
Plasma Processing of Low Vapor Pressure Liquids to Generate Functional Surfaces



Abstract
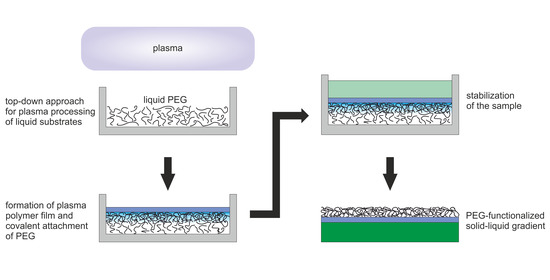
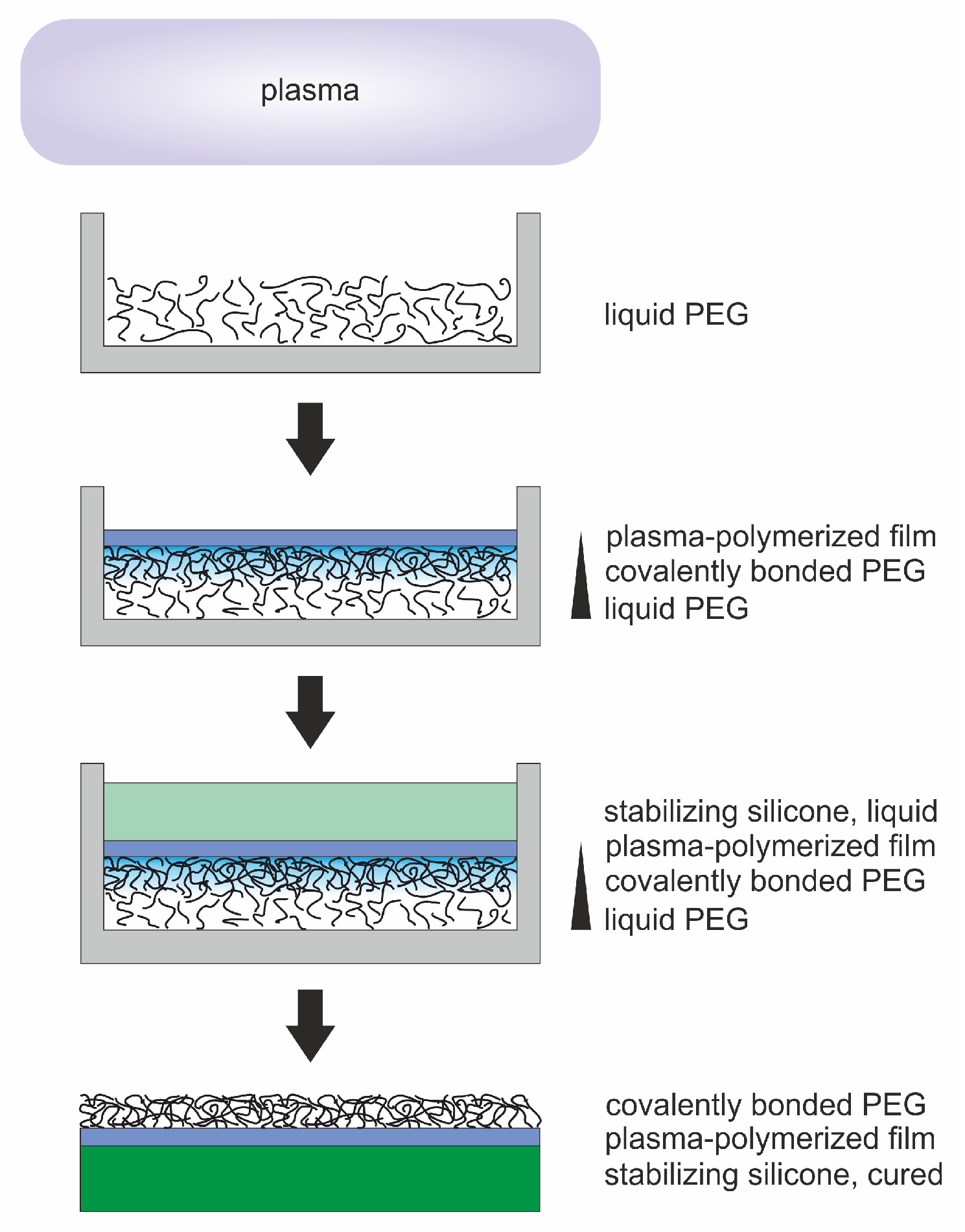
:
1. Introduction
2. Results
