. [Google Scholar] [CrossRef]
- Patil, Y.; Shmeeda, H.; Amitay, Y.; Ohana, P.; Kumar, S.; Gabizon, A. Targeting of folate-conjugated liposomes with co-entrapped drugs to prostate cancer cells via prostate-specific membrane antigen (PSMA). Nanomed.-Nanotechnol. 2018, 14, 1407–1416. [Google Scholar] [CrossRef]
- Beltrán-Gracia, E.; López-Camacho, A.; Higuera-Ciapara, I.; Velázquez-Fernández, J.B.; Vallejo-Cardona, A.A. Nanomedicine review: Clinical developments in liposomal applications. Cancer Nanotechnol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742. [Google Scholar]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef]
- Gonda, A.; Zhao, N.; Shah, J.V.; Calvelli, H.R.; Kantamneni, H.; Francis, N.L.; Ganapathy, V. Engineering tumor-targeting nanoparticles as vehicles for precision nanomedicine. Med One 2019, 4, e190021. [Google Scholar]
- Zhang, H. Onivyde for the therapy of multiple solid tumors. Onco. Targets Ther. 2016, 9, 3001. [Google Scholar] [CrossRef]
- Dicko, A.; Kwak, S.; Frazier, A.A.; Mayer, L.D.; Liboiron, B.D. Biophysical characterization of a liposomal formulation of cytarabine and daunorubicin. Int. J. Pharm. 2010, 391, 248–259. [Google Scholar] [CrossRef]
- Blair, H.A. Daunorubicin/cytarabine liposome: A review in acute myeloid leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Urits, I.; Swanson, D.; Swett, M.C.; Patel, A.; Berardino, K.; Amgalan, A.; Berger, A.A.; Kassem, H.; Kaye, A.; Viswanath, O. A review of patisiran (ONPATTRO®) for the treatment of polyneuropathy in people with hereditary transthyretin amyloidosis. Neurol. Ther. 2020, 9, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Greil, R.; Greil-Ressler, S.; Weiss, L.; Schönlieb, C.; Magnes, T.; Radl, B.; Bolger, G.T.; Vcelar, B.; Sordillo, P.P. A phase 1 dose-escalation study on the safety, tolerability and activity of liposomal curcumin (Lipocurc™) in patients with locally advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Barba, A.A.; Bochicchio, S.; Dalmoro, A.; Lamberti, G. Lipid delivery systems for nucleic-acid-based-drugs: From production to clinical applications. Pharmaceutics 2019, 11, 360. [Google Scholar] [CrossRef]
- Xia, Y.; Tian, J.; Chen, X. Effect of surface properties on liposomal siRNA delivery. Biomaterials 2016, 79, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Strumberg, D.; Kuhlmann, J.; Wolf, M.; Link, K.; Seufferlein, T.; Kaufmann, J.; Gebhardt, F.; Bruyniks, N.; Pelzer, U. A phase Ib/IIa study of combination therapy with gemcitabine and Atu027 in patients with locally advanced or metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2016, 34, 385. [Google Scholar] [CrossRef]
- Yamamoto, N.; Sato, J.; Koyama, T.; Iwasa, S.; Shimomura, A.; Kondo, S.; Kitano, S.; Yonemori, K.; Fujiwara, Y.; Tamura, K.; et al. Phase I study of liposomal formulation of eribulin (E7389-LF) in patients (pts) with advanced solid tumours: Primary results of dose-escalation part. Ann. Oncol. 2019, 30, v125. [Google Scholar] [CrossRef]
- Eskandari, Z.; Bahadori, F.; Celik, B.; Onyuksel, H. Targeted nanomedicines for cancer therapy, from basics to clinical trials. J. Pharm. Pharm. 2020, 23, 132–157. [Google Scholar] [CrossRef] [PubMed]
- Lyon, P.C.; Griffiths, L.F.; Lee, J.; Chung, D.; Carlisle, R.; Wu, F.; Middleton, M.R.; Gleeson, F.V.; Coussios, C.C. Clinical trial protocol for TARDOX: A phase I study to investigate the feasibility of targeted release of lyso-thermosensitive liposomal doxorubicin (ThermoDox®) using focused ultrasound in patients with liver tumours. J. Ther. Ultrasound 2017, 5, 1–8. [Google Scholar] [CrossRef]
- Shin, M.D.; Shukla, S.; Chung, Y.H.; Beiss, V.; Chan, S.K.; Ortega-Rivera, O.A.; Wirth, D.M.; Chen, A.; Sack, M.; Pokorski, J.K.; et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol. 2020, 15, 646–655. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, B.; Nag, O.K.; Rogers, K.E.; Delehanty, J.B. Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery. Molecules 2020, 25, 5672. https://doi.org/10.3390/molecules25235672
Almeida B, Nag OK, Rogers KE, Delehanty JB. Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery. Molecules. 2020; 25(23):5672. https://doi.org/10.3390/molecules25235672
Chicago/Turabian StyleAlmeida, Bethany, Okhil K. Nag, Katherine E. Rogers, and James B. Delehanty. 2020. "Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery" Molecules 25, no. 23: 5672. https://doi.org/10.3390/molecules25235672
APA StyleAlmeida, B., Nag, O. K., Rogers, K. E., & Delehanty, J. B. (2020). Recent Progress in Bioconjugation Strategies for Liposome-Mediated Drug Delivery. Molecules, 25(23), 5672. https://doi.org/10.3390/molecules25235672