
Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge

 ,
,  ,
,  and
and 
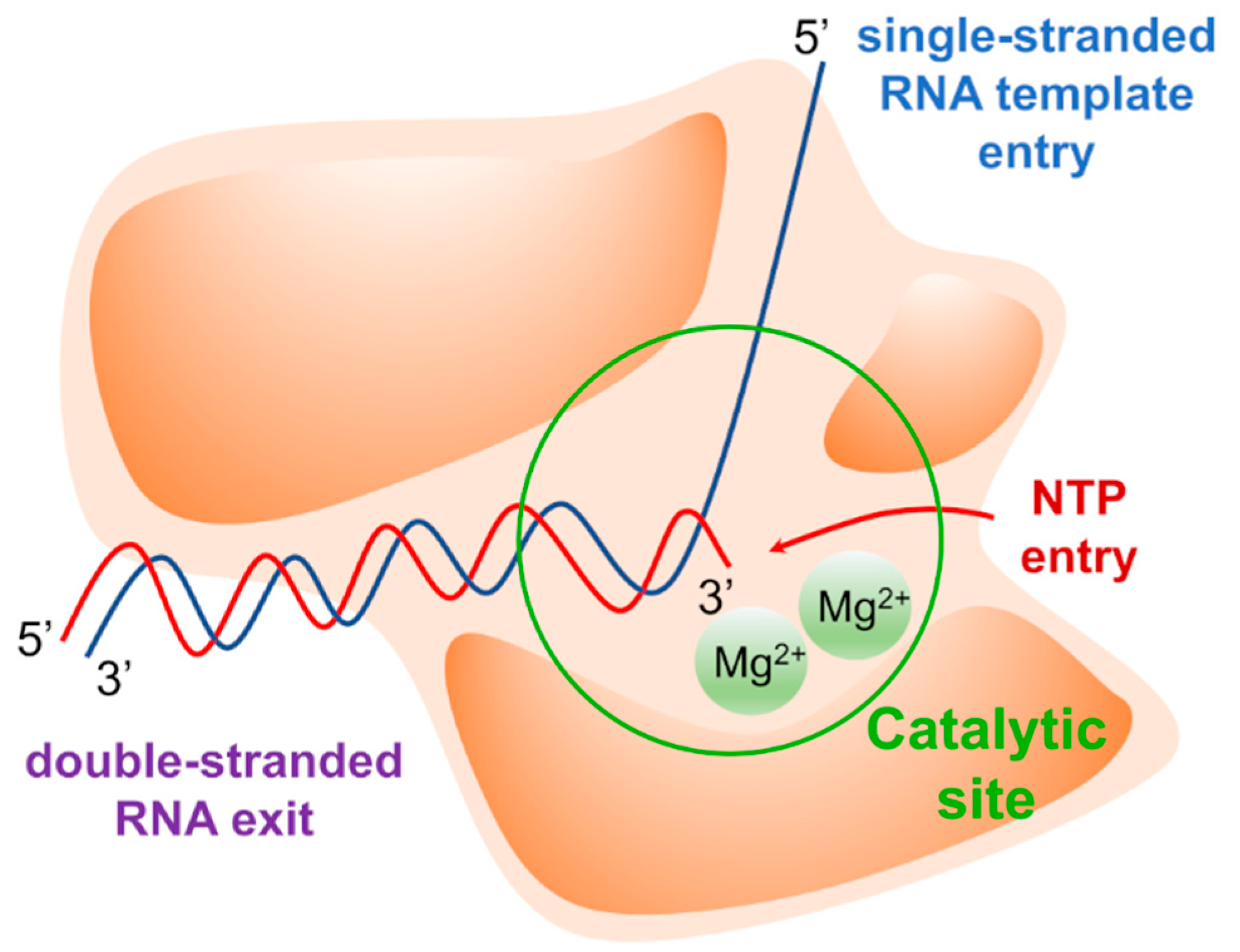{kind=link}
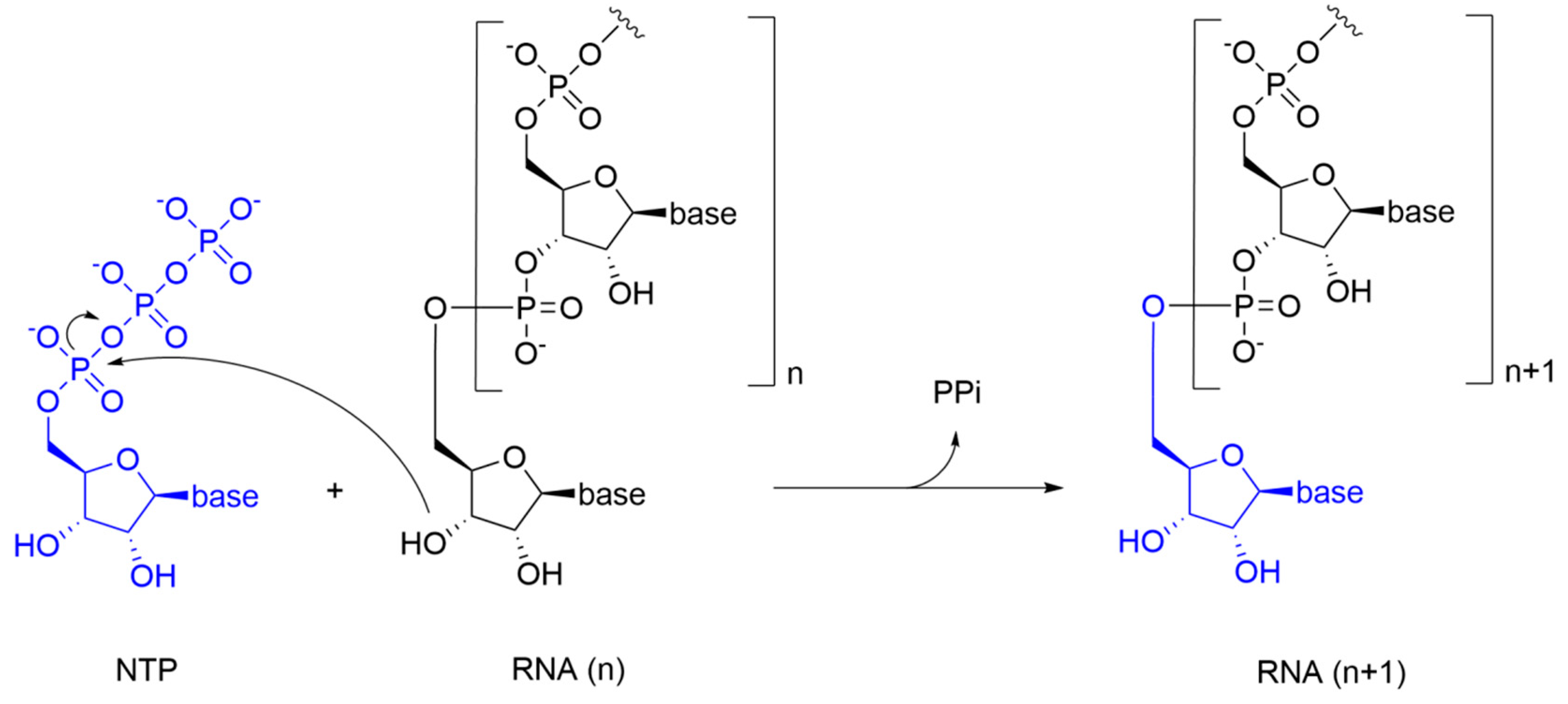{kind=link}
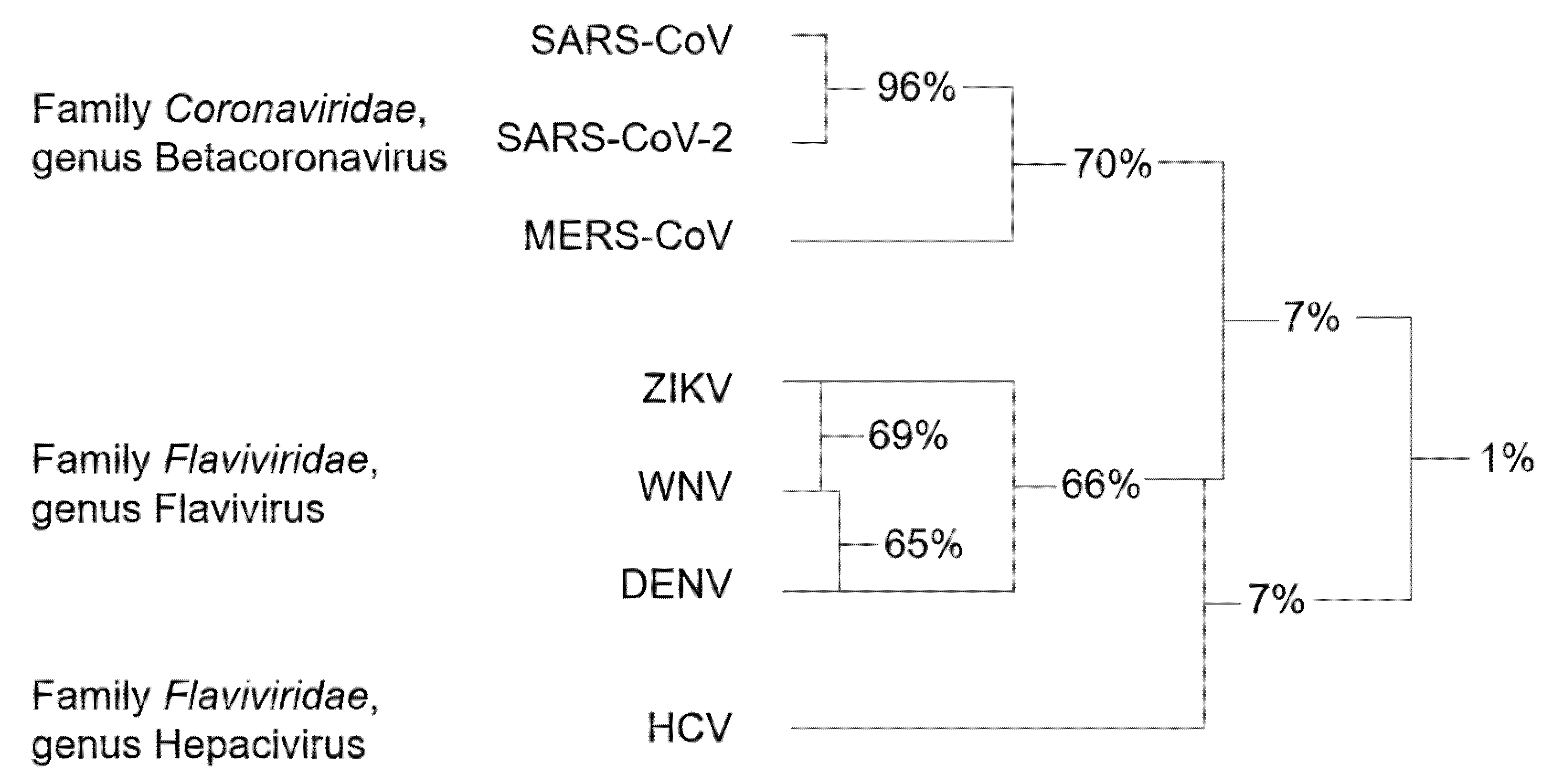{kind=link}
{kind=link}
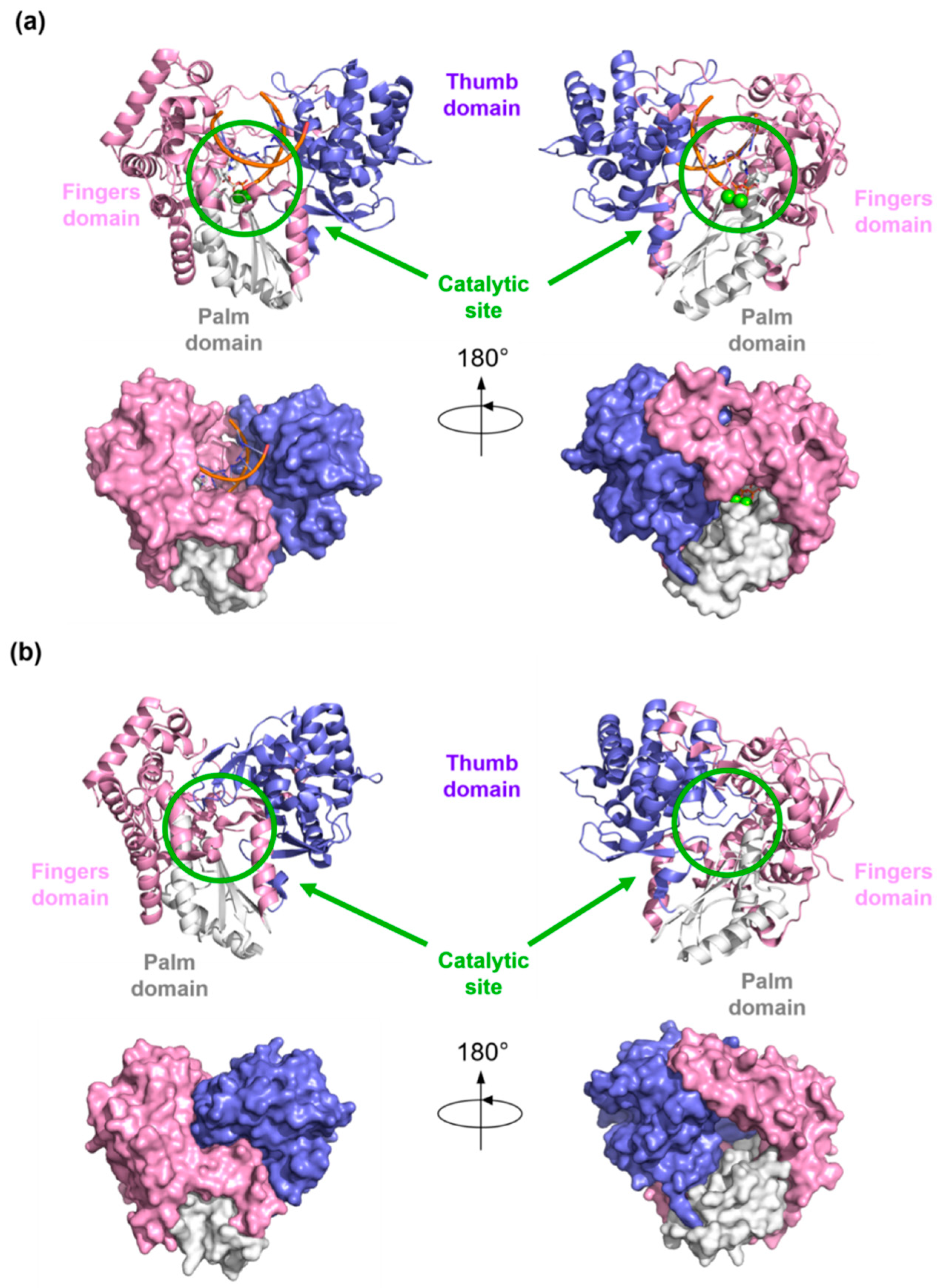{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. RNA Virus Outbreaks
2.1. Coronaviruses’ Outbreaks
2.2. Flaviviruses’ Outbreaks
2.3. HCV Outbreak
3. Sequence of Target RNA Viruses
4. Structural Features of RdRp from Emerging RNA Viruses
4.1. Structure of Coronaviruses RdRp
4.2. Structure of Flaviviruses RdRp
4.3. Structure of HCV RdRp
5. Strategies for the Development of Small Molecule RdRp Inhibitors
5.1. Small Molecule Inhibitors of Coronaviruses’ RdRp
5.2. Small Molecule Inhibitors of Flaviviruses’ RdRp
5.3. Small Molecules Inhibitors of HCV RdRp
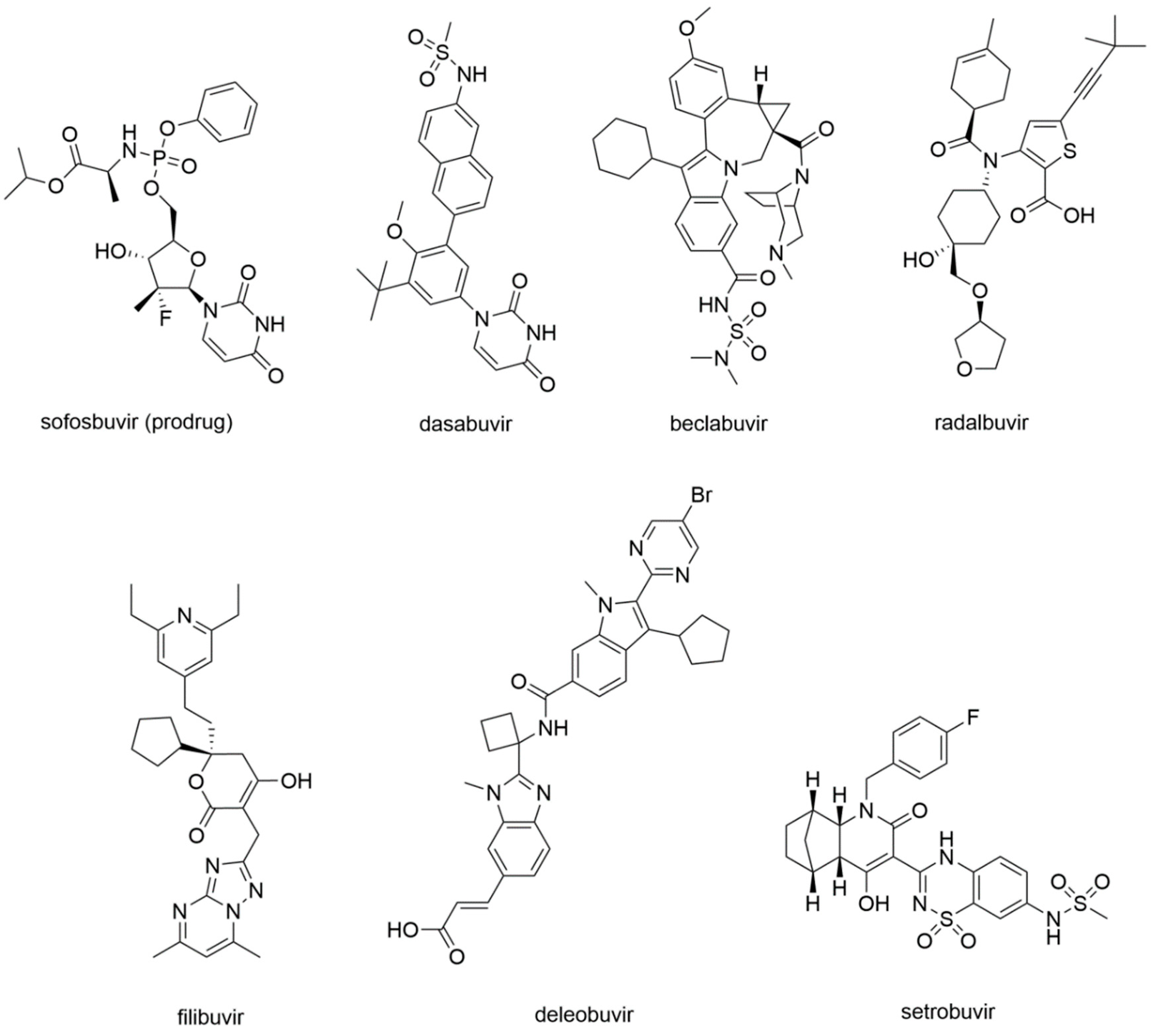
5.3.1. RdRp Inhibitors Approved for the Treatment of HCV Infection
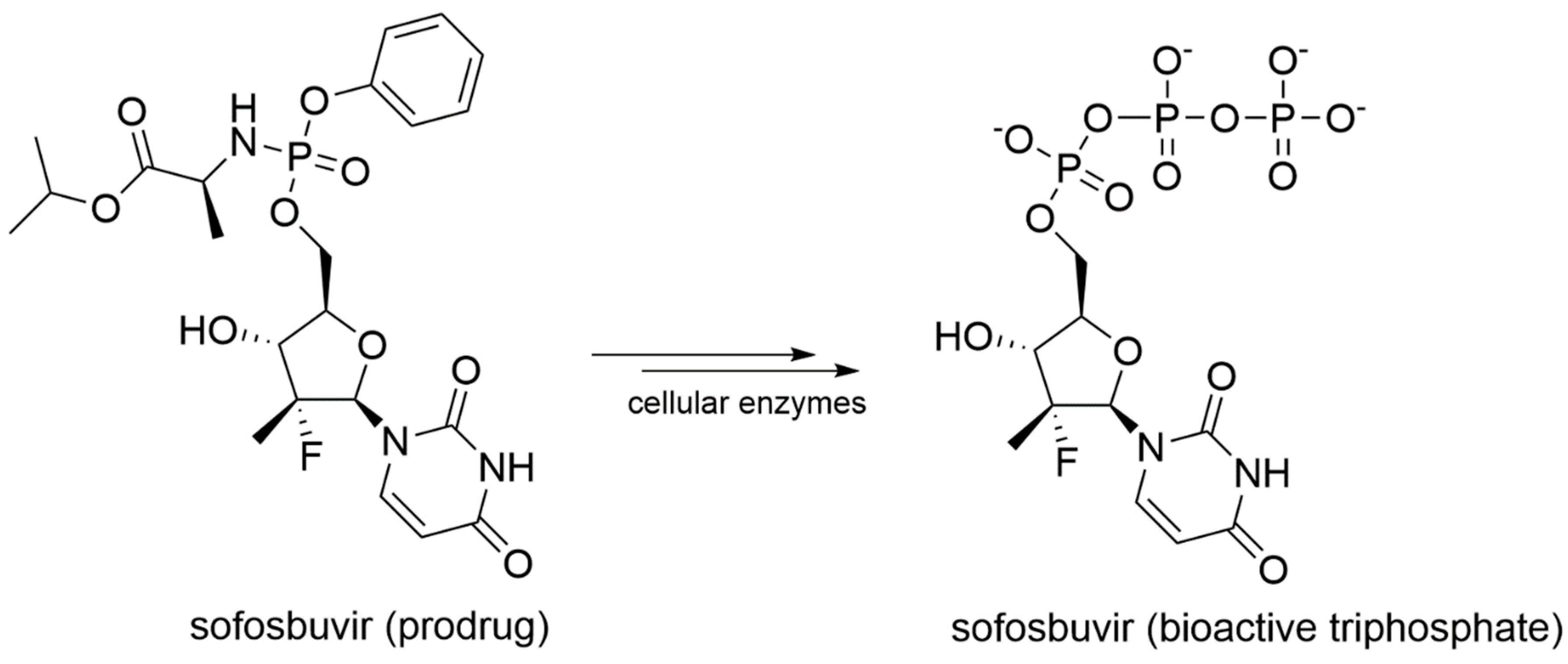
Sofosbuvir
Dasabuvir
5.3.2. Investigational and Discontinued HCV RdRp Inhibitors
Beclabuvir
Radalbuvir
Filibuvir
Deleobuvir and Setrobuvir
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Černý, J.; Bolfíková, B.Č.; Valdés, J.J.; Grubhoffer, L.; Růžek, D. Evolution of Tertiary Structure of Viral RNA Dependent Polymerases. PLoS ONE 2014, 9, e96070. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Malet, H.; Massé, N.; Selisko, B.; Romette, J.-L.; Alvarez, K.; Guillemot, J.C.; Tolou, H.; Yap, T.L.; Vasudevan, S.G.; Lescar, J.; et al. The flavivirus polymerase as a target for drug discovery. Antivir. Res. 2008, 80, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Soh, T.S.; Zheng, J.; Chan, K.W.K.; Phoo, W.W.; Lee, C.C.; Tay, M.Y.F.; Swaminathan, K.; Cornvik, T.C.; Lim, S.P.; et al. A Crystal Structure of the Dengue Virus NS5 Protein Reveals a Novel Inter-domain Interface Essential for Protein Flexibility and Virus Replication. PLoS Pathog. 2015, 11, e1004682. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.T.P.; Fernandes, P.A.; Ramos, M.J. The Catalytic Mechanism of RNA Polymerase II. J. Chem. Theory Comput. 2011, 7, 1177–1188. [Google Scholar] [CrossRef]
- Buck, K.W. Comparison of the Replication of Positive-Stranded Rna Viruses of Plants and Animals. Adv. Virus Res. 1996, 47, 159–251. [Google Scholar] [CrossRef]
- Siegel, R.W.; Bellon, L.; Beigelman, L.; Kao, C.C. Moieties in an RNA promoter specifically recognized by a viral RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 1998, 95, 11613–11618. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Shaevitz, J.W.; Abbondanzieri, E.A.; Landick, R.; Block, S.M. Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature 2003, 426, 684–687. [Google Scholar] [CrossRef]
- Dulin, D.; Vilfan, I.D.; Berghuis, B.A.; Poranen, M.M.; Depken, M.; Dekker, N.H. Backtracking behavior in viral RNA-dependent RNA polymerase provides the basis for a second initiation site. Nucleic Acids Res. 2015, 43, 10421–10429. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- de Farias, S.T.; dos Santos Junior, A.P.; Rêgo, T.G.; José, M.V. Origin and Evolution of RNA-Dependent RNA Polymerase. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Chan-yeung, M.; Xu, R. SARS: Epidemiology. Respirology 2003, 8, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.S.; Bakhrebah, M.A.; Meo, S.A.; Alsuabeyl, M.S.; Zaher, W.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infection: Epidemiology, pathogenesis and clinical characteristics. Eur. Rev. 2018, 22, 4956–4961. [Google Scholar]
