Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes


Abstract
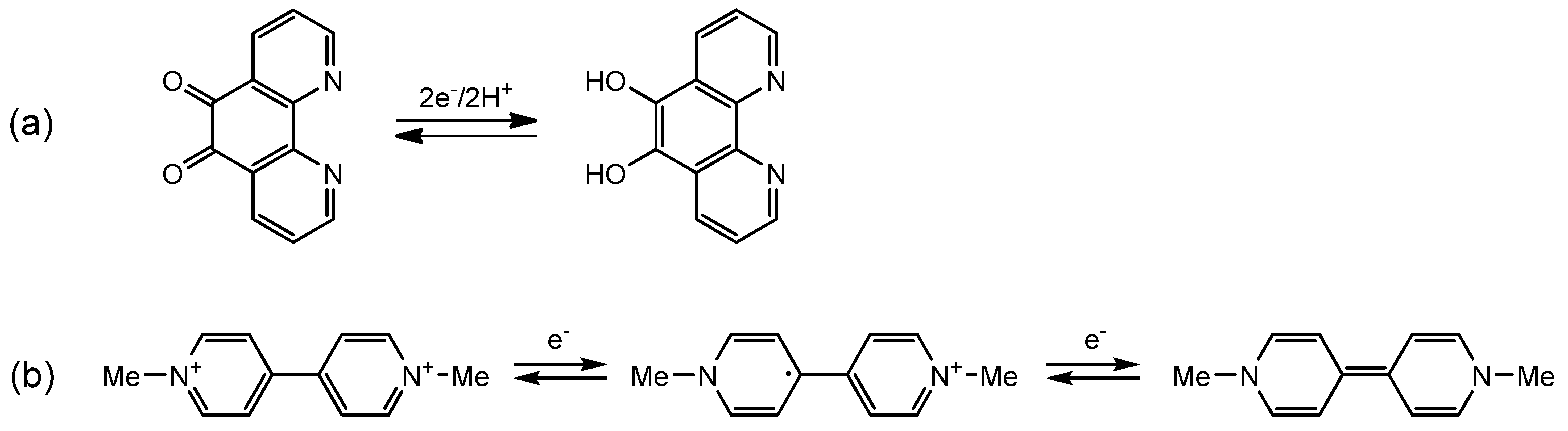
:1. Introduction
2. Results and Discussion
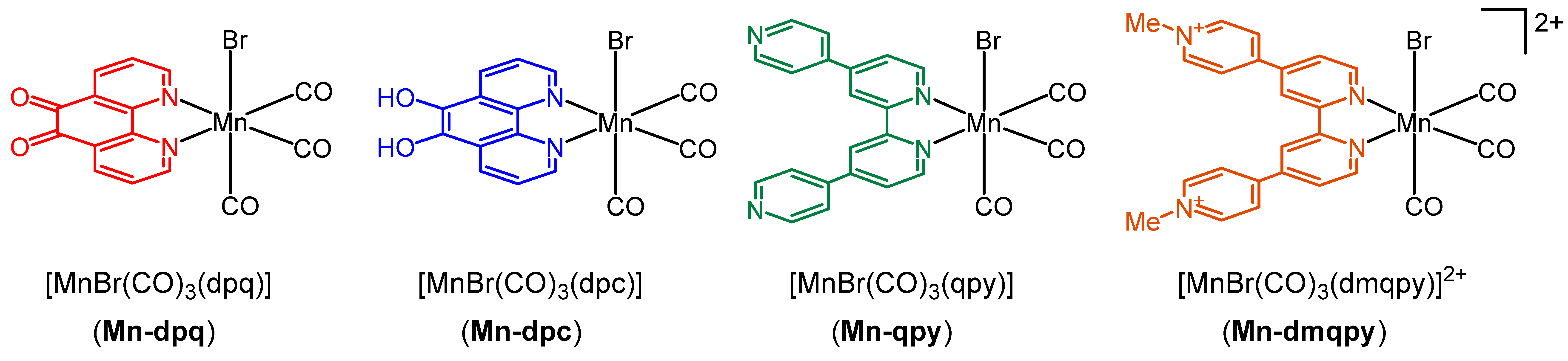
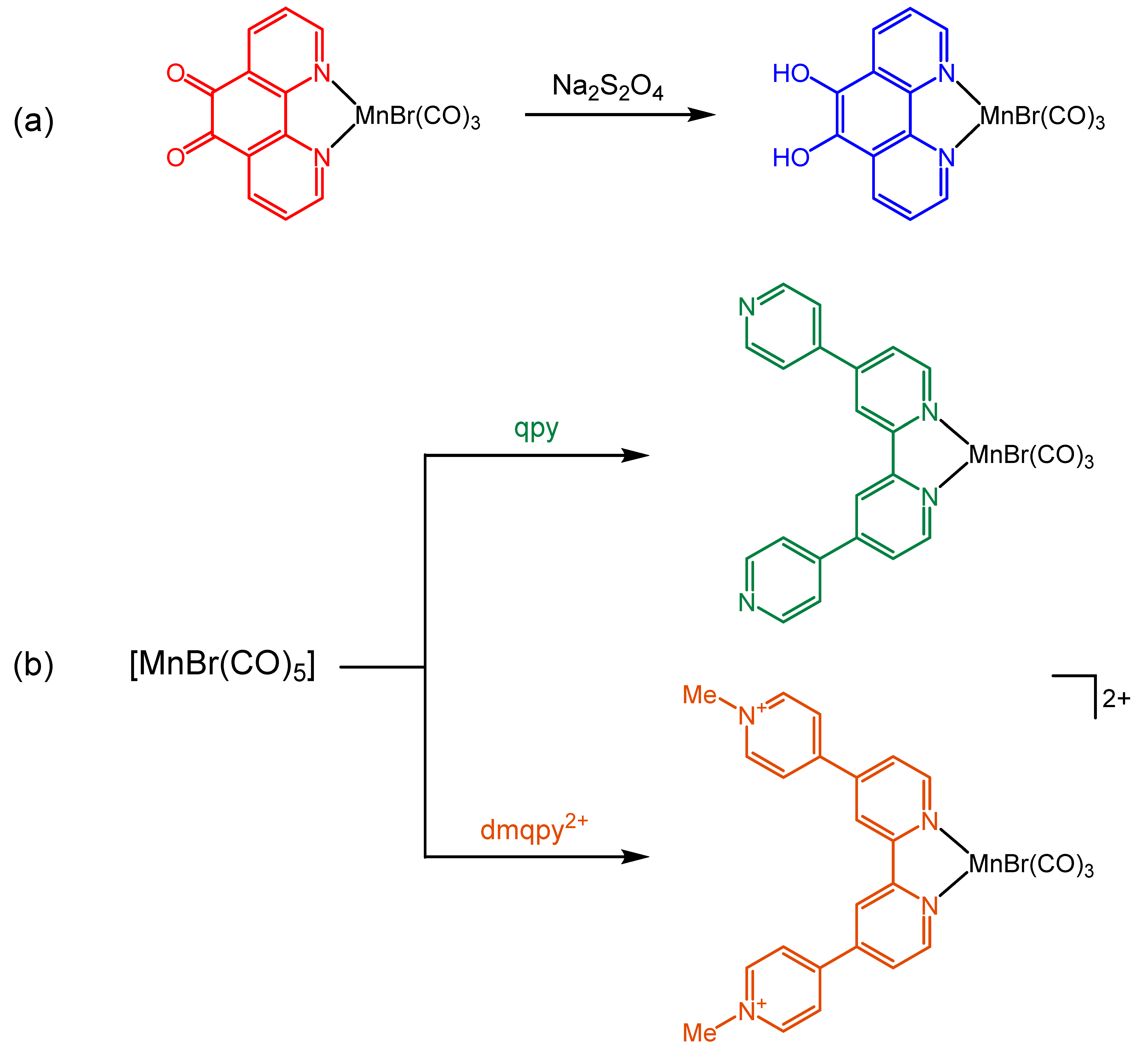
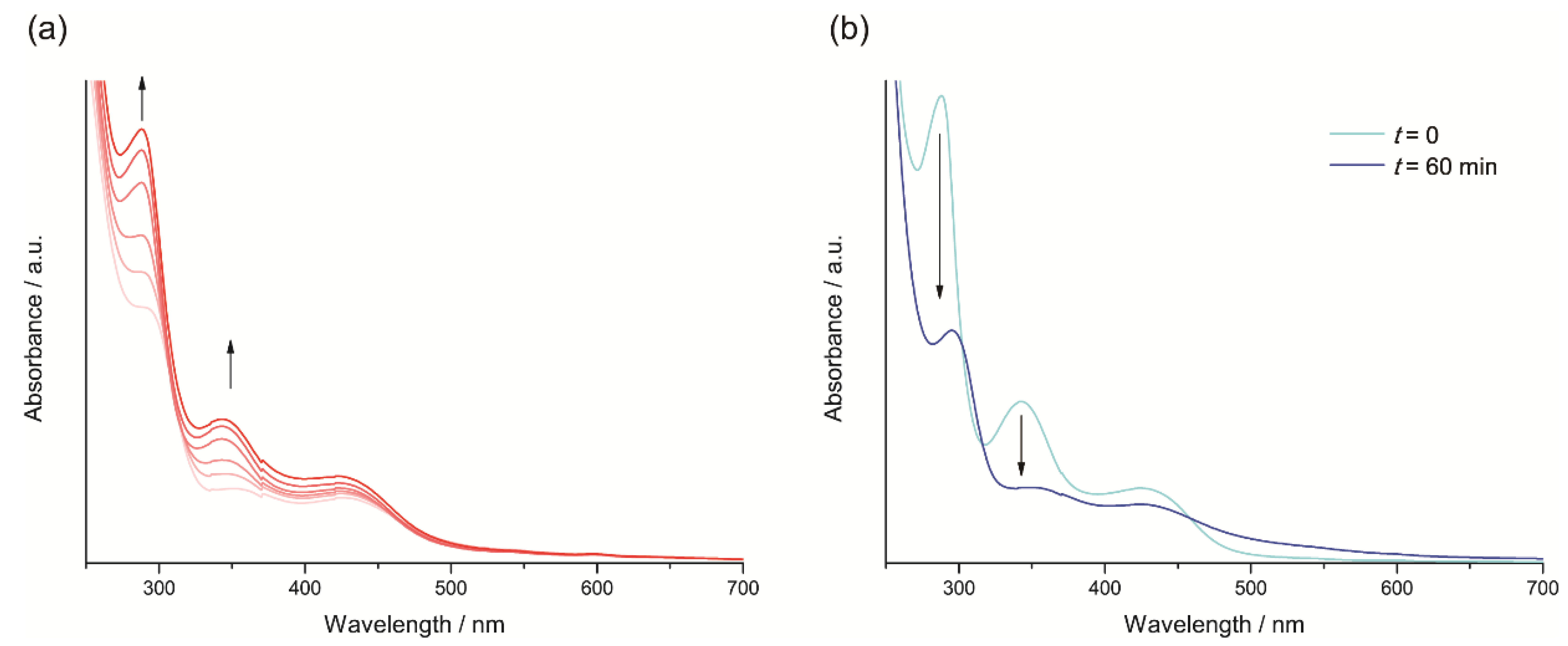
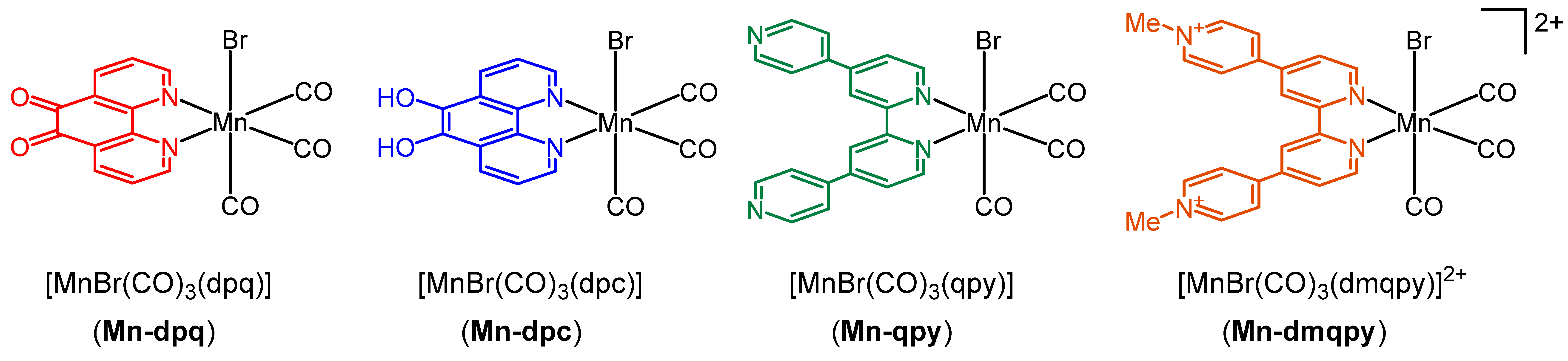
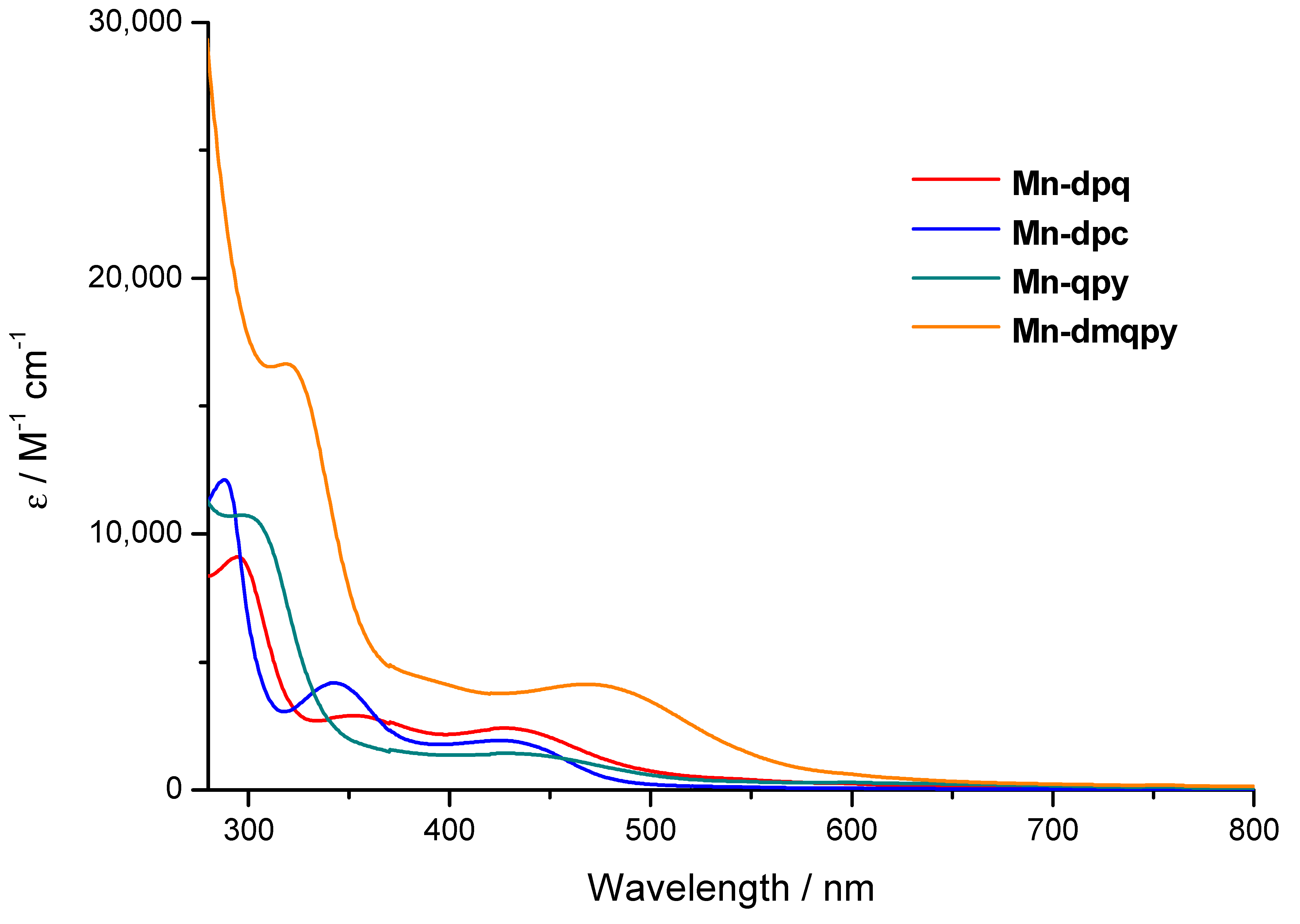
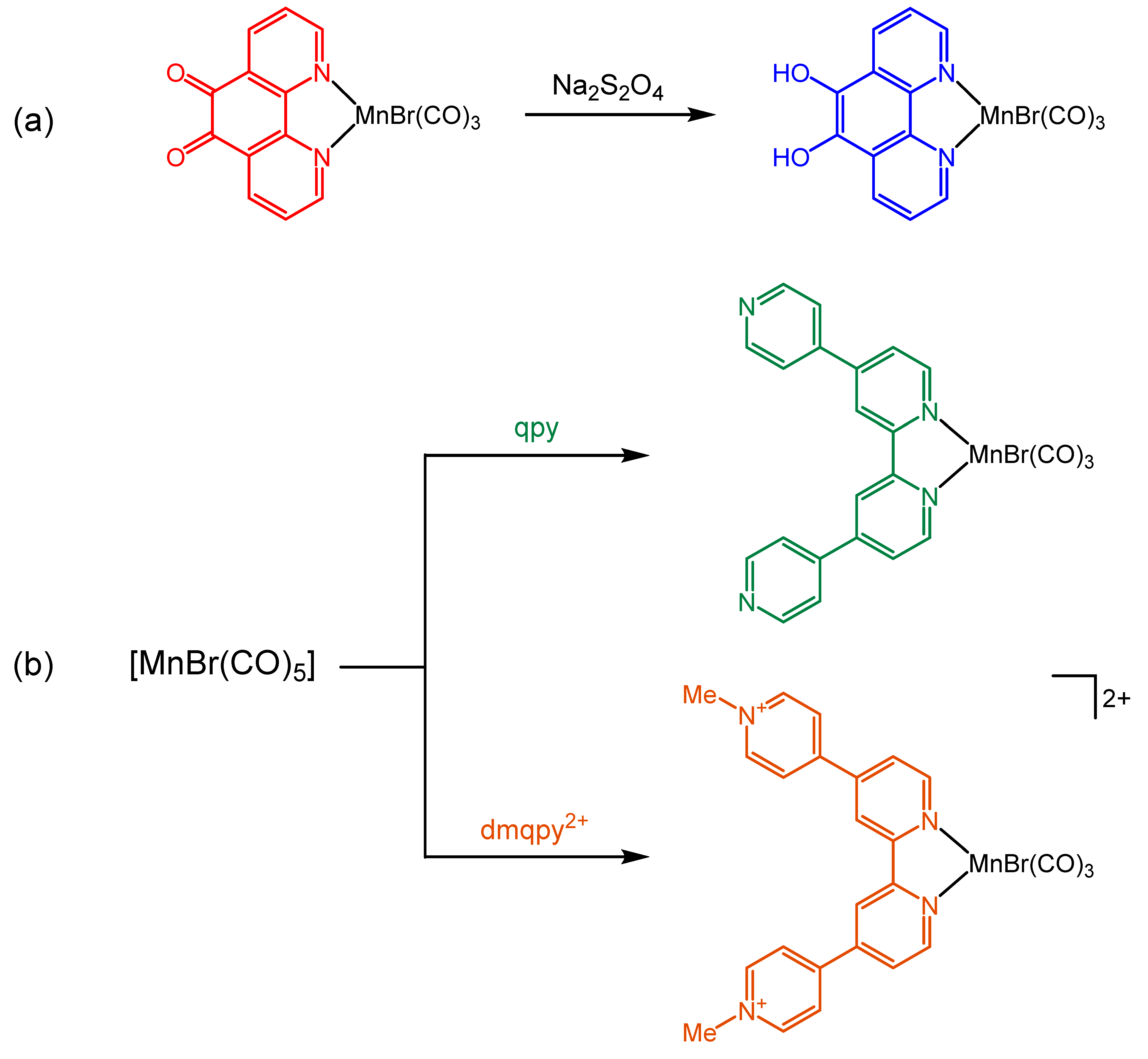
2.1. Synthesis and Spectroscopic Characterization of the Tricarbonylmanganese Complexes
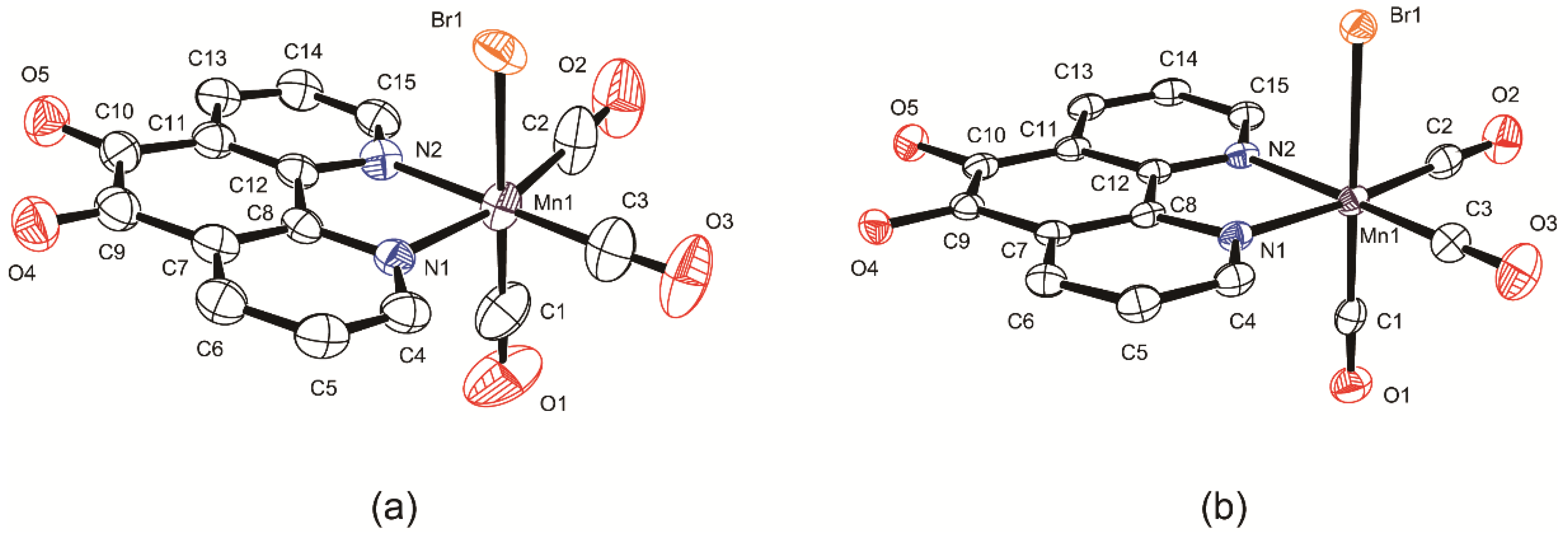
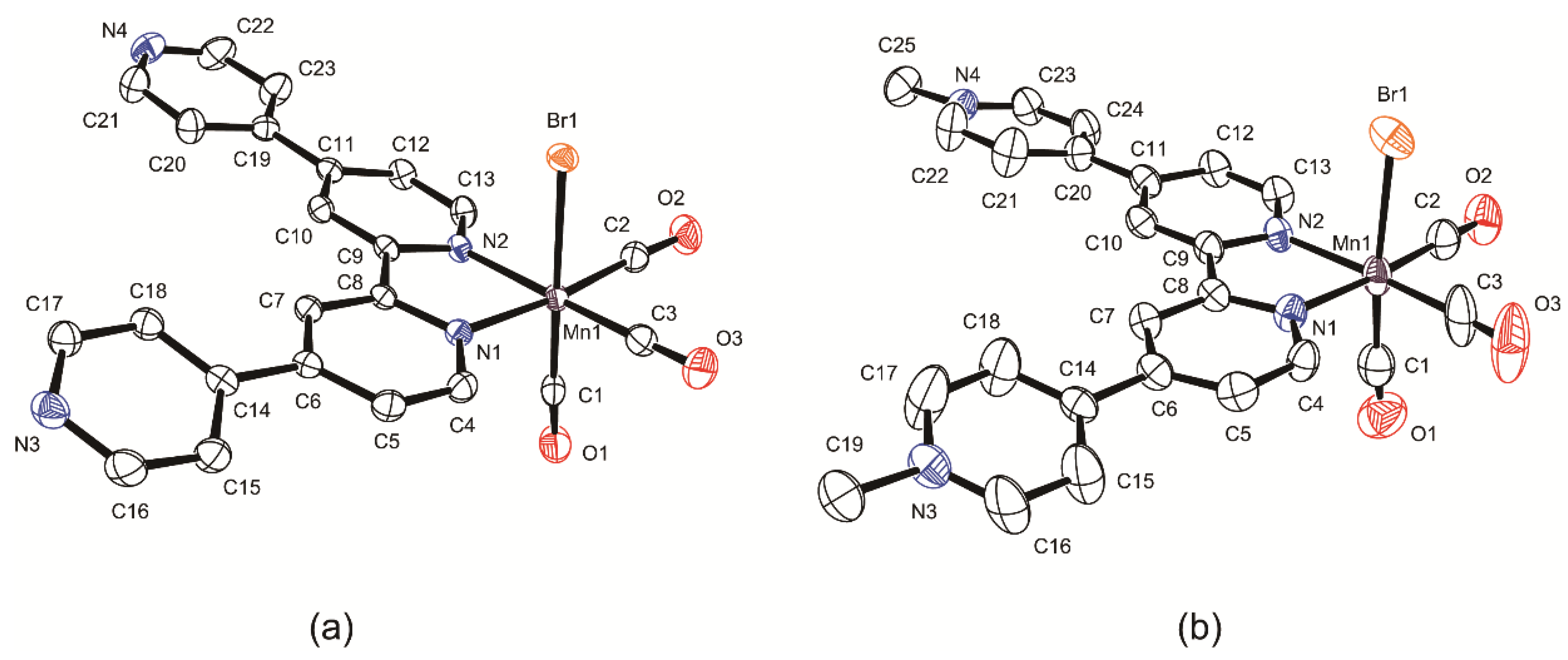
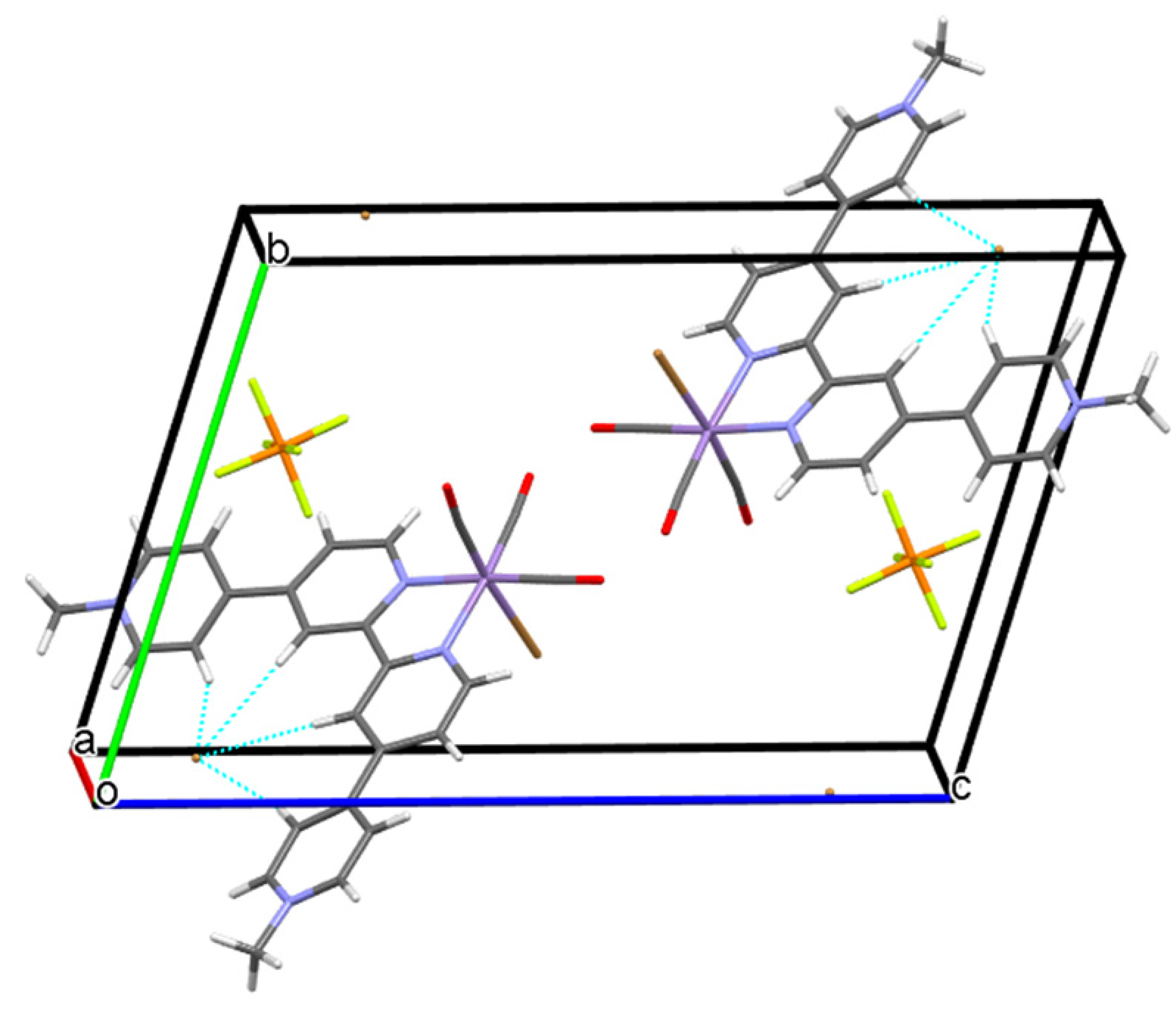
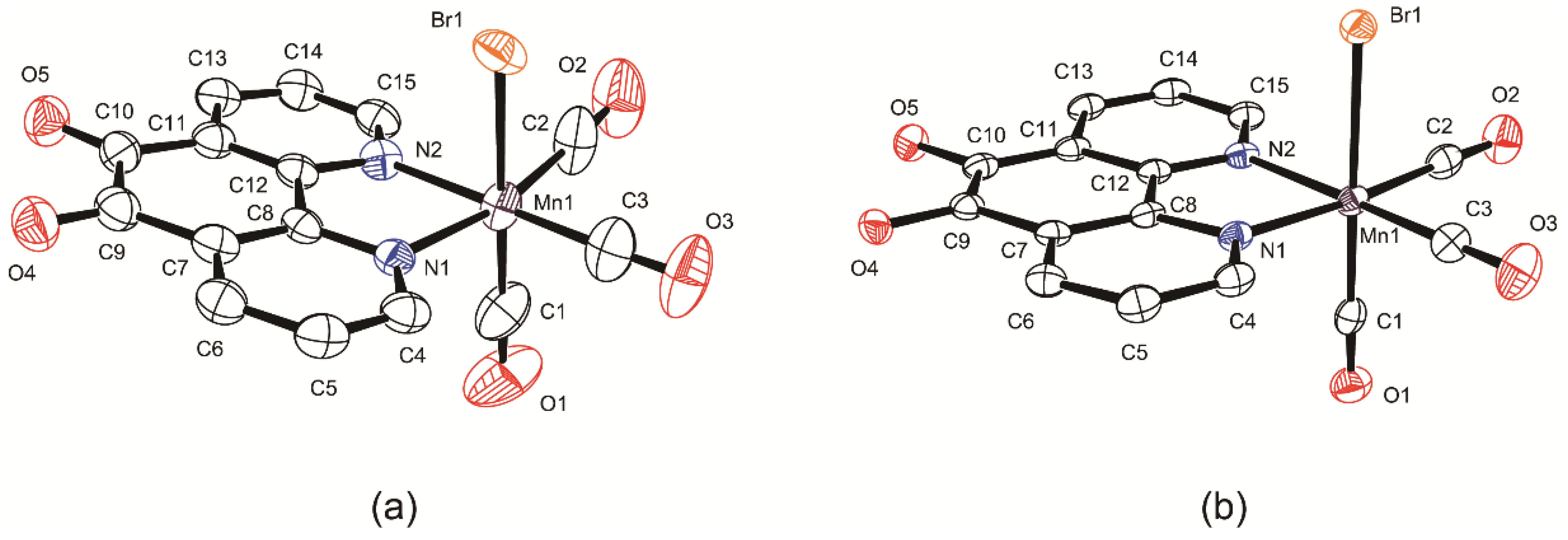
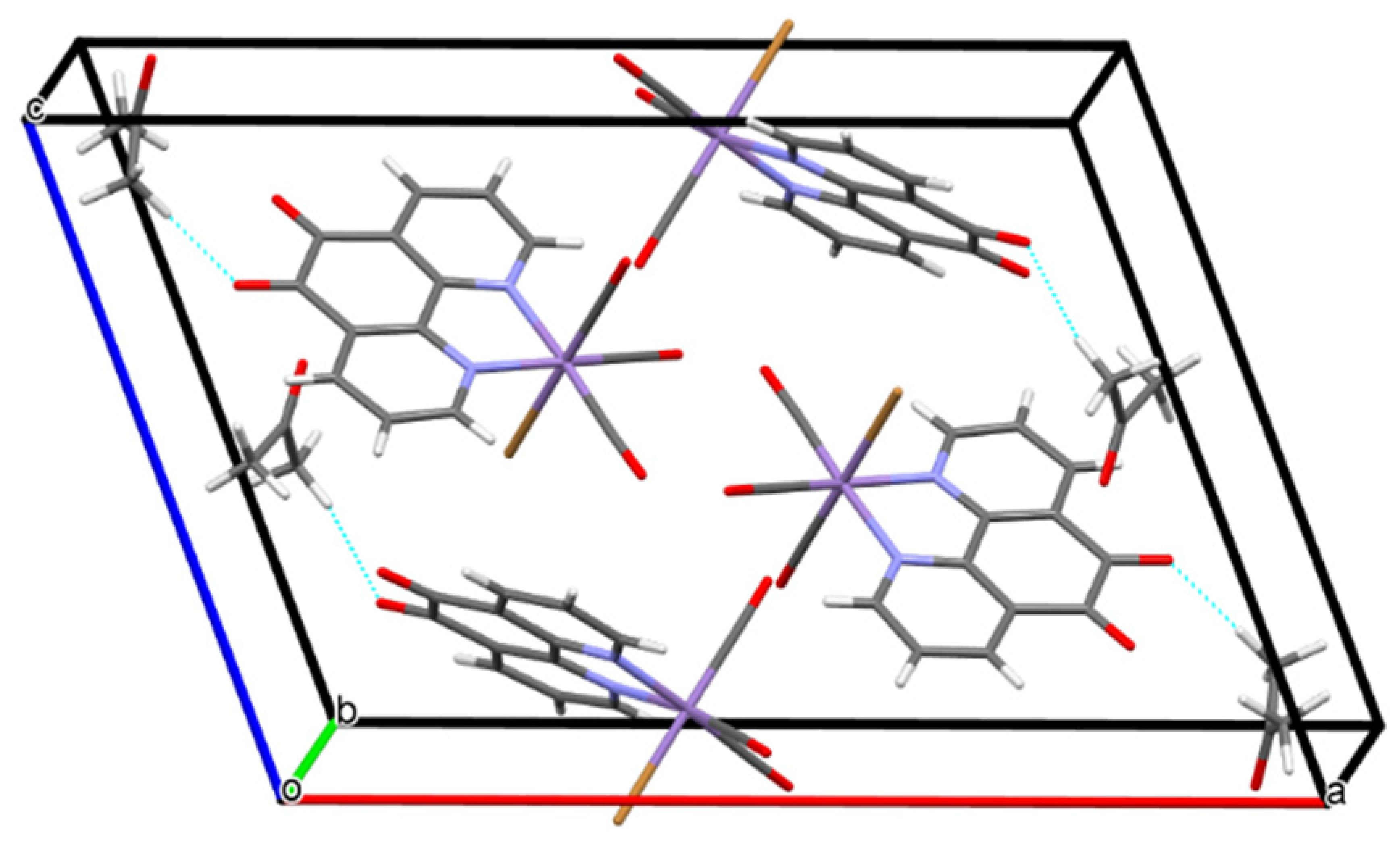
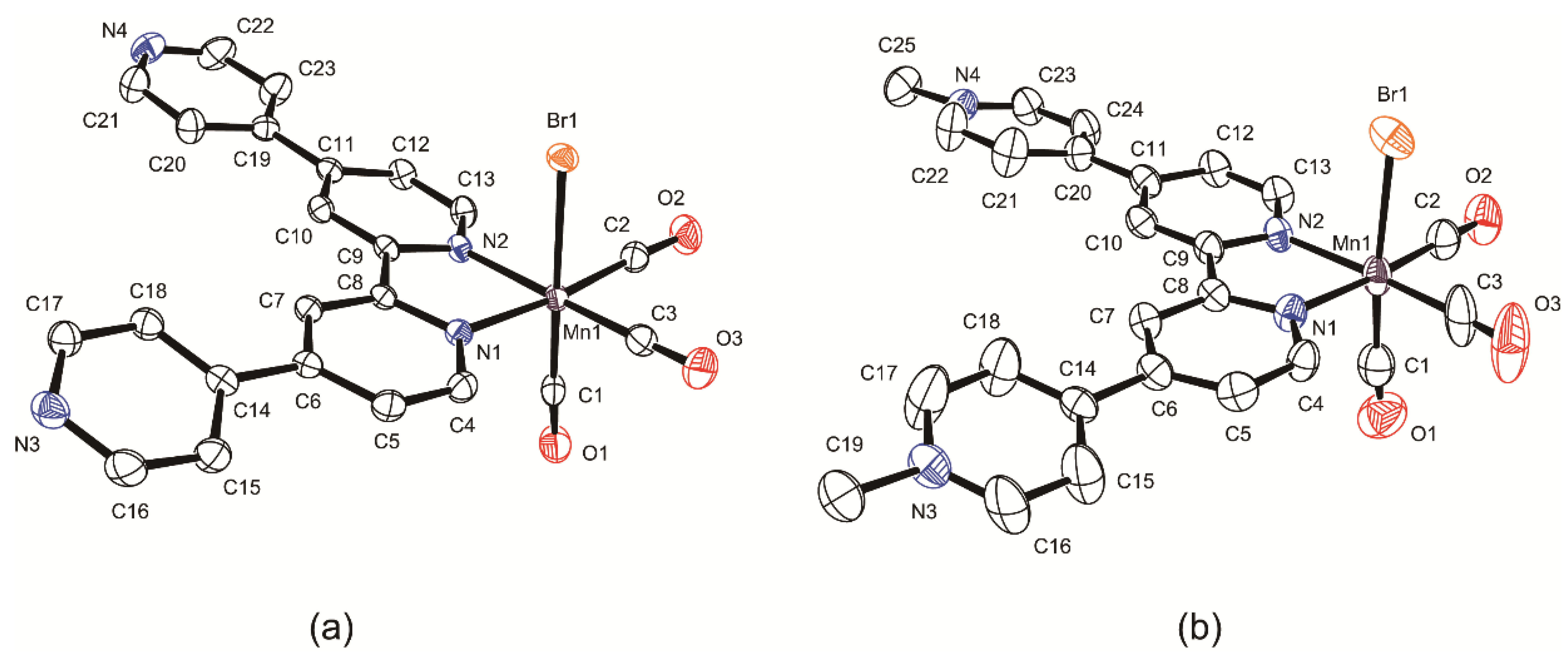
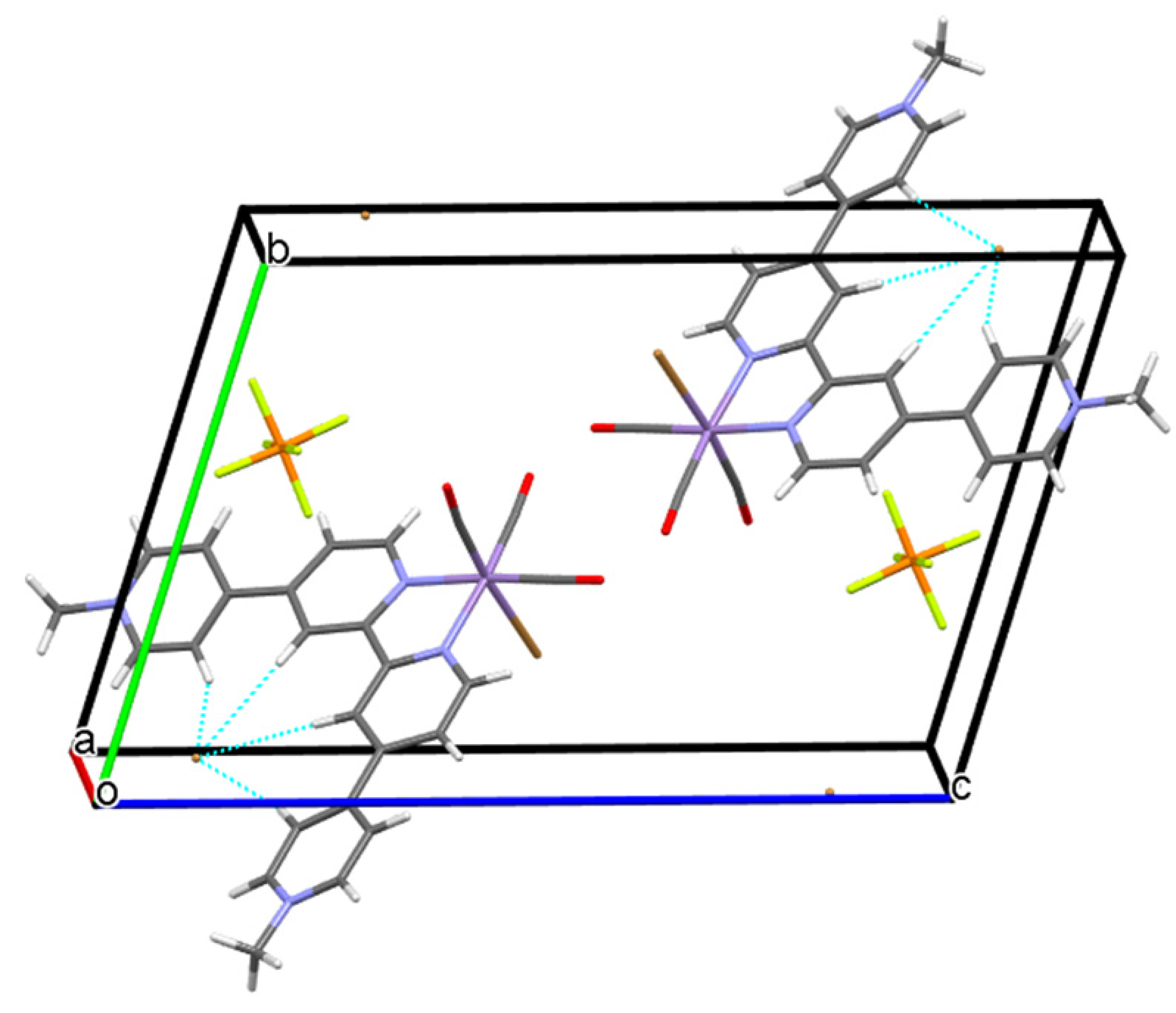
2.2. Structural Characterization of the Tricarbonylmanganese Complexes
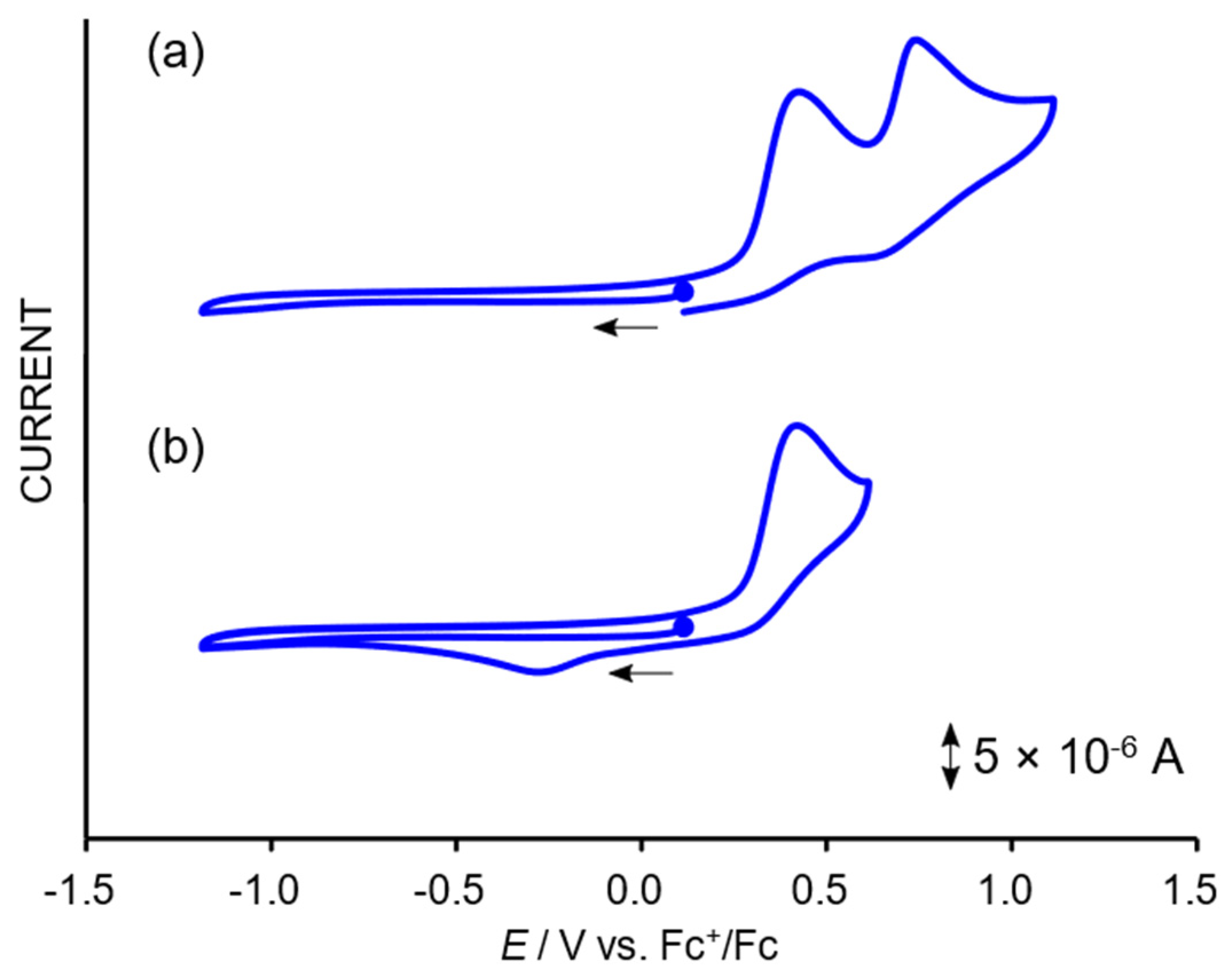
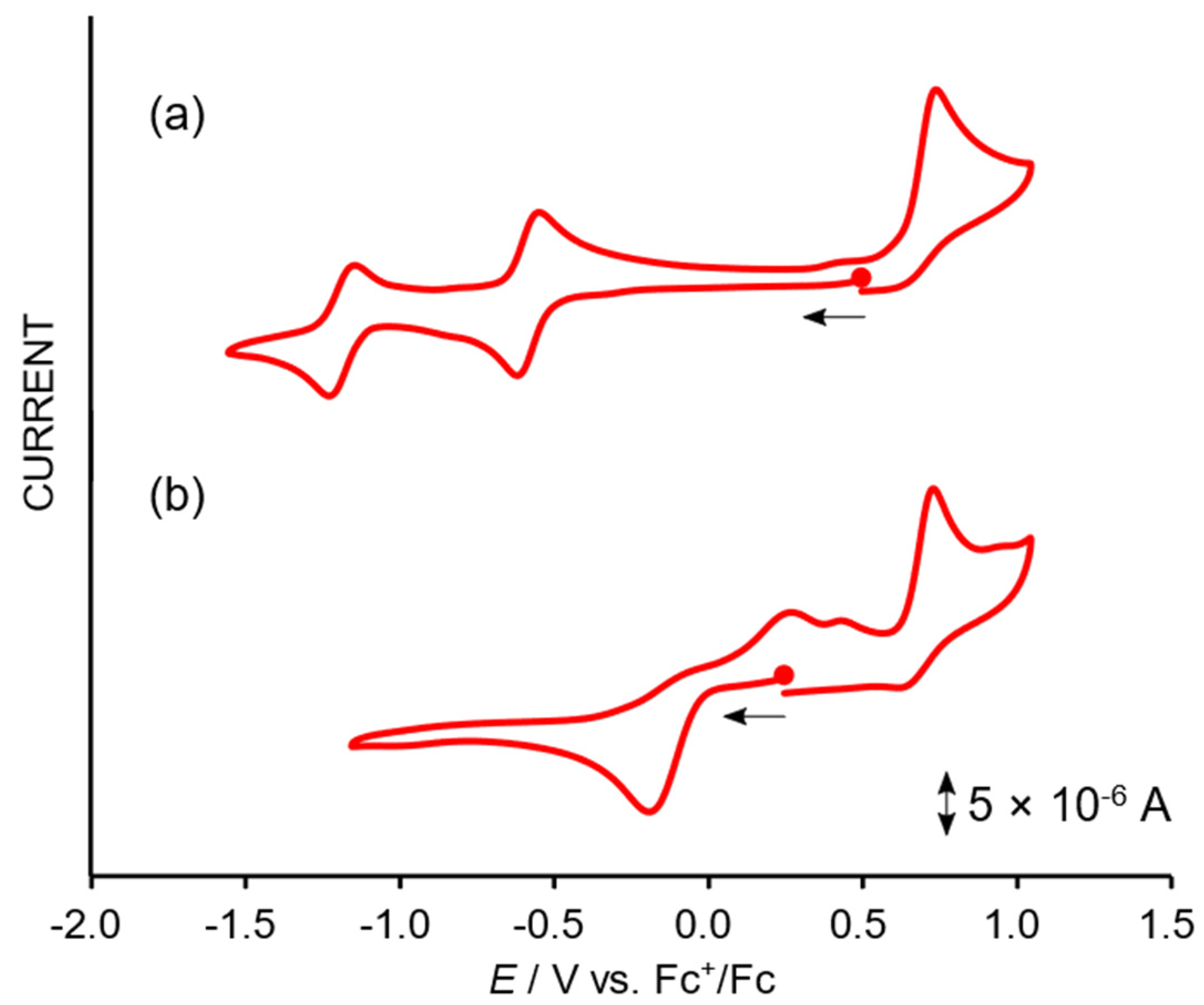
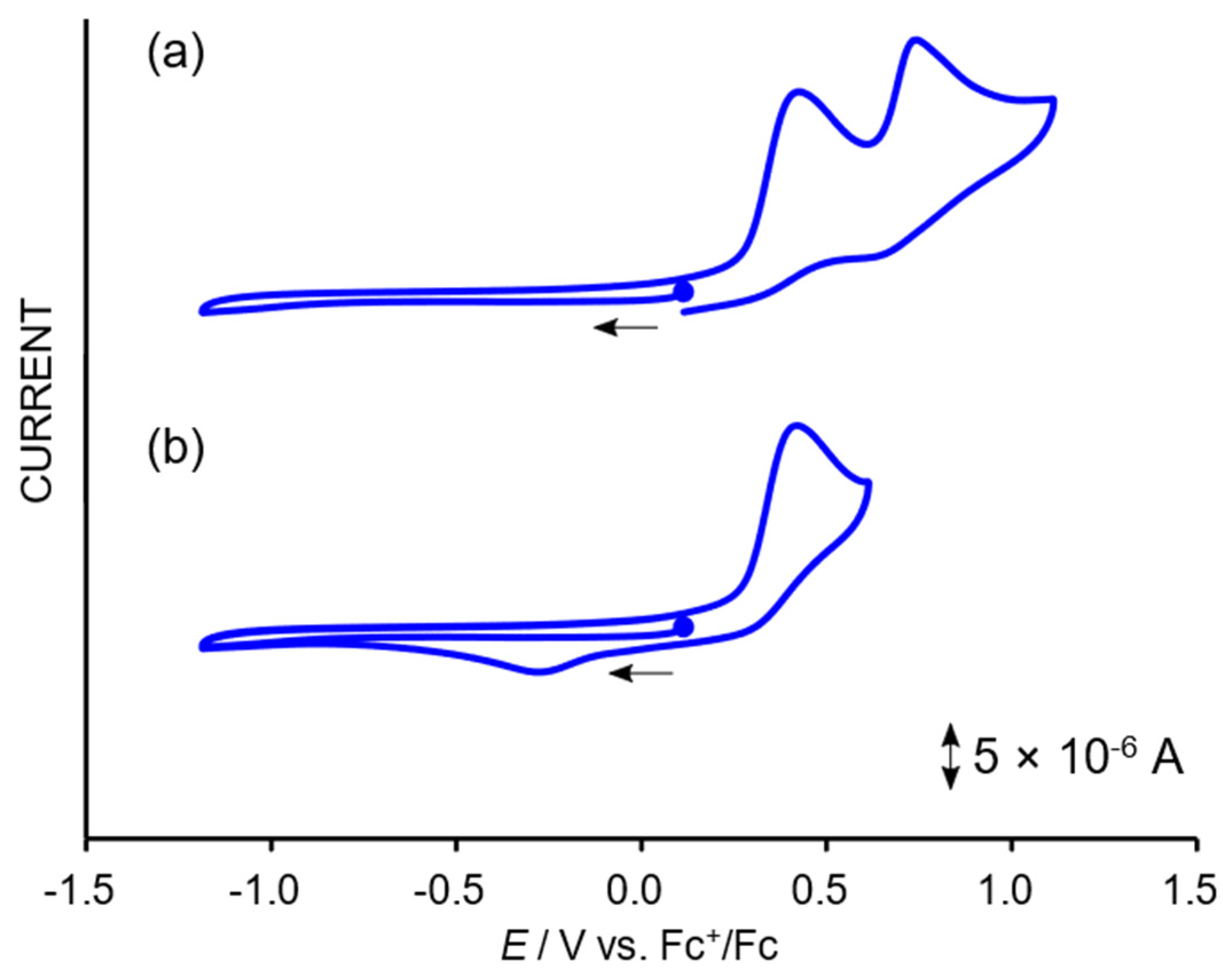
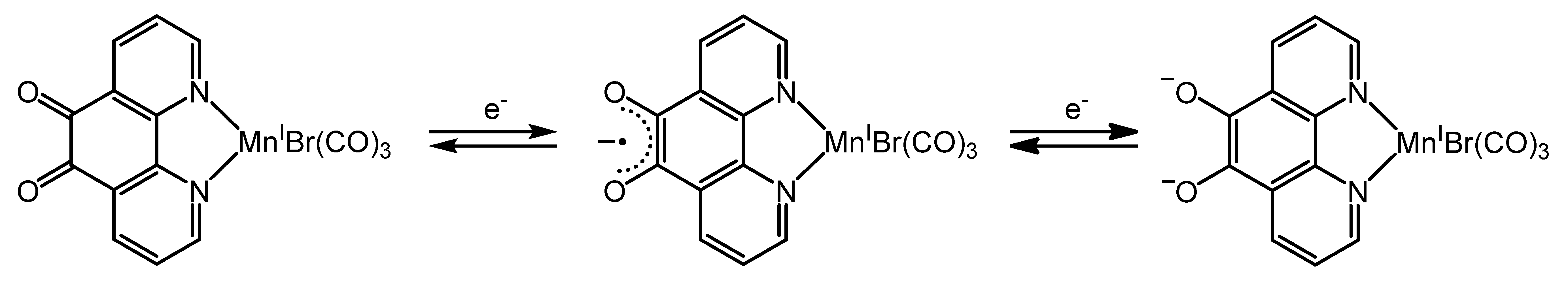
2.3. Redox Properties of the Tricarbonylmanganese Complexes
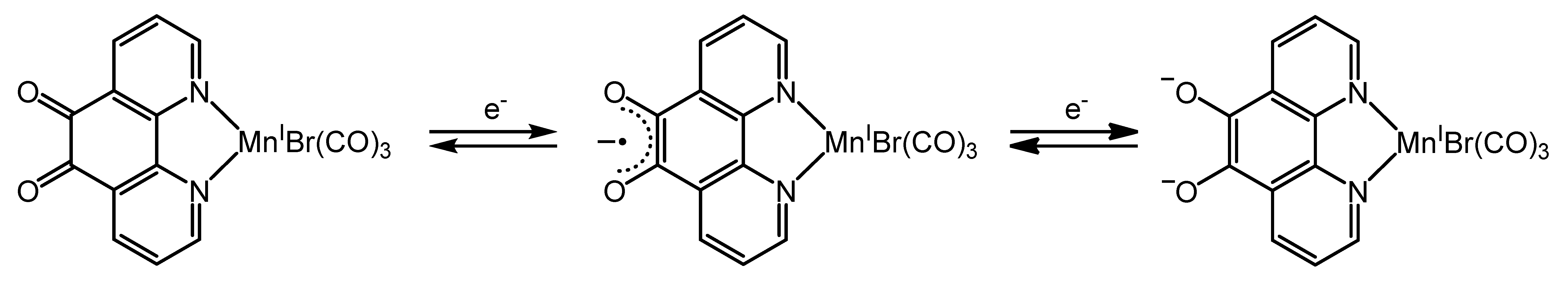
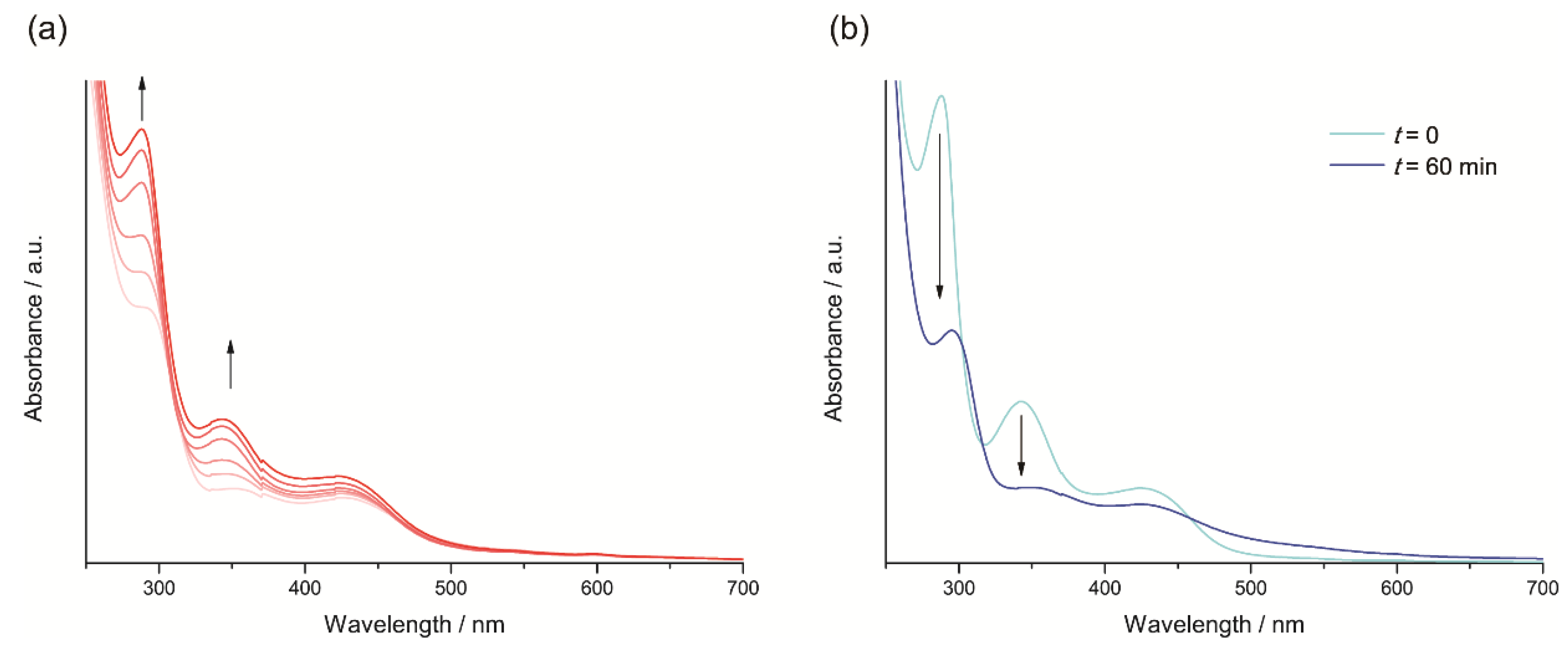
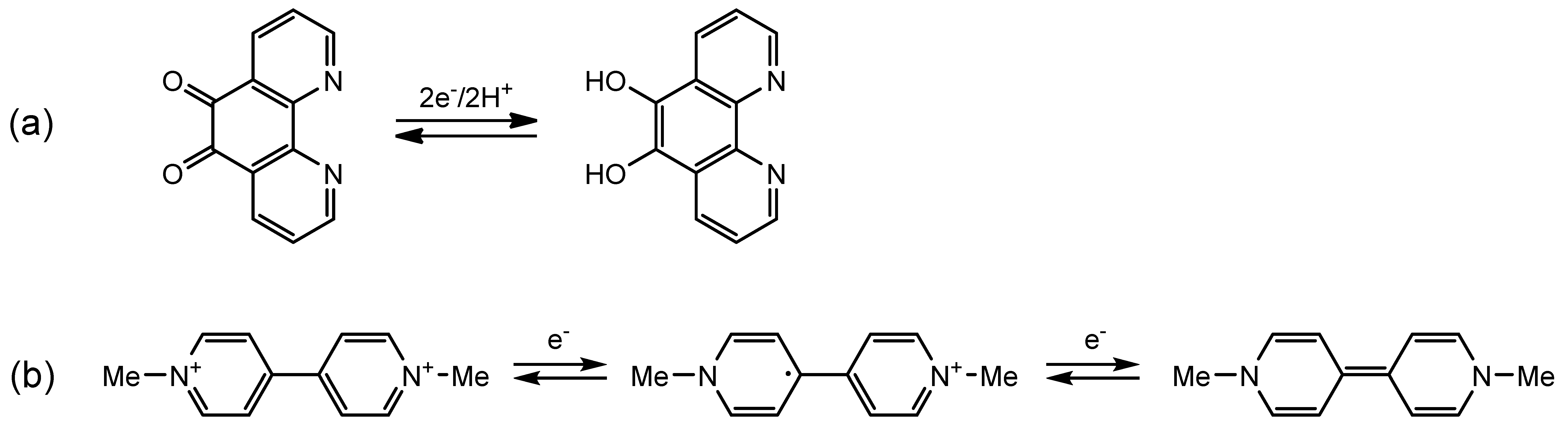
2.3.1. Phenanthroline-Based Complexes: Interconversion between Quinone and Catechol Units
2.3.2. Bipyridine-Based Complexes
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of the Complexes
3.2.1. Synthesis of fac-[MnBr(CO)3(dpc)]
3.2.2. Synthesis of fac-[MnBr(CO)3(qpy)] and fac-[MnBr(CO)3(dmqpy)](PF6)2
