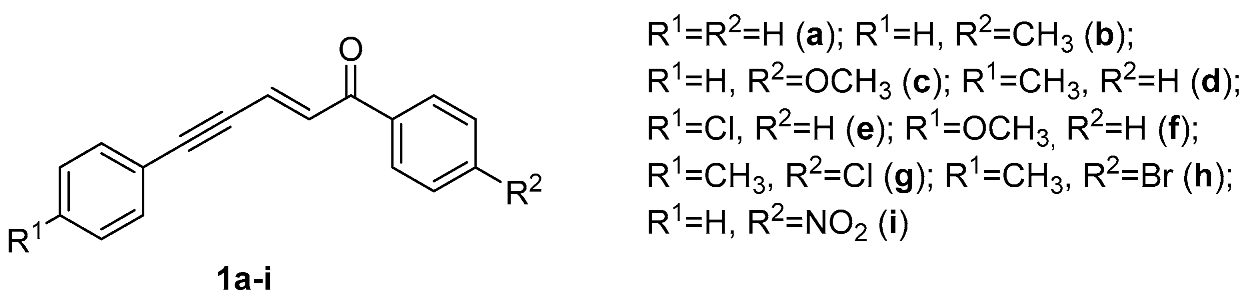
Stereoselective Synthesis of Multisubstituted Cyclohexanes by Reaction of Conjugated Enynones with Malononitrile in the Presence of LDA



Abstract
:
1. Introduction
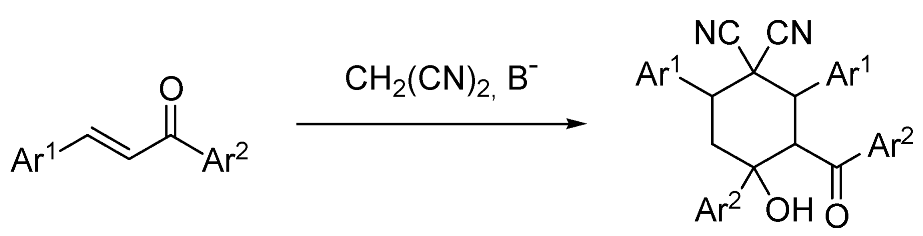
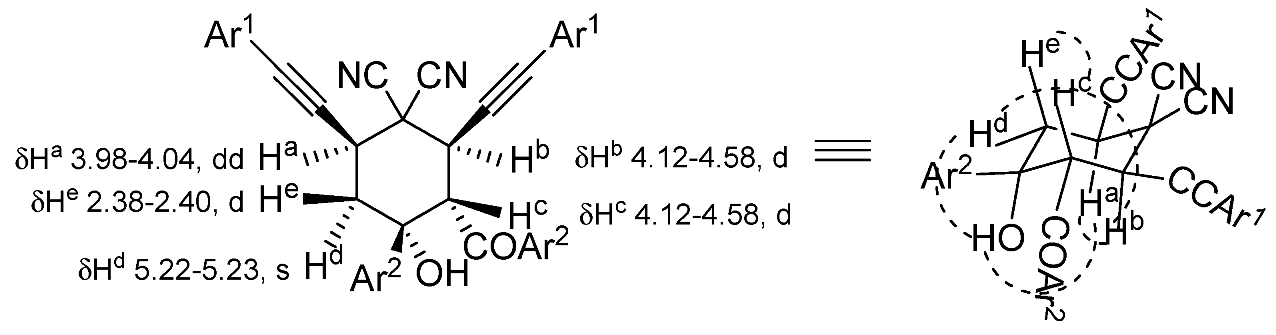
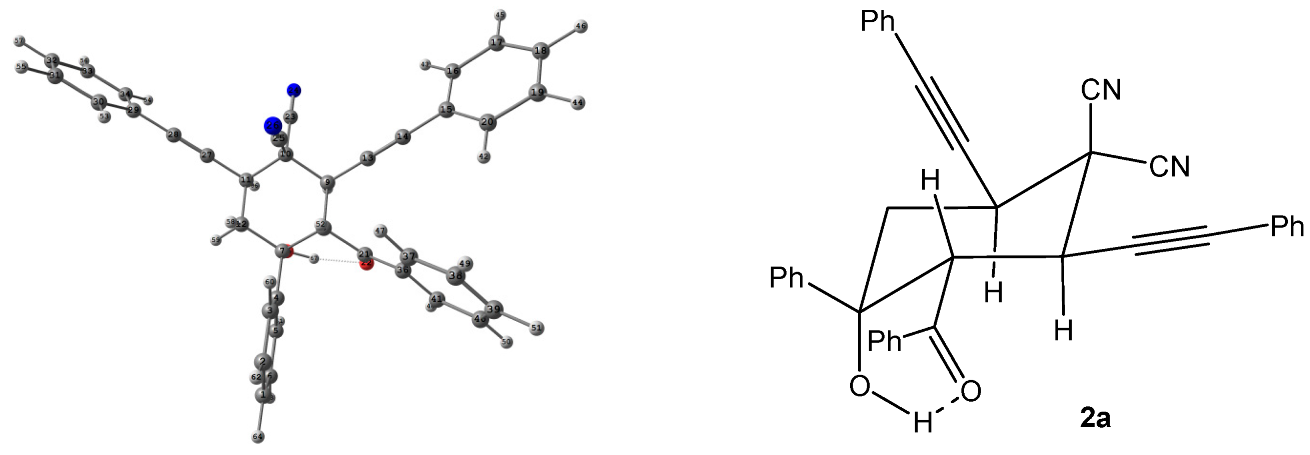
2. Results and Discussion
3. Conclusions
3.1. Experimental Section
3.2. X-ray Diffraction Study
3.3. DFT Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Golovanov, A.A.; Odin, I.S.; Zlotskii, S.S. Conjugated enynones: Preparation, properties and applications in organic synthesis. Russ. Chem. Rev. 2019, 88, 280. [Google Scholar] [CrossRef]
- Golovanov, A.A.; Gusev, D.M.; Odin, I.S.; Zlotskii, S.S. Conjugated 2,4,1- and 1,4,3-enynones as polycentric electrophiles in synthesis of heterocyclic compounds. Chem. Heterocycl. Compd. 2019, 55, 333. [Google Scholar] [CrossRef]
- Kuznetcova, A.V.; Odin, I.S.; Golovanov, A.A.; Grigorev, I.M.; Vasilyev, A.V. Multicomponent reaction of conjugated enynones with malononitrile and sodium alkoxides: Complex reaction mechanism of the formation of pyridine derivatives. Tetrahedron 2019, 75, 4516. [Google Scholar] [CrossRef]
- Hegazy, M.-E.F.; Elshamy, A.I.; Mohamed, T.A.; Hussien, T.A.; Helaly, S.E.; Abdel-Azim, N.S.; Shams, K.A.; Shahat, A.A.; Tawfik, W.A.; Shahen, A.M.; et al. Terpenoid bio-transformations and applications via cell/organ cultures: A systematic review. Crit. Rev. Biotechnol. 2020, 40, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Lin, M.; Su, X.; Li, C.; Wang, H.; Li, B.; Chen, R.; Kang, J. Anti-inflammatory terpenes from Schefflera rubriflora C. J. Tseng & G. Hoo with their TNF-α and IL-6 inhibitory activities. Phytochemistry 2019, 163, 23. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.N.; Satyal, P.; Mayo, J.A.; McFeeters, H.; McFeeters, R.L. Bigger Data Approach to Analysis of Essential Oils and Their Antifungal Activity against Aspergillus niger, Candida albicans, and Cryptococcus neoformans. Molecules 2019, 24, 2868. [Google Scholar] [CrossRef] [Green Version]
- Mitić, Z.S.; Jovanović, B.; Jovanović, S.Č.; Stojanović-Radić, Z.Z.; Mihajilov-Krstev, T.; Jovanović, N.M.; Nikolić, B.M.; Marin, P.D.; Zlatković, B.K.; Stojanović, G.S. Essential oils of Pinus halepensis and P. heldreichii: Chemical composition, antimicrobial and insect larvicidal activity. Ind. Crops Prod. 2019, 140, 111702. [Google Scholar] [CrossRef]
- Deng, W.; Liu, K.; Cao, S.; Sun, J.; Zhong, B.; Chun, J. Chemical Composition, Antimicrobial, Antioxidant, and Antiproliferative Properties of Grapefruit Essential Oil Prepared by Molecular Distillation. Molecules 2020, 25, 217. [Google Scholar] [CrossRef] [Green Version]
- Cikoš, A.-M.; Jurin, M.; Čož-Rakovac, R.C.; Jokic, S.; Jerkovic, I. Update on Monoterpenes from Red Macroalgae: Isolation, Analysis, and Bioactivity. Mar. Drugs 2019, 17, 537. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, J.L.; Campos, E.V.R.; Germano-Costa, T.; Lima, R.; Vechia, J.F.D.; Soares, S.T.; de Andrade, D.J.; Goncalves, K.C.; do Nascimento, J.; Polanczyk, R.A.; et al. Association of zein nanoparticles with botanical compounds for effective pest control systems. Pest Manag. Sci. 2019, 75, 1855. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Feng, Y.-X.; Qi, X.-J.; Wang, Y.; Almaz, B.; Xi, C.; Du, S.-S. Toxicity and repellent activity of essential oil from Mentha piperita Linn. leaves and its major monoterpenoids against three stored product insects. Environ. Sci. Pollut. Res. 2020, 27, 7618. [Google Scholar] [CrossRef] [PubMed]