- Mori, M.; Capasso, C.; Carta, F.; Donald, W.A.; Supuran, C.T. A deadly spillover: SARS-CoV-2 outbreak. Expert Opin. Ther. Pat. 2020, 30, 481–485. [Google Scholar] [CrossRef]
- de Paula Freitas, B.; Ventura, C.V.; Maia, M.; Belfort, R.J. Zika virus and the eye. Curr. Opin. Ophthalmol. 2017, 28, 595–599. [Google Scholar] [CrossRef]
- Medin, C.L.; Rothman, A.L. Zika Virus: The Agent and Its Biology, With Relevance to Pathology. Arch. Pathol. Lab. Med. 2017, 141, 33–42. [Google Scholar] [CrossRef]
- Dick, G.W.A.; Kitchen, S.F.; Haddow, A.J. Zika Virus (I). Isolations and serological specificity. Trans R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- de Oliveira Dias, J.R.; Ventura, C.V.; de Paula Freitas, B.; Prazeres, J.; Ventura, L.O.; Bravo-Filho, V.; Aleman, T.; Ko, A.I.; Zin, A.; Belfort, R.; et al. Zika and the Eye: Pieces of a Puzzle. Prog. Retin. Eye Res. 2018, 66, 85–106. [Google Scholar] [CrossRef]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.-Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef]
- Salehuddin, A.R.; Haslan, H.; Mamikutty, N.; Zaidun, N.H.; Azmi, M.F.; Senin, M.M.; Syed Ahmad Fuad, S.B.; Thent, Z.C. Zika virus infection and its emerging trends in Southeast Asia. Asian Pac. J. Trop. Med. 2017, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef]
- Carlson, C.J.; Dougherty, E.R.; Getz, W. An Ecological Assessment of the Pandemic Threat of Zika Virus. PLoS Negl. Trop. Dis. 2016, 10, e0004968. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, N.; Bradbury, R.S.; Taylor-Robinson, A.W. The global spread of Zika virus: Is public and media concern justified in regions currently unaffected? Infect. Dis. Poverty 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, T.; Rosenberger, K.D.; Brito, C.; Brady, O.; Brasil, P.; Marques, E.T. Risk of microcephaly after Zika virus infection in Brazil, 2015 to 2016. Bull. World Health Organ. 2017, 95, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Pereira, J.P.; Moreira, M.E.; Nogueira, R.M.R.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.-A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- Harapan, H.; Michie, A.; Sasmono, R.T.; Imrie, A. Dengue: A Minireview. Viruses 2020, 12, 829. [Google Scholar] [CrossRef]
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; et al. Global spread of dengue virus types: Mapping the 70 year history. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef]
- Hotta, S. Experimental Studies on DengueI. Isolation, Identification and Modification of the Virus. J. Infect. Dis. 1952, 90, 1–9. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue, Urbanization and Globalization: The Unholy Trinity of the 21st Century. Trop. Med. Health 2011, 39, S3–S11. [Google Scholar] [CrossRef]
- Schaffner, F.; Mathis, A. Dengue and dengue vectors in the WHO European region: Past, present, and scenarios for the future. Lancet Infect. Dis. 2014, 14, 1271–1280. [Google Scholar] [CrossRef]
- Zaayman, D.; Venter, M. West Nile Virus Neurologic Disease in Humans, South Africa, September 2008–May 2009-Volume 18, Number 12—December. Emerg. Infect. Dis. J. CDC 2012, 18, 2051. [Google Scholar] [CrossRef] [PubMed]
- Popescu, C.P.; Florescu, S.A.; Cotar, A.I.; Badescu, D.; Ceianu, C.S.; Zaharia, M.; Tardei, G.; Codreanu, D.; Ceausu, E.; Ruta, S.M. Re-emergence of severe West Nile virus neuroinvasive disease in humans in Romania, 2012 to 2017–implications for travel medicine. Travel Med. Infect. Dis. 2018, 22, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Magurano, F.; Remoli, M.E.; Baggieri, M.; Fortuna, C.; Marchi, A.; Fiorentini, C.; Bucci, P.; Benedetti, E.; Ciufolini, M.G.; Rizzo, C.; et al. Circulation of West Nile virus lineage 1 and 2 during an outbreak in Italy. Clin. Microbiol. Infect. 2012, 18, E545–E547. [Google Scholar] [CrossRef] [PubMed]