3.3. X-ray Crystallographic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a substitute for rhenium in CO2 reduction catalysts: The importance of acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Riplinger, C.; Sampson, M.D.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic contrasts between manganese and rhenium bipyridine electrocatalysts for the reduction of carbon dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef]
- Takeda, H.; Koizumi, H.; Okamoto, K.; Ishitani, O. Photocatalytic CO2 reduction using a Mn complex as a catalyst. Chem. Commun. 2014, 50, 1491–1493. [Google Scholar] [CrossRef]
- Torralba-Peñalver, E.; Luo, Y.; Compain, J.-D.; Chardon-Noblat, S.; Fabre, B. Selective catalytic electroreduction of CO2 at silicon nanowires (SiNWs) photocathodes using non-noble metal-based manganese carbonyl bipyridyl molecular catalysts in solution and grafted onto SiNWs. ACS Catal. 2015, 5, 6138–6147. [Google Scholar] [CrossRef] [Green Version]
- Sampson, M.D.; Kubiak, C.P. Electrocatalytic dihydrogen production by an earth-abundant manganese bipyridine catalyst. Inorg. Chem. 2015, 54, 6674–6676. [Google Scholar] [CrossRef]
- Fei, H.; Sampson, M.D.; Lee, Y.; Kubiak, C.P.; Cohen, S.M. Photocatalytic CO2 reduction to formate using a Mn(I) molecular catalyst in a robust metal-organic framework. Inorg. Chem. 2015, 54, 6821–6828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-X.; Hu, C.-Y.; Wang, W.; Wang, H.; Bian, Z.