- Da Fonsêca, D.V.; Filho, C.S.M.B.; Lima, T.C.; de Almeida, R.N.; de Sousa, D.P. Anticonvulsant Essential Oils and Their Relationship with Oxidative Stress in Epilepsy. Biomoecules. 2019, 9, 835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quassinti, L.; Maggi, F.; Ortolani, F.; Lupidi, G.; Petrelli, D.; Vitali, L.A.; Miano, A.; Bramucci, M. Exploring new applications of tulip tree (Liriodendron tulipifera L.): Leaf essential oil as apoptotic agent for human glioblastoma. Environ. Sci. Pollut. Res. 2019, 26, 30485. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Badarau, A.S.; Swamy, M.K.; Shaw, S.; Maggi, F.; da Silva, L.E.; López, V.; Yeung, A.W.K.; Mocan, A.; Atanasov, A.G. Arctium Species Secondary Metabolites Chemodiversity and Bioactivities. Front. Plant. Sci. 2019, 10, 834. [Google Scholar] [CrossRef] [PubMed]
- Gejn, V.L.; Vagapov, A.V.; Nosova, N.V.; Voronina, E.V.; Vahrin, M.I.; Krivenko, A.P. Synthesis and antimicrobial activity of 2-acetyl-5-hydroxy-5-methyl-3-phenyl-1cyclohexanone and alkyl-4-hydroxy-4-methyl2-oxo-6-phenylcyclohexane-1-carboxylates. Pharm. Chem. J. 2010, 44, 245. [Google Scholar] [CrossRef]
- Vagapov, A.V.; Nosova, N.V.; Gejn, V.L.; Syropyatov, B.Y.; Vahrin, M.I.; Danilova, N.V. Methyl 4-hydroxy-4-methyl-6-phenyl-2-cyanoacetylhydrazonocuclohexane-1-carboxylate, obladauschiy analgeticheskoy activnostyu. Patent RF no. 2446150, C 07 C 255/66, A 61 P 29/00.2012. Available online: http://www.pfa.ru/files/AUTOREF_VAGAPOV.pdf (accessed on 10 December 2020).