- Nash, D.; Mostashari, F.; Fine, A.; Miller, J.; O’Leary, D.; Murray, K.; Huang, A.; Rosenberg, A.; Greenberg, A.; Sherman, M.; et al. The Outbreak of West Nile Virus Infect. in the New York City Area in 1999. N. Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Roehrig, J.T.; Deubel, V.; Smith, J.; Parker, M.; Steele, K.; Crise, B.; Volpe, K.E.; Crabtree, M.B.; Scherret, J.H.; et al. Origin of the West Nile Virus Responsible for an Outbreak of Encephalitis in the Northeastern United States. Science 1999, 286, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.J.; Zhang, J.; Edmonds, J.H.; Prow, N.A.; Gu, X.; Davis, R.; Hornitzky, C.; Arzey, K.E.; Finlaison, D.; Hick, P.; et al. Characterization of Virulent West Nile Virus Kunjin Strain, Australia, 2011-Volume 18, Number 5—May. Emerg. Infect. Dis. J. CDC 2012, 18, 792. [Google Scholar] [CrossRef]
- Musso, D.; Nilles, E.J.; Cao-Lormeau, V.-M. Rapid spread of emerging Zika virus in the Pacific area. Clin. Microbiol. Infect. 2014, 20, O595–O596. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.D. Hepatitis C virus: Molecular biology & current therapeutic options. Indian J. Med. Res. 2010, 131, 17–34. [Google Scholar] [PubMed]
- Maheshwari, A.; Thuluvath, P.J. Management of Acute Hepatitis C. Clin. Liver Dis. 2010, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Zelenev, A.; Jianghong, L.; Mazhnaya, A.; Basu, S.; Altice, F.L. Hepatitis C virus treatment as prevention in an extended network of people who inject drugs in the USA: A modelling study. Lancet Infect. Dis. 2018, 18, 215–224. [Google Scholar] [CrossRef]
- Janiak, M.; Caraballo Cortes, K.; Demkow, U.; Radkowski, M. Spontaneous Elimination of Hepatitis C Virus Infection. In Current Concepts in Medical Research and Practice; Pokorski, M., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; pp. 45–54. ISBN 978-3-319-74150-5. [Google Scholar]
- Rosen, H. Clinical practice. Chronic hepatitis C infection. N. Engl. J. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Shrivastava, S.; Puri, V.; Dilley, K.A.; Ngouajio, E.; Shifflett, J.; Oldfield, L.M.; Fedorova, N.B.; Hu, L.; Williams, T.; Durbin, A.; et al. Whole genome sequencing, variant analysis, phylogenetics, and deep sequencing of Zika virus strains. Sci. Rep. 2018, 8, 15843. [Google Scholar] [CrossRef]
- Marra, M.A.; Jones, S.J.M.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.N.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome Sequence of the SARS-Associated Coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef]
- Richter, J.; Tryfonos, C.; Tourvas, A.; Floridou, D.; Paphitou, N.I.; Christodoulou, C. Complete Genome Sequence of West Nile Virus (WNV) from the First Human Case of Neuroinvasive WNV Infect. in Cyprus. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Kandeil, A.; Shehata, M.M.; El Shesheny, R.; Gomaa, M.R.; Ali, M.A.; Kayali, G. Complete Genome Sequence of Middle East Respiratory Syndrome Coronavirus Isolated from a Dromedary Camel in Egypt. Genome Announc. 2016, 4. [Google Scholar] [CrossRef]
- Sah, R.; Rodriguez-Morales, A.J.; Jha, R.; Chu, D.K.W.; Gu, H.; Peiris, M.; Bastola, A.; Lal, B.K.; Ojha, H.C.; Rabaan, A.A.; et al. Complete Genome Sequence of a 2019 Novel Coronavirus (SARS-CoV-2) Strain Isolated in Nepal. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef]