-Y. Visible light driven reduction of CO2 catalyzed by an abundant manganese catalyst with zinc porphyrin photosensitizer. Appl. Catal. A Gen. 2016, 522, 145–151. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubiak, C.P. Manganese electrocatalysts with bulky bipyridine ligands: Utilizing Lewis acids to promote carbon dioxide reduction at low overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Stanbury, M.; Compain, J.-D.; Trejo, M.; Smith, P.; Gouré, E.; Chardon-Noblat, S. Mn-carbonyl molecular catalysts containing a redox-active phenanthroline-5,6-dione for selective electro- and photoreduction of CO2 to CO or HCOOH. Electrochim. Acta 2017, 240, 288–299. [Google Scholar] [CrossRef]
- Morgan, R.J.; Baker, A.D. 2,2′:4,4′’:4′,4′’’-Quaterpyridyl: A building block for the preparation of novel redox reagents. 1. Preparation and quaternization. J. Org. Chem. 1990, 55, 1986–1993. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Eckert, T.S.; Bruice, T.C. Chemical properties of phenanthrolinequinones and the mechanism of amine oxidation by o-quinones of medium redox potentials. J. Am. Chem. Soc. 1983, 105, 4431–4441. [Google Scholar] [CrossRef]
- Wu, J.-Z.; Li, H.; Zhang, J.-G.; Xu, J.-H. Synthesis and DNA binding behavior of a dipyridocatecholate bridged dicopper(II) complex. Inorg. Chem. Commun. 2002, 5, 71–75. [Google Scholar] [CrossRef]
- Liu, Y.; Hammitt, R.; Lutterman, D.A.; Thummel, R.P.; Turro, C. Marked differences in light-switch behavior of Ru(II) complexes possessing a tridentate DNA intercalating ligand. Inorg. Chem. 2007, 46, 6011–6021. [Google Scholar] [CrossRef]
