Efficient Synthesis of New Fluorinated ?-Amino Acid Enantiomers through Lipase-Catalyzed Hydrolysis



Abstract
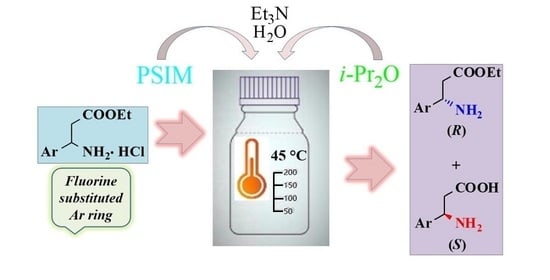
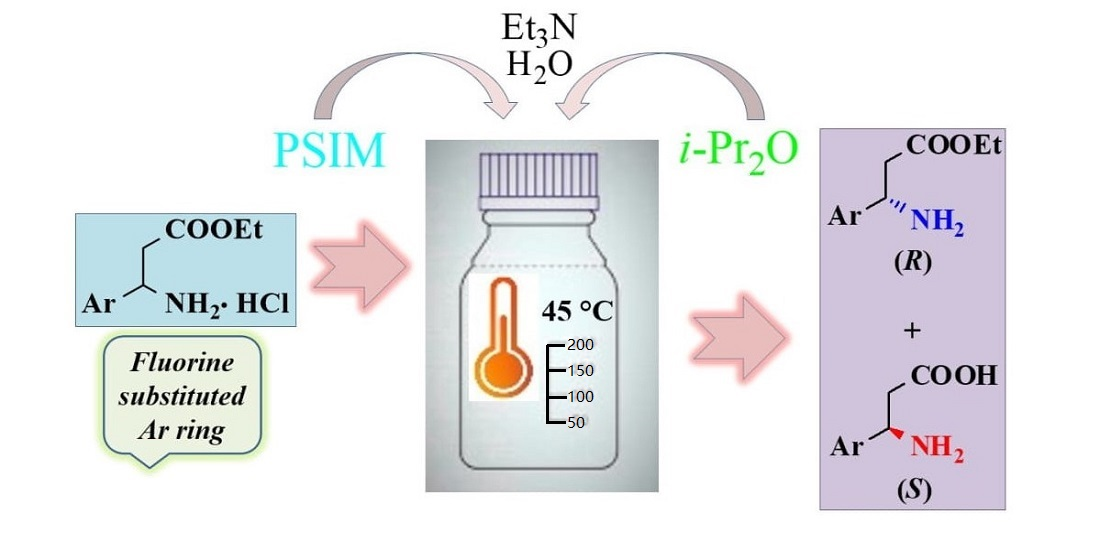
:
1. Introduction
2. Results
2.1. Synthesis of Ethyl 3-Amino-3-Arylpropanoate Hydrochloride Salts (±)-3a–e
2.2. Enzyme-Catalyzed Hydrolysis of (±)-3a–e
2.2.1. Preliminary Experiments
2.2.2. Preparative-Scale Resolutions of (±)-3a–e
2.2.3. Determination of Absolute Configurations
3. Experimental Section
3.1. General Methods
3.2. General Procedure for the Syntheses of Racemic β-Amino Acids 2a–e
3.2.1. (±)-3-Amino-3-(3,4-Difluorophenyl) Propionic Acid 2a
3.2.2. (±)-3-Amino-3-(3,5-Difluorophenyl) Propionic Acid 2b
3.2.3. (±)-3-Amino-3-(4-Fluorophenyl) Propionic Acid 2c
3.2.4. (±)-3-Amino-3-(2-Fluoro-4-Triflouromethylphenyl) Propionic Acid 2d
3.2.5. (±)-3-Amino-3-(2-Fluoro-4-Methylphenyl) Propionic Acid 2e
3.3. General Procedure for the Syntheses of Racemic β-Amino Carboxylic Ester Hydrochloride Salts 3a–e
3.3.1. Hydrochloride Salt of Ethyl (±)-3-Amino-3-(3,4-Difluorophenyl) Propanoate 3a. HCl
3.3.2. Hydrochloride Salt of Ethyl (±)-3-Amino-3-(3,5-Difluorophenyl) Propanoate 3b. HCl
3.3.3. Hydrochloride Salt of Ethyl (±)-3-Amino-3-(4-Flourophenyl) Propanoate 3c. HCl
3.3.4. Hydrochloride Salt of Ethyl (±)-3-Amino-3-(2-Fluoro-4-Trifluoromethylphenyl) Propanoate 3d. HCl
3.3.5. Hydrochloride Salt of Ethyl (±)-3-Amino-3-(2-Fluoro-4-Methylphenyl) Propanoate 3e. HCl
3.4. General Procedure for the Preparative-Scale Resolutions of (±) 3a–e
3.4.1. (R)-Ethyl 3-Amino-3-(3,4-Difluorophenyl) Propanoate 4a
3.4.2. (R)-Ethyl 3-Amino-3-(3,5-Difluorophenyl) Propanoate 4b
3.4.3. (R)-Ethyl 3-Amino-3-(4-Fluorophenyl) Propanoate 4c
3.4.4. (R)-Ethyl 3-Amino-3-(2-Fluoro-4-Triflouromethylphenyl) Propanoate 4d
3.4.5. (R)-Ethyl 3-Amino-3-(2-Fluoro-4-Methylphenyl) Propanoate 4e
3.4.6. (S)-3-Amino-3-(3,4-Difluorophenyl) Propionic Acid 5a
3.4.7. (S)-3-Amino-3-(3,5-Difluorophenyl) Propionic Acid 5b
3.4.8. (S)-3-Amino-3-(4-Fluorophenyl) Propionic Acid 5c
3.4.9. (S)-3-Amino-3-(2-Fluoro-4-Trifluoromethylphenyl) Propionic Acid 5d
3.4.10. (S)-3-Amino-3-(2-Fluoro-4-Methylphenyl) Propionic Acid 5e
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wasserman, H.H.; Berger, G.D. The use of β-lactams in the synthesis of spermine and spermidine alkaloids. Tetrahedron 1983, 39, 2459–2464. [Google Scholar] [CrossRef]
- Renault, O.; Guillon, J.; Dallemagne, P.; Rault, S. Efficient synthesis of 2-aryl-6-methyl-2,3-dihydro-1H-pyridin-4-ones. Tetrahedron Lett. 2000, 41, 681–683. [Google Scholar] [CrossRef]