- Dang, T.T.; Pham, M.H.; Bui, H.V.; Le, D.V. Whole genome sequencing and genetic variations in several dengue virus type 1 strains from unusual dengue epidemic of 2017 in Vietnam. Virol. J. 2020, 17. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F. High-throughput sequencing (HTS) for the analysis of viral populations. Infect. Genet. Evol. 2020, 80, 104208. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Y.; Weiss, S.; Arnold, E.; Sarafianos, S.G.; Ding, J. Molecular model of SARS coronavirus polymerase: Implications for biochemical functions and drug design. Nucleic Acids Res. 2003, 31, 7117–7130. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Lu, G.; Gong, P. Crystal Structure of the Full-Length Japanese Encephalitis Virus NS5 Reveals a Conserved Methyltransferase-Polymerase Interface. PLoS Pathog. 2013, 9, e1003549. [Google Scholar] [CrossRef]
- Thompson, A.A.; Peersen, O.B. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 2004, 23, 3462–3471. [Google Scholar] [CrossRef]
- Xu, H.-T.; Hassounah, S.A.; Colby-Germinario, S.P.; Oliveira, M.; Fogarty, C.; Quan, Y.; Han, Y.; Golubkov, O.; Ibanescu, I.; Brenner, B.; et al. Purification of Zika virus RNA-dependent RNA polymerase and its use to identify small-molecule Zika inhibitors. J. Antimicrob. Chemother. 2017, 72, 727–734. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, A.; Guichou, J.-F.; Brillet, R.; Ahnou, N.; Hernandez, E.; Pallier, C.; Pawlotsky, J.-M. Inhibition of RNA binding to hepatitis C virus RNA-dependent RNA polymerase: A new mechanism for antiviral intervention. Nucleic Acids Res. 2014, 42, 9399–9409. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, T.; Zhang, J.; Zhan, P.; Liu, X. Influenza a virus polymerase: An attractive target for next-generation anti-influenza therapeutics. Drug Discov. Today 2018, 23, 503–518. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Agudo, R.; Domingo, E.; Verdaguer, N. Structural insights into replication initiation and elongation processes by the FMDV RNA-dependent RNA polymerase. Curr. Opin. Struct. Biol. 2009, 19, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.C.; Ruben, M.; Overkleeft, H.S.; van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, J.; Wang, H.; Gao, Y.; Liu, Q.; Mu, A.; Ji, W.; Yan, L.; Zhu, Y.; Zhu, C.; et al. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Tham, H.-W.; Balasubramaniam, V.R.M.T.; Chew, M.-F.; Ahmad, H.; Hassan, S.S. Protein-protein interactions between A. aegypti midgut and dengue virus 2: Two-hybrid screens using the midgut cDNA library. J. Infect. Dev. Ctries. 2015, 9, 1338–1349. [Google Scholar] [CrossRef]
- Šebera, J.; Dubankova, A.; Sychrovský, V.; Ruzek, D.; Boura, E.; Nencka, R. The structural model of Zika virus RNA-dependent RNA polymerase in complex with RNA for rational design of novel nucleotide inhibitors. Sci. Rep. 2018, 8, 11132. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Arias, A.; Escarmís, C.; Verdaguer, N. A comparison of viral RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2006, 16, 27–34. [Google Scholar] [CrossRef]
- Butcher, S.J.; Grimes, J.M.; Makeyev, E.V.; Bamford, D.H.; Stuart, D.I. A mechanism for initiating RNA-dependent RNA polymerization. Nature 2001, 410, 235–240. [Google Scholar] [CrossRef]
- Piorkowski, G.; Richard, P.; Baronti, C.; Gallian, P.; Charrel, R.; Leparc-Goffart, I.; de Lamballerie, X. Complete coding sequence of Zika virus from Martinique outbreak in 2015. New Microbes New Infect. 2016, 11, 52–53. [Google Scholar] [CrossRef]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
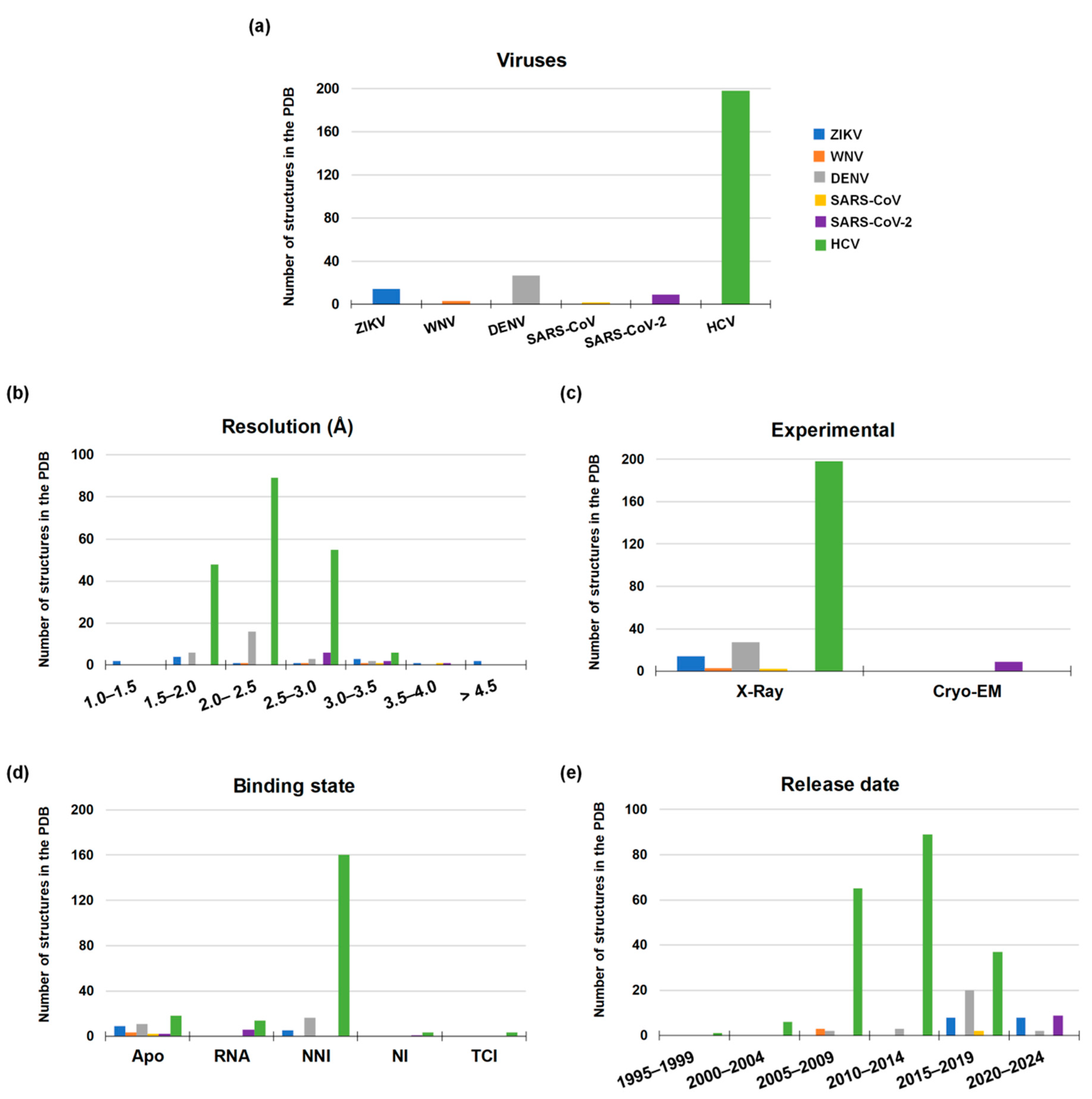