- Victory, P.; Borrell, J.I.; Vidal-Ferran, A. The Reaction of Malononitrile with Chalcone: A Controversial Chemical Process. Tetrahedron Lett. 1991, 32, 5375. [Google Scholar] [CrossRef]
- Rong, L.-C.; Li, X.-Y.; Yang, F.; Wang, H.-Y.; Shi, D.-Q. 3-Benzoyl-4-hydroxy-2,4,6-triphenylcyclohexane-1,1-dicarbonitrile. Acta Cryst. 2006, E62, o1766. [Google Scholar] [CrossRef]
- Wang, A.-Q.; Jin, T.-S.; Liu, L.-B.; Cheng, Z.-L.; Li, T.-S. A Clean and Efficient Method for the Synthesis of 3-Aroyl-2,4,6-triaryl-4-hydroxy-1,1-cyclohexanedicarbonitriles in Water. Asian J. Chem. 2010, 22, 1977. [Google Scholar]
- Gejn, V.L.; Nosova, N.V.; Vagapov, A.V.; Gejn, L.F. Synthesis of 2,6-diaryl-3-benzoyl-4-hydroxy-4-phenyl-1,1- cyclohexanedinitriles. Russ. J. Org. Chem. 2011, 47, 1247. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Comerford, J.W.; Heath, S.; Jones, O.; Morcillo, M.; North, M. Quinine catalysed asymmetric Michael additions in a sustainable solvent. RSC Adv. 2015, 5, 3678. [Google Scholar] [CrossRef]
- Lu, G.; Cai, C. Synthesis of a series of highly substituted cyclohexanols via Michael addition in an aqueous medium. J. Chem. Res. 2011, 35, 147. [Google Scholar] [CrossRef]
- Natoli, S.N.; Hight, L.M.; Zeller, M.; McMillin, D.R. Photophysical Properties of Pt (II) Polypyridines with Five-versus Six-Membered Chelate Rings: Trade-Offs in Angle Strain. J. Inorg. Chem. 2018, 57, 6521. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Patel, J.S. A Stereoselective Synthesis of the Carbon Backbone of Phoslactomycin B. Eur. J. Org. Chem. 2017, 20, 2981. [Google Scholar] [CrossRef]
- Le Fouler, V.; Duret, G.; Bisseret, P.; Blanchard, N. Copper-mediated synthesis of N-vinyl ynamides from N-vinyl carbamates. Tetrahedron Lett. 2018, 59, 3349. [Google Scholar] [CrossRef]
- Lu, B.; Li, C.; Zhang, L. Gold-Catalyzed Highly Regioselective Oxidation of C−C Triple Bonds without Acid Additives: Propargyl Moieties as Masked α,β-Unsaturated Carbonyls. J. Am. Chem. Soc. 2010, 132, 14070. [Google Scholar] [CrossRef] [Green Version]