- Juaristi, E.; Soloshonok, V.A. Enantioselective Synthesis of β-Amino Acids, 2nd ed.; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumore agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Fischbach, M.A.; Clardy, J. A biosynthetic gene cluster for the acetyl-CoA carboxylase inhibitor andrimid. J. Am. Chem. Soc. 2006, 128, 10660–10661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardillo, G.; Gentilucci, L.; Melchiorre, P.; Spampinato, S. Synthesis and binding activity of endomorphin-1-analogues containing β-amino acids. Bioorg. Med. Chem. Lett. 2000, 10, 2755–2758. [Google Scholar] [CrossRef]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Thornberry, N.; Weber, A. Discovery of JANUVIA™ (Sitagiliptin) a selective dipeptidyl peptidase IV inhibitor for the treatment of type2 diabetes. Curr. Top. Med. Chem. 2007, 7, 557–568. [Google Scholar] [CrossRef]
- Pepin, J.; Guern, C.; Milord, F.; Schechter, P.J. Difluoromethylornithine for arseno-resistant trypanosome brucei gambiense sleeping sickness. Lancet 1987, 330, 1431–1433. [Google Scholar] [CrossRef]
- Wolf, J.E.; Shander, D.; Huber, F.; Jackson, J.; Lin, C.-S.; Mathes, B.M.; Schrode, K. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair: Eflornithine treatment for unwanted facial hair. Int. J. Dermatol. 2007, 46, 94–98. [Google Scholar] [CrossRef]
- Cimarelli, C.; Palmieri, G.; Volpini, E. An improved synthesis of enantiopure β-amino acids. Synth. Commun. 2001, 31, 2943–2953. [Google Scholar] [CrossRef]
- Sivakumar, A.V.; Babu, G.S.; Bhat, S.V. Asymmetric synthesis of β-amino acids through application of chiral sulfoxide. Tetrahedron Asymmetry 2001, 12, 1095–1099. [Google Scholar] [CrossRef]
- Wenzel, A.G.; Jacobsen, E.N. Asymmetric catalytic mannich reactions catalyzed by urea derivatives: Enantioselective synthesis of β-aryl-β-amino acids. J. Am. Chem. Soc. 2002, 124, 12964–12965. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Hesmondhalgh, L.; Hu, S. A chemoenzymatic synthesis of an androgen receptor antagonist. Org. Process Res. Dev. 2007, 11, 903–906. [Google Scholar] [CrossRef]
- Allwein, S.P.; Roemmele, R.C.; Haley, J.J.; Mowrey, D.R.; Petrillo, D.E.; Reif, J.J.; Gingrich, D.E.; Bakale, R.P. Development and scale-up of an optimized route to the ALK inhibitor CEP-28122. Org. Process Res. Dev. 2012, 16, 148–155. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Recent lipase-catalyzed hydrolytic approaches to pharmacologically important β-and γ-amino acids. Curr. Med. Chem. 2012, 19, 6178–6187. [Google Scholar] [PubMed]