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, F.; Boccuto, A.; Picarazzi, F.; Giannini, A.; Giammarino, F.; Saladini, F.; Mori, M.; Mastrangelo, E.; Zazzi, M.; Vicenti, I. Evaluation of sofosbuvir activity and resistance profile against West Nile virus in vitro. Antivir. Res. 2020, 175, 104708. [Google Scholar] [CrossRef] [PubMed]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D.; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Hamatake, R.K.; Mathis, D.M.; Racela, J.; Rigat, K.L.; Lemm, J.; Colonno, R.J. De Novo Initiation of RNA Synthesis by the RNA-Dependent RNA Polymerase (NS5B) of Hepatitis C Virus. J. Virol. 2000, 74, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Serpi, M.; Pertusati, F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir Chem. Chemother. 2018, 26. [Google Scholar] [CrossRef]
- Xu, H.-T.; Colby-Germinario, S.P.; Hassounah, S.A.; Fogarty, C.; Osman, N.; Palanisamy, N.; Han, Y.; Oliveira, M.; Quan, Y.; Wainberg, M.A. Evaluation of Sofosbuvir (β-D-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine) as an inhibitor of Dengue virus replication. Sci. Rep. 2017, 7, 6345. [Google Scholar] [CrossRef]
- Eltahla, A.A.; Luciani, F.; White, P.A.; Lloyd, A.R.; Bull, R.A. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses 2015, 7, 5206–5224. [Google Scholar] [CrossRef]
- Winquist, J.; Abdurakhmanov, E.; Baraznenok, V.; Henderson, I.; Vrang, L.; Danielson, U.H. Resolution of the interaction mechanisms and characteristics of non-nucleoside inhibitors of hepatitis C virus polymerase. Antivir. Res. 2013, 97, 356–368. [Google Scholar] [CrossRef]
- Mosley, R.T.; Edwards, T.E.; Murakami, E.; Lam, A.M.; Grice, R.L.; Du, J.; Sofia, M.J.; Furman, P.A.; Otto, M.J. Structure of Hepatitis C Virus Polymerase in Complex with Primer-Template RNA. J. Virol. 2012, 86, 6503–6511. [Google Scholar] [CrossRef]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Bisbocci, M.; Bailey, C.; Bosserman, M.; Cellucci, A.; Forte, E.; Incitti, I.; Orsatti, L.; et al. Characterization of the Inhibition of Hepatitis C Virus RNA Replication by Nonnucleosides. J. Virol. 2004, 78, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Takahashi, C.; Moreland, N.J.; Chang, Y.-T.; Sawasaki, T.; Ryo, A.; Vasudevan, S.G.; Suzuki, Y.; Yamamoto, N. Establishment of a robust dengue virus NS3–NS5 binding assay for identification of protein–protein interaction inhibitors. Antivir. Res. 2012, 96, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Kuo, M.D.; Chien, L.J.; Hsu, S.L.; Wang, Y.M.; Lin, J.H. RNA-protein interactions: Involvement of NS3, NS5, and 3’ noncoding regions of Japanese encephalitis virus genomic RNA. J. Virol. 1997, 71, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 Proteins of Dengue Virus Type 2 in the Putative RNA Replicase Is Linked to Differential Phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar] [CrossRef]
- Johansson, M.; Brooks, A.J.; Jans, D.A.; Vasudevan, S.G. A small region of the dengue virus-encoded RNA-dependent RNA polymerase, NS5, confers interaction with both the nuclear transport receptor importin-β and the viral helicase, NS3. J. Gen. Virol. 2001, 82, 735–745. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- McDonald, A.G.; Tipton, K.F. Enzymes: Irreversible Inhibition. In eLS; American Cancer Society: Atlanta, GA, USA, 2020; pp. 1–17. ISBN 978-0-470-01590-2. [Google Scholar]
- Coleman, C.I.; Limone, B.; Sobieraj, D.M.; Lee, S.; Roberts, M.S.; Kaur, R.; Alam, T. Dosing Frequency and Medication Adherence in Chronic Dis. JMCP 2012, 18, 527–539. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Van Arnum, P. Drug Repurposing and Repositioning: Making New Out of Old. Available online: https://www.dcatvci.org/11-value-chain-insights/114-drug-repurposing-and-repositioning-making-new-out-of-old# (accessed on 6 November 2020).