- Paw, W.; Eisenberg, R. Synthesis, characterization, and spectroscopy of dipyridocatecholate complexes of platinum. Inorg. Chem. 1997, 36, 2287–2293. [Google Scholar] [CrossRef]
- Ambroise, A.; Maiya, B.G. Ruthenium(II) complexes of 6,7-dicyanodipyridoquinoxaline: Synthesis, luminescence studies, and DNA interaction. Inorg. Chem. 2000, 39, 4264–4272. [Google Scholar] [CrossRef]
- Fujihara, T.; Okamura, R.; Wada, T.; Tanaka, K. Coordination ability of 1,10-phenanthroline-5,6-dione: Syntheses and redox behavior of a Ru(II) complex with an o-quinoid moiety and of bridged Ru(II)–M(II) complexes (M = Pd, Pt). Dalton Trans. 2003, 3221–3226. [Google Scholar] [CrossRef]
- We could not observe signals derived from the OH groups of the catechol units even in DMSO-d6.
- Shi, A.; Pokhrel, M.R.; Bossmann, S.H. Synthesis of highly charged ruthenium(II)-quaterpyridinium complexes: A bottom-up approach to monodisperse nanostructures. Synthesis 2007, 4, 505–514. [Google Scholar]
- Coe, B.J.; Harper, E.C.; Helliwell, M.; Ta, Y.T. Syntheses and properties of complexes with bis(2,2′-bipyridyl)ruthenium(II) moieties coordinated to 4,4′:2′,2′’:4′’,4′’’-quaterpyridinium ligands. Polyhedron 2011, 30, 1830–1841. [Google Scholar] [CrossRef]
- Kurtz, D.A.; Dhakal, B.; Hulme, R.J.; Nichol, G.S.; Felton, G.A.N. Correlations between photophysical and electrochemical properties for a series of new Mn carbonyl complexes containing substituted phenanthroline ligands. Inorg. Chim. Acta 2015, 427, 22–26. [Google Scholar] [CrossRef]