- Rodríguez-Mata, M.; García-Urdiales, E.; Gotor-Fernández, V.; Gotor, V. Stereoselective chemoenzymatic preparation of β-amino esters: Molecular modelling considerations in lipase-mediated processes and application to the synthesis of (S)-dapoxetine. Adv. Synth. Catal. 2010, 352, 395–406. [Google Scholar] [CrossRef]
- Tasnádi, G.; Forró, E.; Fülöp, F. An efficient new enzymatic method for the preparation of β-aryl-β-amino acid enantiomers. Tetrahedron Asymmetry 2008, 19, 2072–2077. [Google Scholar] [CrossRef]
- Tasnádi, G.; Forró, E.; Fülöp, F. Burkholderia cepacia lipase is an excellent enzyme for the enantioselective hydrolysis of β-heteroaryl-β-amino esters. Tetrahedron Asymmetry 2009, 20, 1771–1777. [Google Scholar] [CrossRef]
- Tasnádi, G.; Forró, E.; Fülöp, F. Improved enzymatic syntheses of valuable β-arylalkyl-β-amino acid enantiomers. Org. Biomol. Chem. 2010, 8, 793–799. [Google Scholar] [CrossRef]
- Forró, E.; Megyesi, R.; Paál, T.A.; Fülöp, F. Efficient dynamic kinetic resolution method for the synthesis of enantiopure 6-hydroxy- and 6-methoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid. Tetrahedron Asymmetry 2016, 27, 1213–1216. [Google Scholar] [CrossRef]
- Megyesi, R.; Mándi, A.; Kurtán, T.; Forró, E.; Fülöp, F. Dynamic kinetic resolution of ethyl 1,2,3,4-tetrahydro-β-carboline-1-carboxylate: Use of different hydrolases for stereocomplementary processes. Eur. J. Org. Chem. 2017, 32, 4713–4718. [Google Scholar] [CrossRef]
- Paál, T.A.; Forró, E.; Fülöp, F.; Liljeblad, A.; Kanerva, L.T. Lipase-catalyzed kinetic resolution of 1,2,3,4-tetrahydroisoquinoline-1-acetic acid esters. Tetrahedron Asymmetry 2008, 19, 2784–2788. [Google Scholar] [CrossRef]
- Paál, T.A.; Forró, E.; Liljeblad, A.; Kanerva, L.T.; Fülöp, F. Lipase-Catalyzed kinetic and dynamic kinetic resolution of 1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid. Tetrahedron Asymmetry 2007, 18, 1428–1433. [Google Scholar] [CrossRef]
- Rangel, H.; Carrillo-Morales, M.; Galindo, J.M.; Castillo, E.; Obregón-Zúñiga, A.; Juaristi, E.; Escalante, J. Structural features of N-benzylated-β-amino acid methyl esters essential for enantiodifferentiation by lipase B from candida antarctica in hydrolytic reactions. Tetrahedron Asymmetry 2015, 26, 325–332. [Google Scholar] [CrossRef]
- Pérez-Venegas, M.; Reyes-Rangel, G.; Neri, A.; Escalante, J.; Juaristi, E. Mechanochemical enzymatic resolution of N-benzylated-β3-amino esters. Beilstein J. Org. Chem. 2017, 13, 1728–1734. [Google Scholar] [CrossRef] [Green Version]