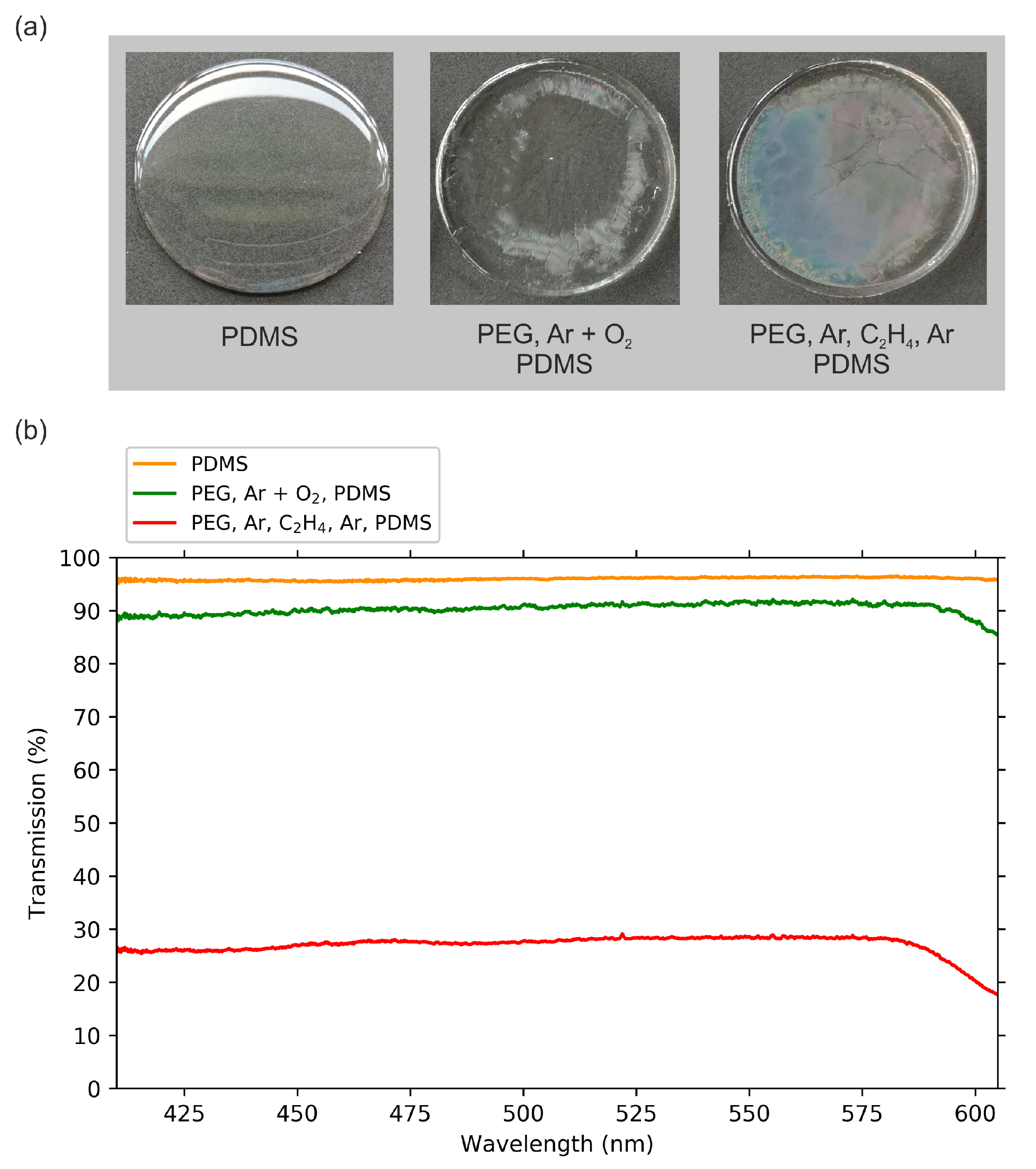
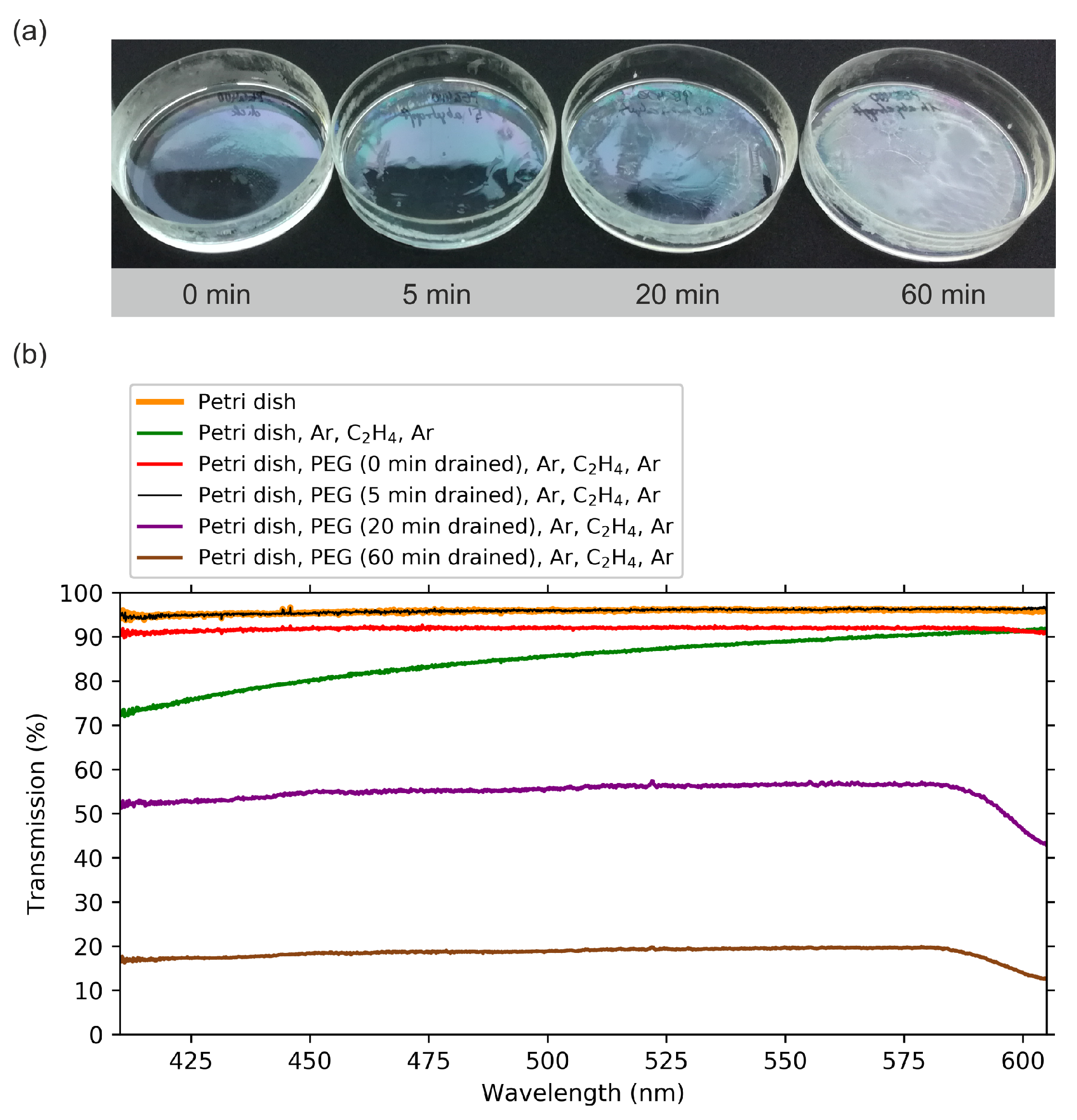
2.1. Optical Transmission Spectroscopy