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug Repurposing (DR): An Emerging Approach in Drug Discovery. Drug Repurposing 2020. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharm. Rep. 2020, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Serafin, M.B.; Bottega, A.; Foletto, V.S.; da Rosa, T.F.; Hörner, A.; Hörner, R. Drug repositioning is an alternative for the treatment of coronavirus COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105969. [Google Scholar] [CrossRef]
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef]
- Sacramento, C.Q.; de Melo, G.R.; de Freitas, C.S.; Rocha, N.; Hoelz, L.V.B.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017, 7, 40920. [Google Scholar] [CrossRef]
- Szabat, M.; Lorent, D.; Czapik, T.; Tomaszewska, M.; Kierzek, E.; Kierzek, R. RNA Secondary Structure as a First Step for Rational Design of the Oligonucleotides towards Inhibition of Influenza A Virus Replication. Pathogens 2020, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Kesy, J.; Patil, K.M.; Kumar, S.R.; Shu, Z.; Yong, H.Y.; Zimmermann, L.; Ong, A.A.L.; Toh, D.-F.K.; Krishna, M.S.; Yang, L.; et al. A Short Chemically modified dsRNA-Binding PNA (dbPNA) Inhibits Influenza Viral Replication by Targeting Viral RNA Panhandle Structure. Bioconjugate Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-S.; Lin, W.-H.; T-A Hsu, J.; Hsieh, H.-P. Antiviral Drug Discovery against SARS-CoV. Curr. Med. Chem. 2006, 13, 2003–2020. [Google Scholar] [CrossRef] [PubMed]
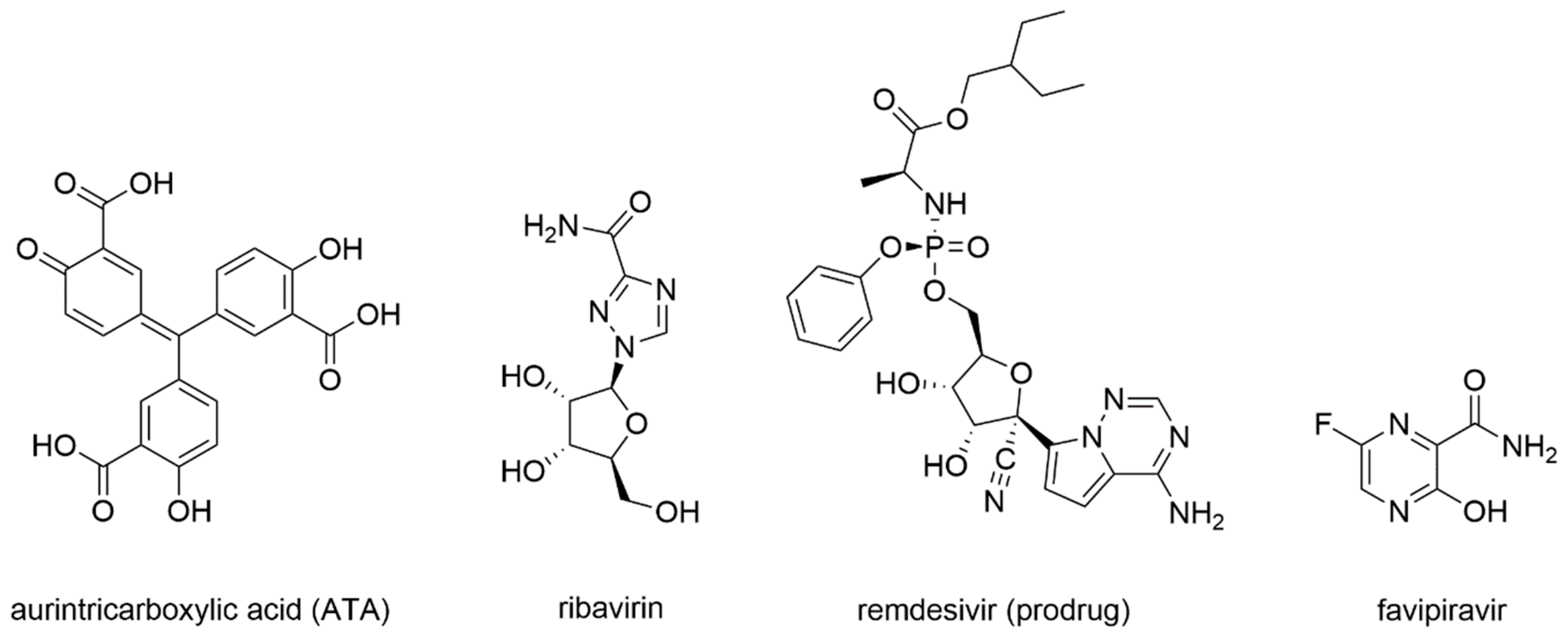
- He, R.; Adonov, A.; Traykova-Adonova, M.; Cao, J.; Cutts, T.; Grudesky, E.; Deschambaul, Y.; Berry, J.; Drebot, M.; Li, X. Potent and selective inhibition of SARS coronavirus replication by aurintricarboxylic acid. Biochem. Biophys. Res. Commun. 2004, 320, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Yap, Y.; Zhang, X.; Andonov, A.; He, R. Structural analysis of inhibition mechanisms of Aurintricarboxylic Acid on SARS-CoV polymerase and other proteins. Comput. Biol. Chem. 2005, 29, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Bavari, S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin. Drug Discov. 2019, 14, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, D.; de Wit, E.; Martellaro, C.; Callison, J.; Munster, V.J.; Feldmann, H. Inhibition of novel β coronavirus replication by a combination of interferon-α2b and ribavirin. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Hart, B.J.; Dyall, J.; Postnikova, E.; Zhou, H.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G.; Frieman, M.B.; Holbrook, M.R.; Jahrling, P.B.; et al. Interferon-β and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J. Gen. Virol. 2014, 95, 571–577. [Google Scholar] [CrossRef]
- Falzarano, D.; de Wit, E.; Rasmussen, A.L.; Feldmann, F.; Okumura, A.; Scott, D.P.; Brining, D.; Bushmaker, T.; Martellaro, C.; Baseler, L.; et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV–infected rhesus macaques. Nat. Med. 2013, 19, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Subissi, L.; Silveira De Morais, A.T.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed]
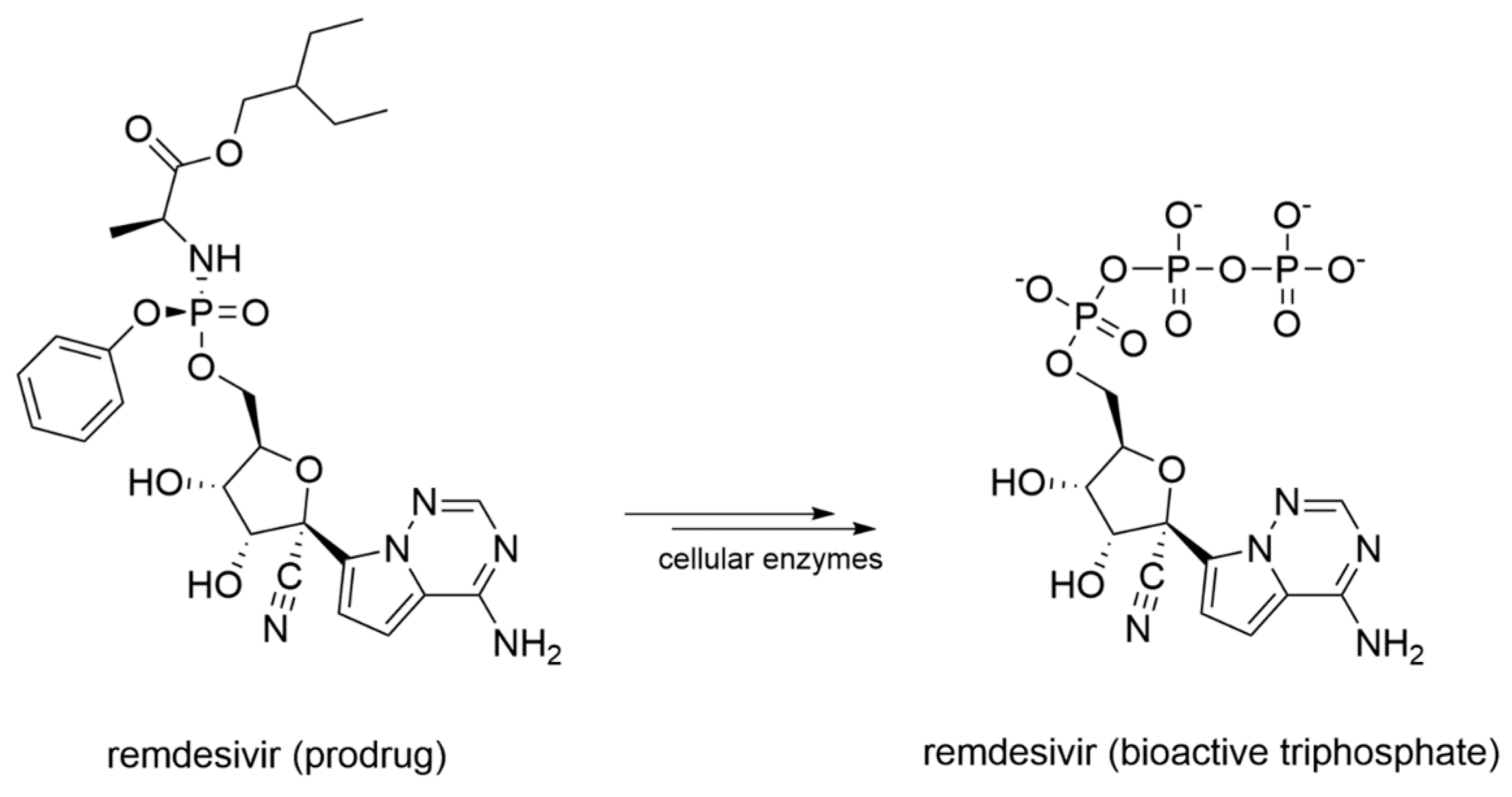
- Siegel, D.; Hui, H.C.; Doerffler, E.; Clarke, M.O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60, 1648–1661. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- De Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. MBio 2018, 9. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent Sci. 2020. [Google Scholar] [CrossRef]
- Li, G.; Clercq, E.D. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
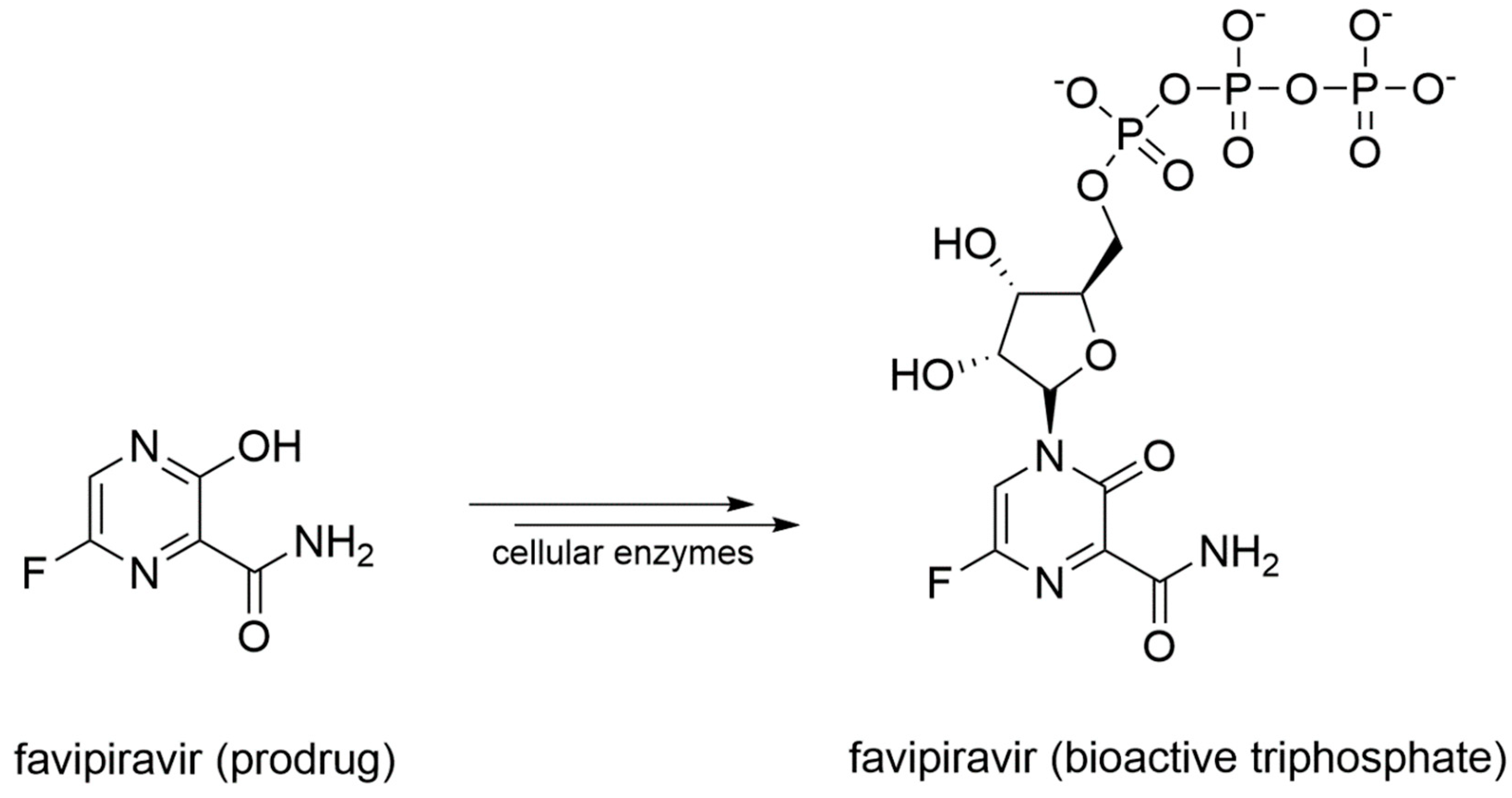