- Henke, W.C.; Otolski, C.J.; Moore, W.N.G.; Elles, C.G.; Blakemore, J.D. Ultrafast spectroscopy of [Mn(CO)3] complexes: Tuning the kinetics of light-driven CO release and solvent binding. Inorg. Chem. 2020, 59, 2178–2187. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Chabolla, S.A.; Dang, T.; Kubiak, C.P. Developing a mechanistic understanding of molecular electrocatalysts for CO2 reduction using infrared spectroelectrochemistry. Organometallics 2014, 33, 4550–4559. [Google Scholar] [CrossRef]
- Jimenez, J.; Chakraborty, I.; Mascharak, P.K. Synthesis and assessment of CO-release capacity of manganese carbonyl complexes derived from rigid α-diimine ligands of varied complexity. Eur. J. Inorg. Chem. 2015, 30, 5021–5026. [Google Scholar] [CrossRef] [Green Version]
- Lense, S.; Guzei, I.A.; Andersen, J.; Thao, K.C. Crystal structures of a manganese(I) and a rhenium(I) complex of a bipyridine ligand with a non-coordinating benzoic acid moiety. Acta Cryst. 2018, E74, 731–736. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.J.; Smith, C.L.; Neri, G.; Whitehead, G.F.S.; Robertson, C.M.; Cowan, A.J. Improving the efficiency of electrochemical CO2 reduction using immobilized manganese complexes. Faraday Discuss. 2015, 183, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Wadayama, K.; Takase, T.; Oyama, D. fac-Bromido/chlorido(0.50/0.50)[3-carbamoyl-1-(1,10-phenanthrolin-2-ylmethyl)pyridinium-κ2N,N’]tricarbonylmanganese(I) 0.49-bromide 0.51-chloride methanol monosolvate. IUCrData 2019, 4, x181792. [Google Scholar] [CrossRef]