- Nagy, B.; Galla, Z.; Bencze, L.C.; Toșa, M.I.; Paizs, C.; Forró, E.; Fülöp, F. Covalently immobilized lipases are efficient stereoselective catalysts for the kinetic resolution of rac-(5-phenylfuran-2-yl)-β-alanine ethyl ester hydrochlorides. Eur. J. Org. Chem. 2017, 20, 2878–2882. [Google Scholar] [CrossRef]
- Patel, R.N. Green Biocatalysis; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Zhang, X.-X.; Gao, Y.; Hu, X.-S.; Ji, C.-B.; Liu, Y.-L.; Yu, J.-S. Recent advances in catalytic enantioselective synthesis of fluorinated α- and β-amino acids. Adv. Synth. Catal. 2020, 362, 4763–4793. [Google Scholar] [CrossRef]
- Zablocki, J.A.; Tjoeng, F.S.; Bovy, P.R.; Miyano, M.; Garland, R.B.; Williams, K.; Schretzman, L.; Zupec, M.E.; Rico, J.G.; Lindmark, R.J.; et al. A novel series of orally active antiplatelet agents. Bioorg. Med. Chem. 1995, 3, 539–551. [Google Scholar] [CrossRef]
- Johnson, T.B.; Livak, J.E. Researches on pyrimidines. CXLIX. The synthesis of aryl substituted dihydrouracils and their conversion to uracil derivatives. J. Am. Chem. Soc. 1936, 58, 299–303. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. New enzymatic two-step cascade reaction for the preparation of a key intermediate for the taxol side-chain. Eur. J. Org. Chem. 2010, 16, 3074–3079. [Google Scholar] [CrossRef]
- Forró, E. New gas chromatographic method for the enantioseparation of β-amino acids by a rapid double derivatization technique. J. Chromatogr. A 2009, 1216, 1025–1029. [Google Scholar] [CrossRef]
- Straathof, A.J.J.; Rekels, J.L.L.; Heijnen, J.J. Mass balancing in kinetic resolution: Calculating yield and enantiomeric excess using chiral balance. Biotechnol. Bioeng. 1995, 45, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.G.; Fletcher, A.M.; Lv, L.; Roberts, P.M.; Thomson, J.E. Asymmetric synthesis of β-fluoroaryl-β-amino acids. Tetrahedron Asymmetry 2012, 23, 910–925. [Google Scholar] [CrossRef]
- Forró, E.; Paál, T.; Tasnádi, G.; Fülöp, F. A new route to enantiopure β-aryl-substituted β-amino acids and 4-aryl-substituted β-lactams through lipase-catalyzed enantioselective ring cleavage of β-lactams. Adv. Synth. Catal. 2006, 348, 917–923. [Google Scholar] [CrossRef]
- Bull, S.D.; Davies, S.G.; Delgado-Ballester, S.; Kelly, P.M.; Kotchie, L.J.; Gianotti, M.; Laderas, M.; Smith, A.D. Asymmetric synthesis of β-haloaryl-β-amino acid derivatives. J. Chem. Soc. Perkin Trans 1 2001, 23, 3112–3121. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Entry | Enzyme | ees (%) b | eep (%) c | Conv. (%) d | Ee |