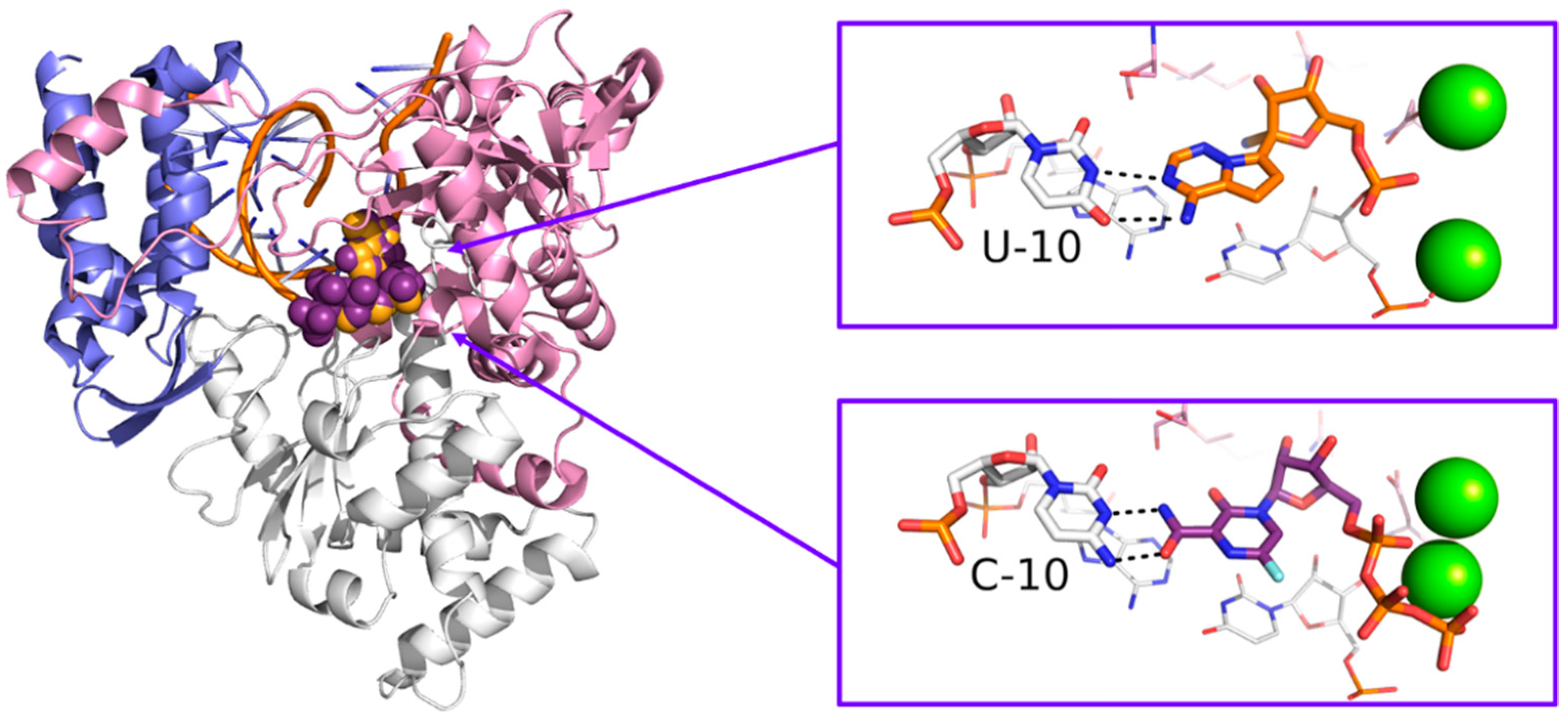
- Shannon, A.; Selisko, B.; Le, N.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Favipiravir strikes the SARS-CoV-2 at its Achilles heel, the RNA polymerase. Biorxiv 2020. [Google Scholar] [CrossRef]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020. [Google Scholar] [CrossRef]
- Götte, M.; Feld, J.J. Direct-acting antiviral agents for hepatitis C: Structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 338–351. [Google Scholar] [CrossRef]
- Lim, S.P.; Noble, C.G.; Seh, C.C.; Soh, T.S.; Sahili, A.E.; Chan, G.K.Y.; Lescar, J.; Arora, R.; Benson, T.; Nilar, S.; et al. Potent Allosteric Dengue Virus NS5 Polymerase Inhibitors: Mechanism of Action and Resistance Profiling. PLoS Pathog. 2016, 12, e1005737. [Google Scholar] [CrossRef]
- Noble, C.G.; Lim, S.P.; Arora, R.; Yokokawa, F.; Nilar, S.; Seh, C.C.; Wright, S.K.; Benson, T.E.; Smith, P.W.; Shi, P.-Y. A Conserved Pocket in the Dengue Virus Polymerase Identified through Fragment-based Screening. J. Biol. Chem. 2016, 291, 8541–8548. [Google Scholar] [CrossRef] [PubMed]
- Gharbi-Ayachi, A.; Santhanakrishnan, S.; Wong, Y.H.; Chan, K.W.K.; Tan, S.T.; Bates, R.W.; Vasudevan, S.G.; El Sahili, A.; Lescar, J. Non-Nucleoside Inhibitors of Zika virus RNA-dependent RNA polymerase. J. Virol. 2020, JVI.00794-20. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Li, Q.; Pang, L.; Wang, Y.; Zhang, Y.; Duan, Z.; Liang, C.; Cen, S. Identification of a Broad-Spectrum Viral Inhibitor Targeting a Novel Allosteric Site in the RNA-Dependent RNA Polymerases of Dengue Virus and Norovirus. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Ki Chan, K.W.; Burali, M.S.; Gwee, C.P.; Wang, S.; Astolfi, A.; Massari, S.; Sabatini, S.; Tabarrini, O.; Mastrangelo, E.; et al. Pyridobenzothiazolones Exert Potent Anti-Dengue Activity by Hampering Multiple Functions of NS5 Polymerase. ACS Med. Chem. Lett. 2020, 11, 773–782. [Google Scholar] [CrossRef]
- Wilkins, T.; Malcolm, J.K.; Raina, D.; Schade, R.R. Hepatitis C: Diagnosis and Treatment. AFP 2010, 81, 1351–1357. [Google Scholar]
- Yee, B.E.; Nguyen, N.H.; Zhang, B.; Lin, D.; Vutien, P.; Wong, C.R.; Lutchman, G.A.; Nguyen, M.H. Sustained virological response and its treatment predictors in hepatitis C virus genotype 4 compared to genotypes 1, 2, and 3: A meta-analysis. BMJ Open Gastroenterol. 2015, 2. [Google Scholar] [CrossRef]
- Pan, Q.; Peppelenbosch, M.P.; Janssen, H.L.; de Knegt, R.J. Telaprevir/boceprevir era: From bench to bed and back. World J. Gastroenterol. 2012, 18, 6183–6188. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From non-A, non-B hepatitis to hepatitis C virus cure. J. Hepatol. 2015, 62, S87–S99. [Google Scholar] [CrossRef]
- McConachie, S.M.; Wilhelm, S.M.; Kale-Pradhan, P.B. New direct-acting antivirals in hepatitis C therapy: A review of sofosbuvir, ledipasvir, daclatasvir, simeprevir, paritaprevir, ombitasvir and dasabuvir. Expert Rev. Clin. Pharmacol. 2016, 9, 287–302. [Google Scholar] [CrossRef]
- Maasoumy, B.; Port, K.; Markova, A.A.; Serrano, B.C.; Rogalska-Taranta, M.; Sollik, L.; Mix, C.; Kirschner, J.; Manns, M.P.; Wedemeyer, H.; et al. Eligibility and Safety of Triple Therapy for Hepatitis C: Lessons Learned from the First Experience in a Real World Setting. PLoS ONE 2013, 8, e55285. [Google Scholar] [CrossRef]
- Kim, A. Hepatitis C Virus. Ann. Intern. Med. 2016, 165, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L.; Luetkemeyer, A.F. Understanding Hepatitis C Virus Drug Resistance: Clinical Implications for Current and Future Regimens. Top Antivir. Med. 2017, 25, 103–109. [Google Scholar] [PubMed]
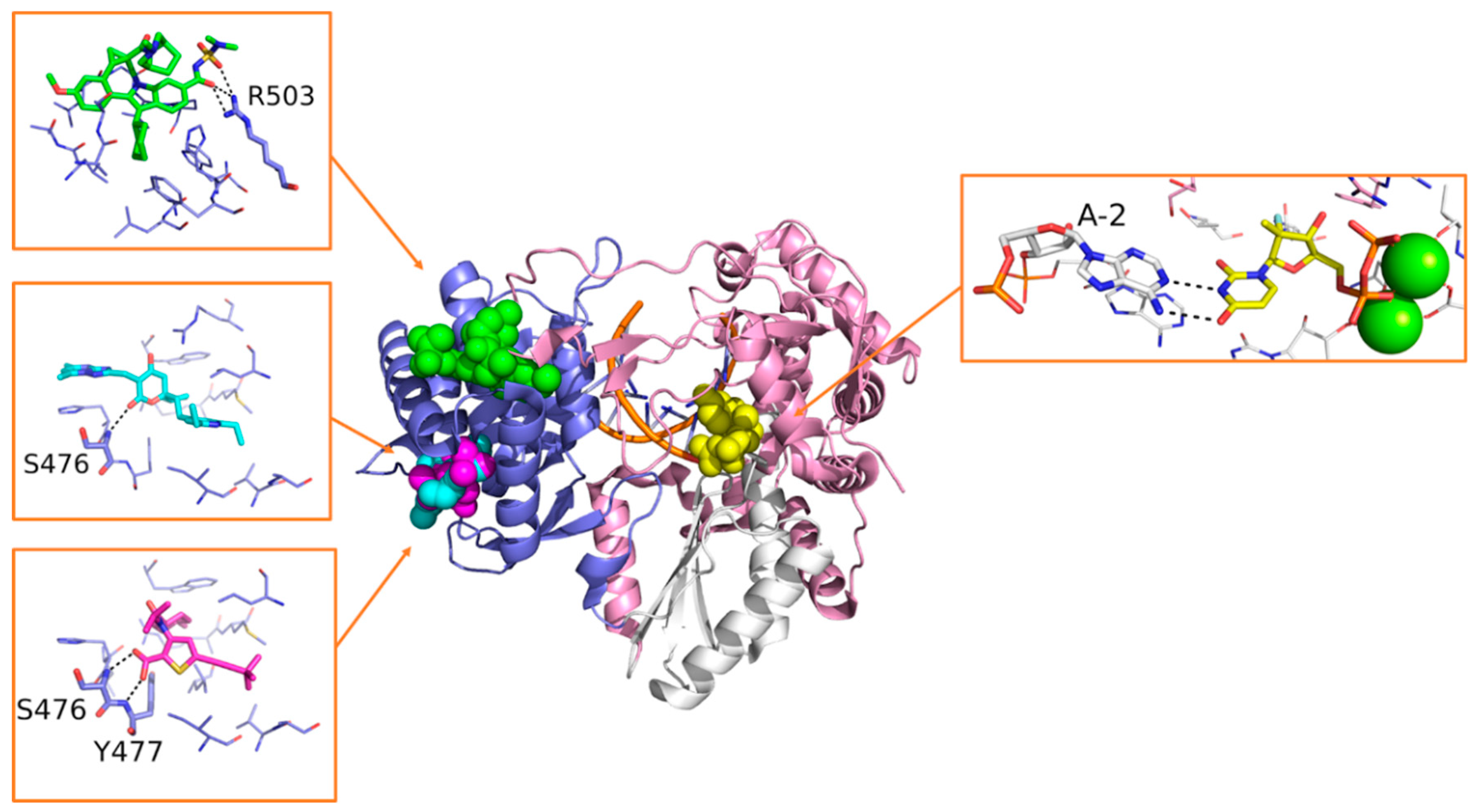