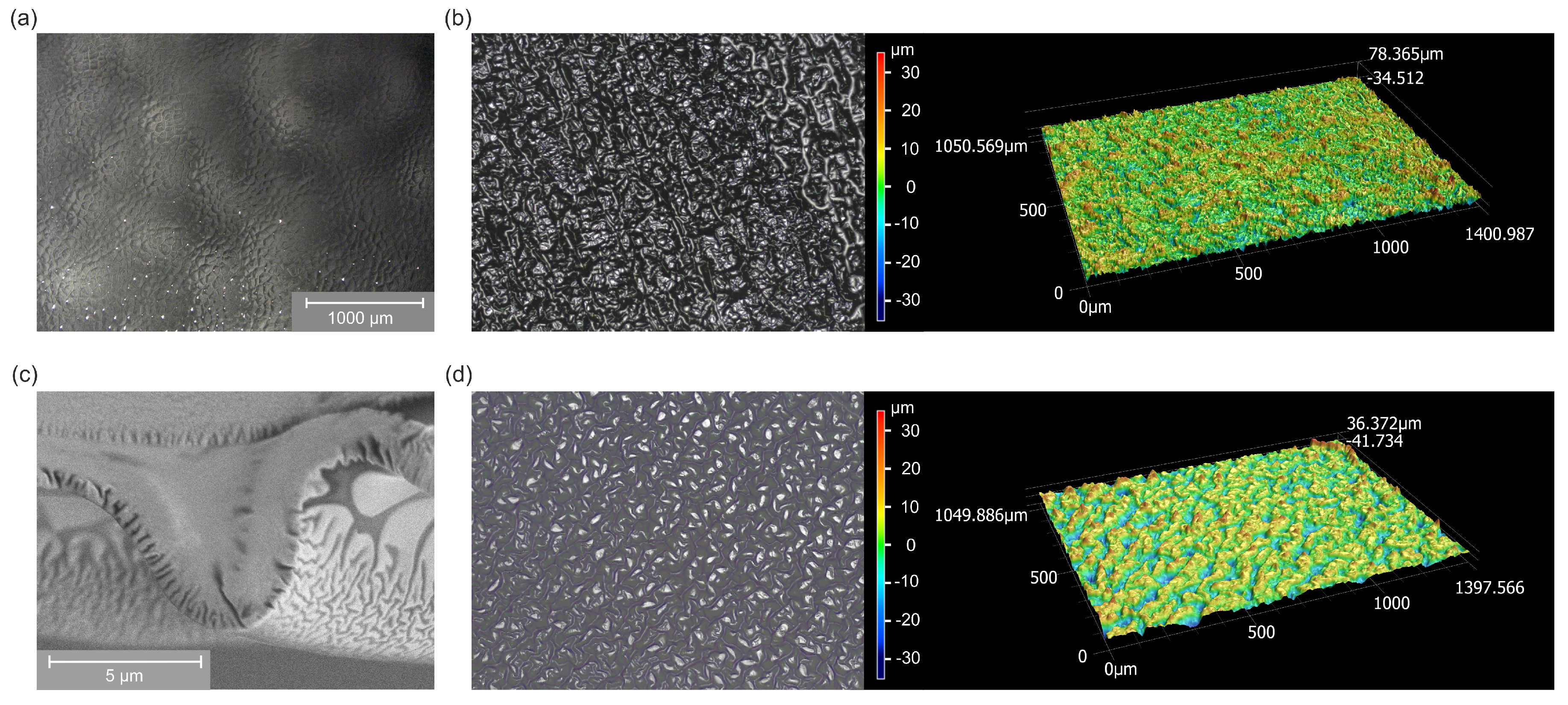
2.2. Surface Morphology
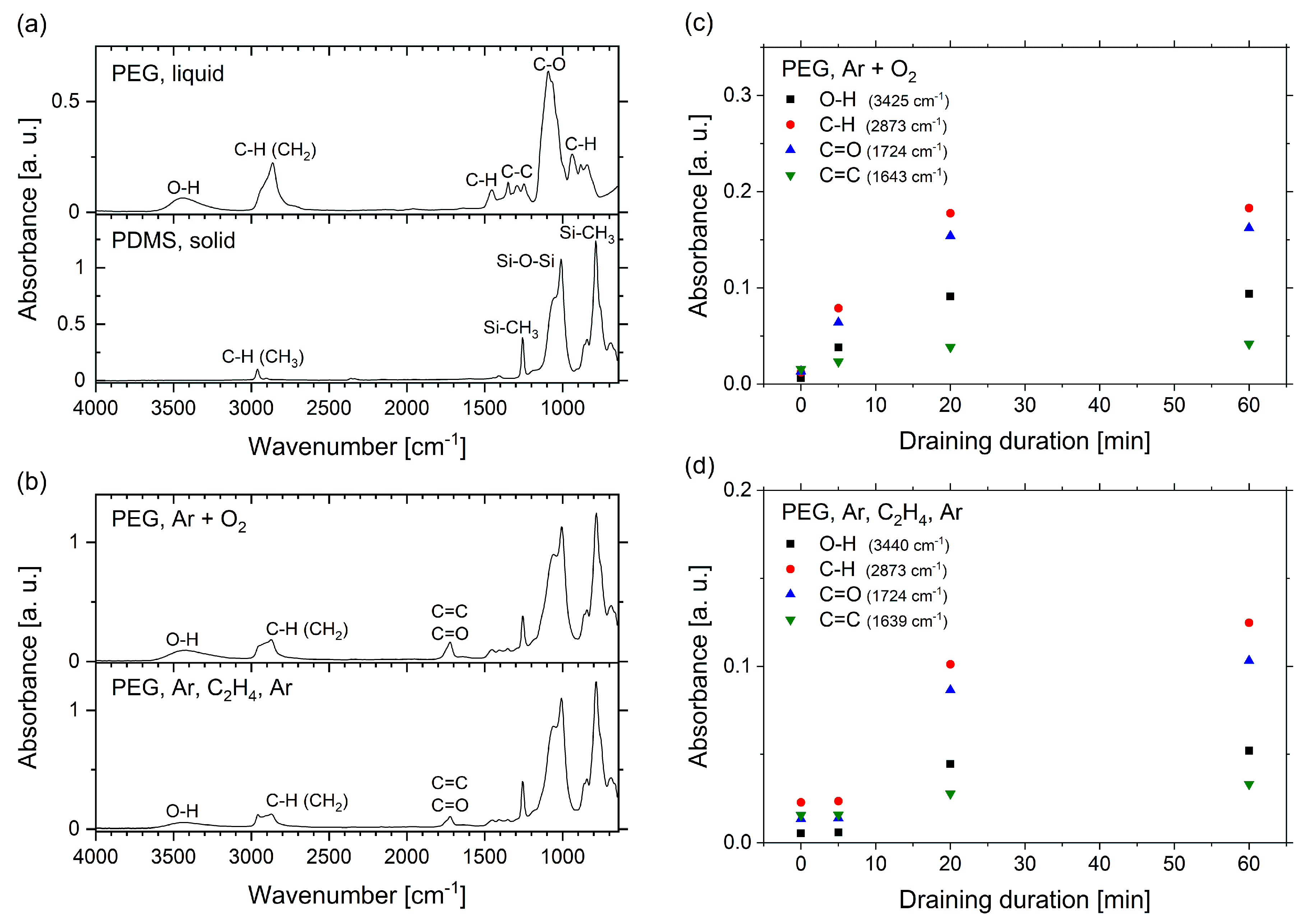