- Wipf, P.; Coleman, C.M.; Janjic, J.M.; Iyer, P.S.; Fodor, M.D.; Shafer, Y.A.; Stephenson, C.R.J.; Kendall, C.; Day, B.W. Microwave-assisted “libraries from libraries” approach toward the synthesis of allyl-and C-cyclopropylalkylamides. J. Comb. Chem. 2005, 7, 322. [Google Scholar] [CrossRef]
- Wang, H.; Lu, G.; Sormunen, G.J.; Malik, H.A.; Liu, P.; Montgomery, J. NHC Ligands Tailored for Simultaneous Regio- and Enantiocontrol in Nickel-Catalyzed Reductive Couplings. J. Am. Chem. Soc. 2017, 139, 9317. [Google Scholar] [CrossRef]
- Fujihara, T.; Horimoto, Y.; Mizoe, T.; Sayyed, F.B.; Tani, Y.; Terao, J.; Sakaki, S.; Tsuji, Y. Nickel-Catalyzed Double Carboxylation of Alkynes Employing Carbon Dioxide. Org. Lett. 2014, 18, 4960. [Google Scholar] [CrossRef]
- Wan, H.; Xu, Q.; Gu, P.; Li, H.; Chen, D.; Li, N.; He, J.; Lu, J. AIE-based fluorescent sensors for low concentration toxic ion detection in water. J. Hazard. Mater. 2020, 403, 123656. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A Found. Crystallogr. 2008, 64, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Nakajima Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Golovanov, A.A.; Latypova, D.R.; Bekin, V.V.; Pisareva, V.S.; Vologzhanina, A.V.; Dokichev, A.V. Synthesis of 1,5-disubstituted (e)-pent-2-en-4-yn-1-ones. Russ. J. Org. Chem. 2013, 49, 1264. [Google Scholar] [CrossRef]
- Saulnier, S.; Golovanov, A.A.; Vasilyev, A.V. A controlled tandem transformation of conjugated enynones with arenes under superelectrophilic activation leading to aryl-substituted dienones and indenes. RSC Adv. 2016, 6, 103546. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
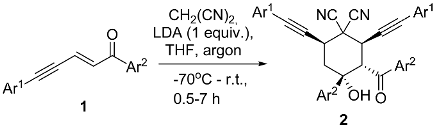
| En-Try | Starting Enynone | Reaction Conditions | Reaction Products | |
|---|---|---|---|---|
| Temperature, Time | Enynone: CH2(CN)2 Ratio | |||
| 1 |  | r.t., 3 h | 2:1 | 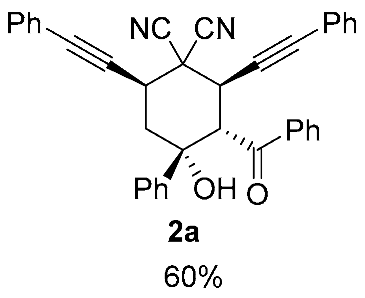 |
| 2 | 1a | r.t., 1 h | 1:1 | 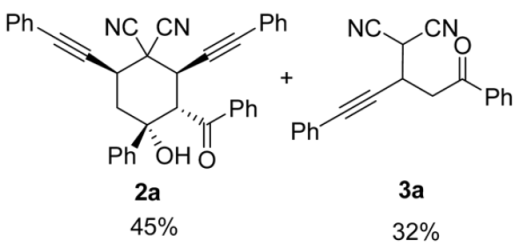 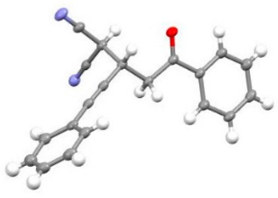 X-ray of 3a (CCDC 1998291) |
| 3 | 1a | −40 °C, 0.5 h | 1:1 | 2a (48%) + 3a (53%) |
| 4 | 1a | −70 °C, 0.5 h | 1:1 | 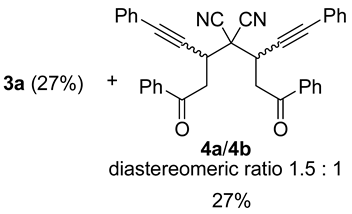 |
| 5 | 1a | −70 °C, 3 h | 1:1 | 2a (17%) + 3a (23%) + 4a/4b (diastereomeric ratio 2:1, 31%) |