- Kanno, T.; Takase, T.; Oyama, D. Synthesis and crystal structures of manganese(I) carbonyl complexes bearing ester-substituted α-diimine ligands. Acta Cryst. 2020, E76, 1433–1436. [Google Scholar]
- Calderazzo, F.; Marchetti, F.; Pampaloni, G.; Passarelli, V. Co-ordination properties of 1,10-phenanthroline-5,6-dione towards group 4 and 5 metals in low and high oxidation states. J. Chem. Soc. Dalton Trans. 1999, 4389–4396. [Google Scholar] [CrossRef]
- Lin, X.-Y.; Tang, S.-J.; Wu, W.-S. 5,6-Dihydroxy-1,10-phenanthrolin-1-ium chloride dihydrate. Acta Cryst. 2009, E65, o2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, C.A.; Abruna, H.D. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorg. Chem. 1985, 24, 4263–4267. [Google Scholar] [CrossRef]
- When Mn-dpq was measured with a Pt working electrode, only an oxidation wave was observed at 0.43 V. Thus, the electrode reactions taking place under these conditions may depend on the material employed (i.e., carbon or platinum); Lei, Y.; Anson, F.C. Hydration of the carbonyl groups in 1,10-phenanthroline-5,6-dione induced by binding protons or metal cations to the pyridine nitrogen sites. J. Am. Chem. Soc. 1995, 117, 9849–9854.
- We could not carry out CV measurements for dpc due to its low solubility in almost all solvents.