|---|---|---|---|---|---|
| 1 | Lipase PSIM | 88 | 95 | 48 | 108 |
| 2 | Lipase AY | 2 | 9 | 18 | 1 |
| 3 | Lipase AK | 18 | 75 | 19 | 8 |
| 4 | PPL | 2 | 29 | 5 | 2 |
| 5 | CAL-B | 2 | 5 | 30 | 1 |
| Entry | Solvent (1 mL) | ees (%) b | eep (%) c | Conv. (%) d | Ee |
|---|---|---|---|---|---|
| 1 | TBME | 95 | 88 | 52 | 59 |
| 2 | 2-Me-THF | 97 | 93 | 51 | 113 |
| 3 | EtOAc | 6 | 52 | 11 | 3 |
| 4 | Propylene carbonate | 92 | 79 | 54 | 27 |
| 5 | no solvent | 90 | 92 | 49 | 74 |
| Entry | Lipase PSIM (mg mL–1) | ees (%) b | eep (%) c | Conv. (%) d | Ee |
|---|---|---|---|---|---|
| 1 | 10 | 97 | 97 | 50 | >200 |
| 2 | 5 | 95 | 98 | 49 | >200 |
| 3 | 2 | 85 | 92 | 48 | 63 |
| Entry | Enzyme Conc. (mg mL–1) | ees (%) b | eep (%) c | Conv. (%) d | Ee |
|---|---|---|---|---|---|
| 1 | 2 | 4 | 81 | 5 | 10 |
| 2 | 5 | 15 | 85 | 15 | 14 |
| 3 | 10 | 21 | 86 | 20 | 17 |
| 4 | 20 | 46 | 92 | 33 | 38 |
| 5 | 40 | 97 | 89 | 52 | 74 |
| Substrate | Rt (h) | Conv. (%) | E | β-Amino Acid (5a–e) | β-Amino Ester (4a–e) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield (%) | Isomer | eeb (%) | (H2O) | Yield (%) | Isomer | eec (%) | (CHCl3) | ||||
| 3a | 8 | 50 | >200 | 48 | (S) | >99 | –3.1 d | 49 | (R) | 97 | +17.9 e |
| 3b | 72 | 49 | >200 | 48 | (S) | >99 | –5 f | 38 | (R) | 94 | +9 g |
| 3c | 18 | 50 | >200 | 49 | (S) | >99 | –3 h | 49 | (R) | >99 | +18.9 i |
| 3d | 26 | 49 | >200 | 49 | (S) | >99 | –11 j | 48 | (R) | >99 | +20.3 k |
| 3e | 23 | 50 | >200 | 48 | (S) | >99 | –13 l | 47 | (R) | >99 | +16 m |
Sample Availability: Samples of the compounds (±)-2a–e, (±)-3a–e, (R)-4a–e and (S)-5a–e are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahmohammadi, S.; Fülöp, F.; Forró, E. Efficient Synthesis of New Fluorinated ?-Amino Acid Enantiomers through Lipase-Catalyzed Hydrolysis. Molecules 2020, 25, 5990. https://doi.org/10.3390/molecules25245990
Shahmohammadi S, Fülöp F, Forró E. Efficient Synthesis of New Fluorinated ?-Amino Acid Enantiomers through Lipase-Catalyzed Hydrolysis. Molecules. 2020; 25(24):5990. https://doi.org/10.3390/molecules25245990
Chicago/Turabian StyleShahmohammadi, Sayeh, Ferenc Fülöp, and Enikő Forró. 2020. "Efficient Synthesis of New Fluorinated ?-Amino Acid Enantiomers through Lipase-Catalyzed Hydrolysis" Molecules 25, no. 24: 5990. https://doi.org/10.3390/molecules25245990
APA StyleShahmohammadi, S., Fülöp, F., & Forró, E. (2020). Efficient Synthesis of New Fluorinated ?-Amino Acid Enantiomers through Lipase-Catalyzed Hydrolysis. Molecules, 25(24), 5990. https://doi.org/10.3390/molecules25245990