| 6 |  | r.t., 3 h | 2:1 |  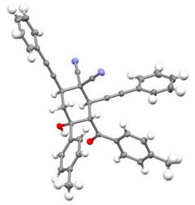 X-ray of 2b (CCDC 1998357) |
| 7 | 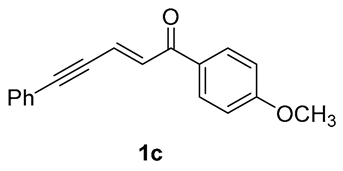 | r.t., 3 h | 2:1 | 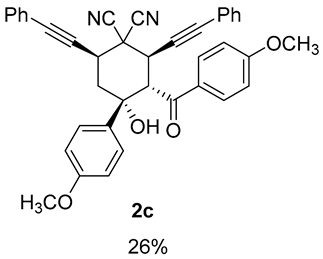 |
| 8 | 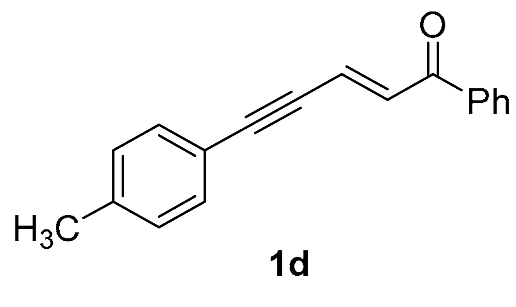 | r.t., 3 h | 2:1 | 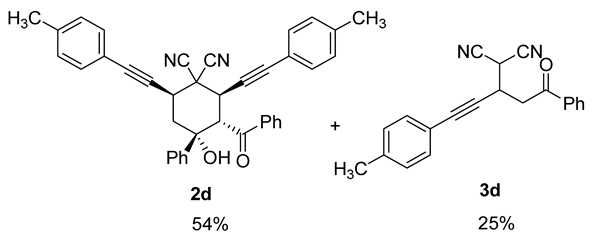 |
| 9 | 1d | r.t., 7 h | 2:1 | 2d (25%) + 3d (23%) |
| 10 | 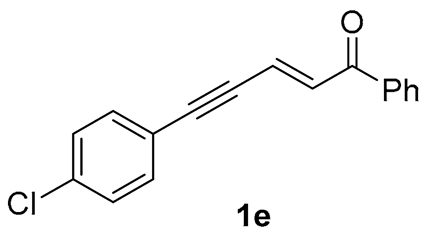 | r.t., 3 h | 2:1 | 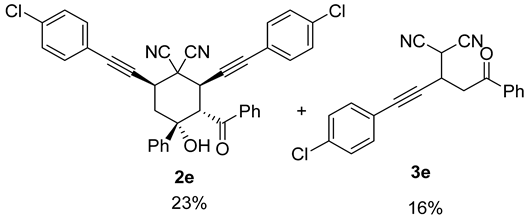 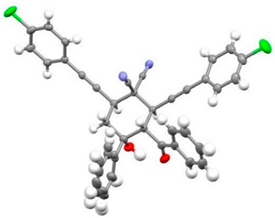 X-ray of 2e (CCDC 1998289) |
| 11 | 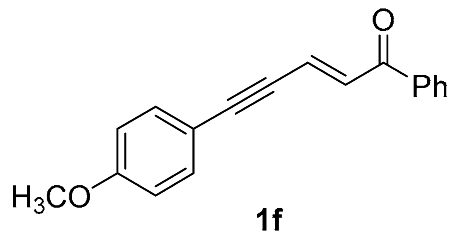 | r.t., 3 h | 2:1 | 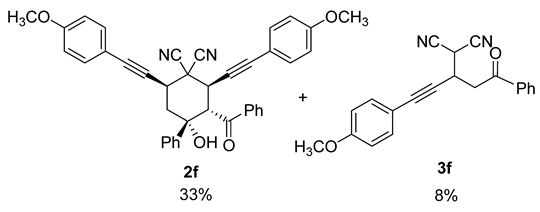 |
| 12 | 1f | r.t., 7 h | 2:1 | 2f (13%) + 3f (2%) |
| 13 | 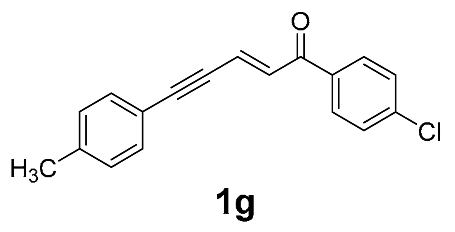 | r.t., 3 h | 2:1 |  |
| 14 |  | r.t., 3 h | 2:1 |  |
| 15 |  | r.t., 3 h | 2:1 | Complex mixture of reaction products. |
Sample Availability: Samples of the compounds 2, 3, 4 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Igushkina, A.V.; Golovanov, A.A.; Boyarskaya, I.A.; Kolesnikov, I.E.; Vasilyev, A.V. Stereoselective Synthesis of Multisubstituted Cyclohexanes by Reaction of Conjugated Enynones with Malononitrile in the Presence of LDA. Molecules 2020, 25, 5920. https://doi.org/10.3390/molecules25245920
Igushkina AV, Golovanov AA, Boyarskaya IA, Kolesnikov IE, Vasilyev AV. Stereoselective Synthesis of Multisubstituted Cyclohexanes by Reaction of Conjugated Enynones with Malononitrile in the Presence of LDA. Molecules. 2020; 25(24):5920. https://doi.org/10.3390/molecules25245920
Chicago/Turabian StyleIgushkina, Anastasiya V., Alexander A. Golovanov, Irina A. Boyarskaya, Ilya E. Kolesnikov, and Aleksander V. Vasilyev. 2020. "Stereoselective Synthesis of Multisubstituted Cyclohexanes by Reaction of Conjugated Enynones with Malononitrile in the Presence of LDA" Molecules 25, no. 24: 5920. https://doi.org/10.3390/molecules25245920
APA StyleIgushkina, A. V., Golovanov, A. A., Boyarskaya, I. A., Kolesnikov, I. E., & Vasilyev, A. V. (2020). Stereoselective Synthesis of Multisubstituted Cyclohexanes by Reaction of Conjugated Enynones with Malononitrile in the Presence of LDA. Molecules, 25(24), 5920. https://doi.org/10.3390/molecules25245920