- Stallings, M.D.; Morrison, M.M.; Sawyer, D.T. Redox chemistry of metal-catechol complexes in aprotic media. 1. Electrochemistry of substituted catechols and their oxidation products. Inorg. Chem. 1981, 20, 2655–2660. [Google Scholar] [CrossRef]
- Pordel, S.; White, J.K. Impact of Mn(I) photoCORM ligand set on photochemical intermediate formation during visible light-activated CO release. Inorg. Chim. Acta 2020, 500, 119206. [Google Scholar] [CrossRef]
- A newly emerged oxidation wave (0.35 V) accompanying the reductions was assignable to the oxidation of Br– since another CV measurement using n-Bu4NBr showed an oxidation wave at 0.34 V under identical conditions. Therefore, the electrochemical results strongly suggest dissociation of the Br ligand and the following Mn-Mn dimerization.
- Yamada, M.; Tanaka, Y.; Yoshimoto, Y.; Kuroda, S.; Shimao, I. Synthesis and properties of diamino-substituted dipyrido [3,2-a:2′,3′-c]phenazine. Bull. Chem. Soc. Jpn. 1992, 65, 1006–1011. [Google Scholar] [CrossRef]
- Chakraborty, I.; Carrington, S.J.; Mascharak, P.K. Photodelivery of CO by designed photoCORMs: Correlation between absorption in the visible region and metal–CO bond labilization in carbonyl complexes. ChemMedChem 2014, 9, 1266–1274. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09W, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- We could not obtain any satisfactory results in the elemental analysis of Mn-dpc, due to its incombustibility.
- This is a service of the International Union of Crystallography (IUCr).