- Liu, Y.; Lim, B.H.; Jiang, W.W.; Flentge, C.A.; Hutchinson, D.K.; Madigan, D.L.; Randolph, J.T.; Wagner, R.; Maring, C.J.; Kati, W.M.; et al. Identification of aryl dihydrouracil derivatives as palm initiation site inhibitors of HCV NS5B polymerase. Bioorganic Med. Chem. Lett. 2012, 22, 3747–3750. [Google Scholar] [CrossRef] [PubMed]
- Kati, W.; Koev, G.; Irvin, M.; Beyer, J.; Liu, Y.; Krishnan, P.; Reisch, T.; Mondal, R.; Wagner, R.; Molla, A.; et al. In Vitro Activity and Resistance Profile of Dasabuvir, a Nonnucleoside Hepatitis C Virus Polymerase Inhibitor. Antimicrob. Agents Chemother. 2015, 59, 1505–1511. [Google Scholar] [CrossRef]
- Gentles, R.G.; Ding, M.; Bender, J.A.; Bergstrom, C.P.; Grant-Young, K.; Hewawasam, P.; Hudyma, T.; Martin, S.; Nickel, A.; Regueiro-Ren, A.; et al. Discovery and Preclinical Characterization of the Cyclopropylindolobenzazepine BMS-791325, A Potent Allosteric Inhibitor of the Hepatitis C Virus NS5B Polymerase. J. Med. Chem. 2014, 57, 1855–1879. [Google Scholar] [CrossRef]
- Lazerwith, S.E.; Lew, W.; Zhang, J.; Morganelli, P.; Liu, Q.; Canales, E.; Clarke, M.O.; Doerffler, E.; Byun, D.; Mertzman, M.; et al. Discovery of GS-9669, a Thumb Site II Non-Nucleoside Inhibitor of NS5B for the Treatment of Genotype 1 Chronic Hepatitis C Infect. J. Med. Chem. 2014, 57, 1893–1901. [Google Scholar] [CrossRef]
- Li, H.; Tatlock, J.; Linton, A.; Gonzalez, J.; Jewell, T.; Patel, L.; Ludlum, S.; Drowns, M.; Rahavendran, S.V.; Skor, H.; et al. Discovery of (R)-6-Cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554) as a Potent and Orally Available Hepatitis C Virus Polymerase Inhibitor. J. Med. Chem. 2009, 52, 1255–1258. [Google Scholar] [CrossRef]
- Jiao, P.; Xue, W.; Shen, Y.; Jin, N.; Liu, H. Understanding the drug resistance mechanism of hepatitis C virus NS5B to PF-00868554 due to mutations of the 423 site: A computational study. Mol. BioSyst. 2014, 10, 767–777. [Google Scholar] [CrossRef]
- Gentile, I.; Buonomo, A.R.; Zappulo, E.; Borgia, G. Discontinued drugs in 2012–2013: Hepatitis C virus infection. Expert Opin. Investig. Drugs 2015, 24, 239–251. [Google Scholar] [CrossRef][Green Version]
- Larrey, D.; Lohse, A.W.; Trepo, C.; Bronowicki, J.-P.; Arastéh, K.; Bourlière, M.; Calleja, J.L.; Stern, J.O.; Nehmiz, G.; Abdallah, N.; et al. Antiviral Effect, Safety, and Pharmacokinetics of Five-Day Oral Administration of Deleobuvir (BI 207127), an Investigational Hepatitis C Virus RNA Polymerase Inhibitor, in Patients with Chronic Hepatitis C. Antimicrob Agents Chemother. 2013, 57, 4727–4735. [Google Scholar] [CrossRef]
- Kapelusznik, L.; Heil, E.L.; Temesgen, Z.; Talwani, R. Setrobuvir: RNA-directed RNA polymerase inhibitor treatment of hepatitis C virus infection. Drugs Future 2012, 37, 725–733. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picarazzi, F.; Vicenti, I.; Saladini, F.; Zazzi, M.; Mori, M. Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge. Molecules 2020, 25, 5695. https://doi.org/10.3390/molecules25235695
Picarazzi F, Vicenti I, Saladini F, Zazzi M, Mori M. Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge. Molecules. 2020; 25(23):5695. https://doi.org/10.3390/molecules25235695
Chicago/Turabian StylePicarazzi, Francesca, Ilaria Vicenti, Francesco Saladini, Maurizio Zazzi, and Mattia Mori. 2020. "Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge" Molecules 25, no. 23: 5695. https://doi.org/10.3390/molecules25235695
APA StylePicarazzi, F., Vicenti, I., Saladini, F., Zazzi, M., & Mori, M. (2020). Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge. Molecules, 25(23), 5695. https://doi.org/10.3390/molecules25235695