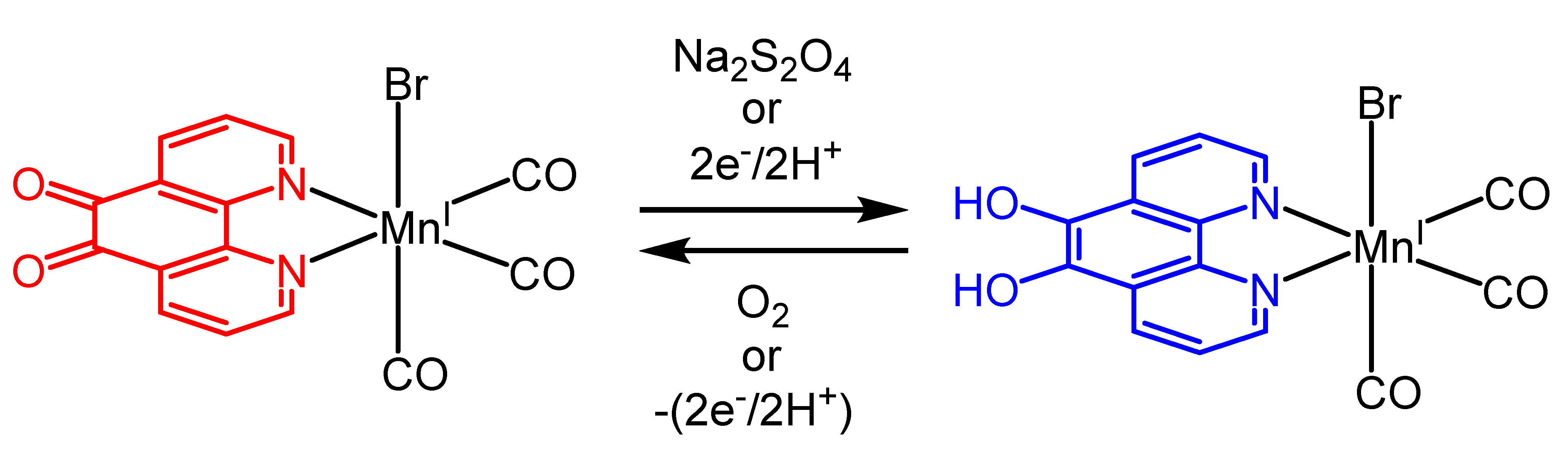

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
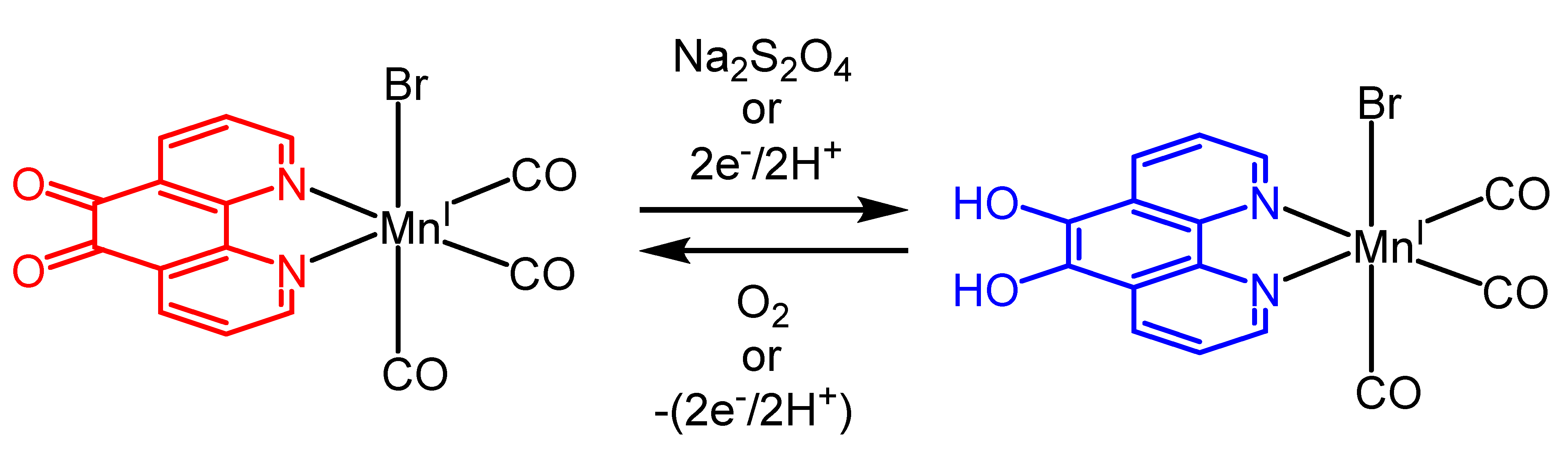{kind=link}
| Technique | Mn-dpq | Mn-dpc | Mn-qpy | Mn-dmqpy |
|---|---|---|---|---|
| IR (νCO/cm−1) | 2033 | 2032 | 2027 | 2029 |
| 1944 1928 1698 | 1961 1938 | 1936 1914 | 1944 1927 | |
| UV-vis (λmax/nm) | 427 (2.4) | 425 (1.9) | 427 (1.4) | 468 (4.1) |
| (ε/×103 M−1cm−1) | 352 (2.9) 294 (9.0) | 343 (4.1) 288 (12) | 297 (11) | 319 (17) |
| Parameter | Mn-dpq | Mn-dpc | Mn-qpy | Mn-dmqpy | ||||
|---|---|---|---|---|---|---|---|---|
| Expt. | Calc. | Expt. | Calc. | Expt. | Calc. | Expt. | Calc. | |
| Mn1-C1 | 1.799(5) | 1.801 | 1.812(2) | 1.797 | 1.813(2) | 1.797 | 1.806(8) | 1.801 |
| Mn1-C2 | 1.817(7) | 1.815 | 1.804(2) | 1.813 | 1.8123(19) | 1.814 | 1.829(8) | 1.817 |
| Mn1-C3 | 1.810(7) | 1.815 | 1.809(2) | 1.813 | 1.8054(17) | 1.815 | 1.834(12) | 1.817 |
| Mn1-N1 | 2.054(4) | 2.077 | 2.0452(17) | 2.080 | 2.0344(15) | 2.066 | 2.051(6) | 2.063 |
| Mn1-N2 | 2.049(4) | 2.077 | 2.0614(19) | 2.080 | 2.0369(12) | 2.065 | 2.045(7) | 2.063 |
| Mn1-Br1 | 2.5385(7) | 2.599 | 2.5628(10) | 2.610 | 2.5250(4) | 2.610 | 2.5617(12) | 2.600 |
| C1-O1 | 1.133(7) | 1.156 | 1.121(3) | 1.157 | 1.120(3) | 1.156 | 1.124(11) | 1.155 |
| C2-O2 | 1.145(9) | 1.154 | 1.141(3) | 1.155 | 1.142(2) | 1.155 | 1.140(11) | 1.154 |
| C3-O3 | 1.151(8) | 1.154 | 1.143(3) | 1.155 | 1.145(2) | 1.155 | 1.091(16) | 1.154 |
| C9-O4 | 1.212(6) | 1.217 | 1.362(2) | 1.353 | ||||
| C10-O5 | 1.220(7) | 1.217 | 1.367(2) | 1.353 | ||||
| Mn1-C1-O1 | 177.5(6) | 179.9 | 178.53(19) | 179.9 | 176.81(17) | 179.9 | 173.1(9) | 180.0 |
| Mn1-C2-O2 | 177.5(6) | 178.9 | 177.09(18) | 178.9 | 176.06(15) | 178.9 | 178.6(7) | 178.8 |
| Mn1-C3-O3 | 176.5(5) | 178.8 | 177.56(18) | 178.9 | 177.8(2) | 178.9 | 176.5(9) | 178.8 |
| R1/R3 1 | 15.52(7) | 30.58 | 16.8(3) | 35.56 | ||||
| R2/R4 2 | 4.63(7) | 30.88 | 12.0(3) | 35.65 | ||||
| Reaction (V vs. Fc+/Fc) | Mn-dpq | Mn-dpc | Mn-qpy | Mn-dmqpy |
|---|---|---|---|---|
| Reductions (ΔE) | −0.58 1 (0.07) | −0.28 3,4 | −1.48 3 | −1.14 1 (0.06) |
| −1.19 1 (0.08) | −1.56 3 | −1.36 3 | ||
| −1.78 1 (0.08) | ||||
| −2.20 1 (0.09) | ||||
| Oxidations | 0.74 2 | 0.43 2 | −1.26 2 | −1.34 2 |
| 0.74 2 | −0.42 2,4 | 0.34 2,5 | ||
| 0.35 2,4 |
| Parameter | Mn-dpq [MnBr(CO)3(dpq)](CH3)2CO | Mn-dpc [MnBr(CO)3(dpc)] | Mn-qpy [MnBr(CO)3(qpy)] | Mn-dmqpy [MnBr(CO)3(dmqpy)]BrPF6 |
|---|---|---|---|---|
| Formula | C18H12BrMnN2O6 | C15H8BrMnN2O5 | C23H14BrMnN4O3 | C25H20Br2F6MnN4O3P |
| Formula weight | 487.14 | 431.08 | 529.23 | 784.17 |
| Temperature (K) | 93 | 93 | 93 | 93 |
| Crystal system | monoclinic | triclinic | monoclinic | triclinic |
| Space group | P21/c | P-1 | P21/c | P-1 |
| a (Å) | 20.3113(11) | 7.077(3) | 11.8530(2) | 6.4688(3) |
| b (Å) | 6.9096(3) | 9.759(4) | 18.0938(3) | 12.4896(6) |
| c (Å) | 14.1250(7) | 11.600(5) | 10.53500(19) | 18.8132(10) |
| α (°) | 90 | 91.966(4) | 90 | 72.4654(14) |
| β (°) | 108.8940(14) | 107.572(3) | 111.2740(7) | 89.3644(14) |
| γ (°) | 90 | 104.311(5) | 90 | 82.1867(13) |
| V (Å3) | 1875.54(16) | 734.9(6) | 2105.43(7) | 1435.18(12) |
| Z | 4 | 2 | 4 | 2 |
| Calcd density (g/cm3) | 1.725 | 1.948 | 1.669 | 1.814 |
| μ (Mo Kα) (mm−1) | 2.879 | 3.655 | 2.564 | 3.383 |
| No. unique reflns | 18452 | 7485 | 21537 | 14809 |
| No. obsd reflns | 4280 | 3303 | 4815 | 6510 |
| Refinement method | Full-matrix least squares on F2 | |||
| Parameters | 255 | 217 | 289 | 379 |
| R (I > 2σ(I)) 1 | 0.0572 | 0.0247 | 0.0254 | 0.0776 |
| wR (all data) 2 | 0.1480 | 0.0667 | 0.0642 | 0.2171 |
| S | 1.042 | 1.065 | 1.072 | 1.025 |
Sample Availability: Samples of the compounds Mn-dpq, Mn-dpc, Mn-qpy, and Mn-dmqpy are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, T.; Takase, T.; Oyama, D. Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes. Molecules 2020, 25, 5921. https://doi.org/10.3390/molecules25245921
Kanno T, Takase T, Oyama D. Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes. Molecules. 2020; 25(24):5921. https://doi.org/10.3390/molecules25245921
Chicago/Turabian StyleKanno, Takatoshi, Tsugiko Takase, and Dai Oyama. 2020. "Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes" Molecules 25, no. 24: 5921. https://doi.org/10.3390/molecules25245921
APA StyleKanno, T., Takase, T., & Oyama, D. (2020). Effects of Chemically-Modified Polypyridyl Ligands on the Structural and Redox Properties of Tricarbonylmanganese(I) Complexes. Molecules, 25(24), 5921. https://doi.org/10.3390/molecules25245921