2.3. FTIR Spectroscopy
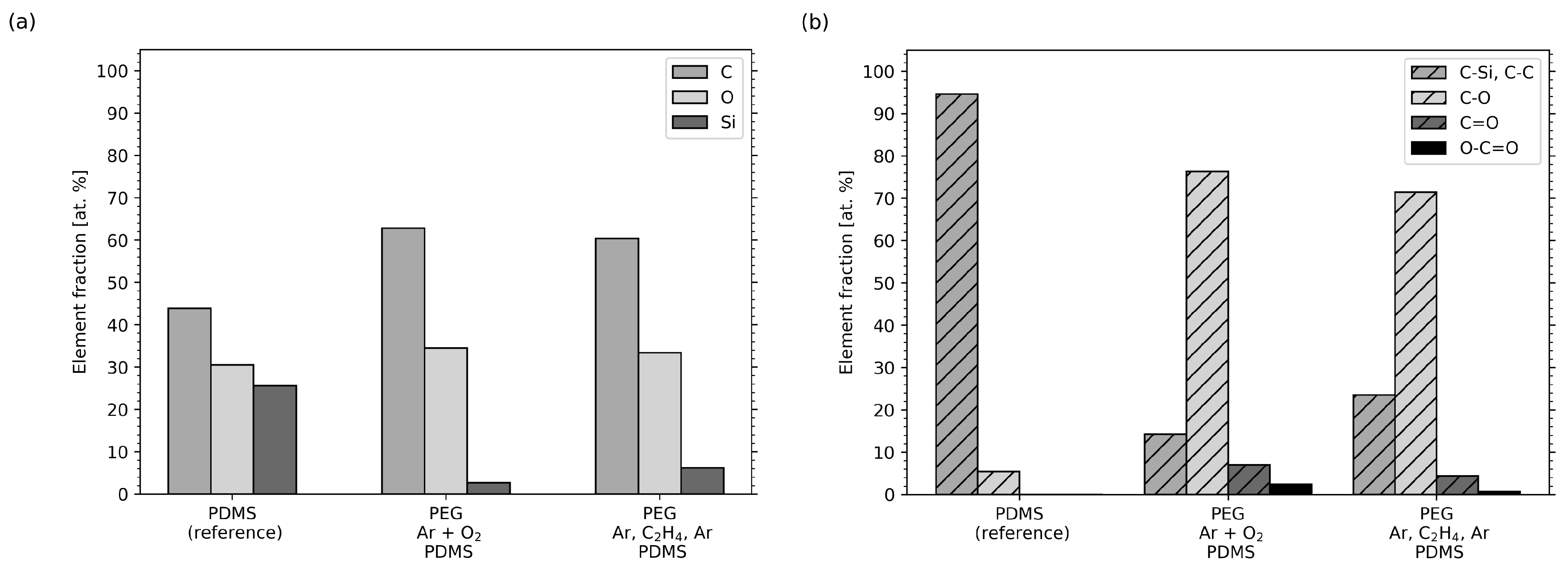
2.4. XPS Measurements
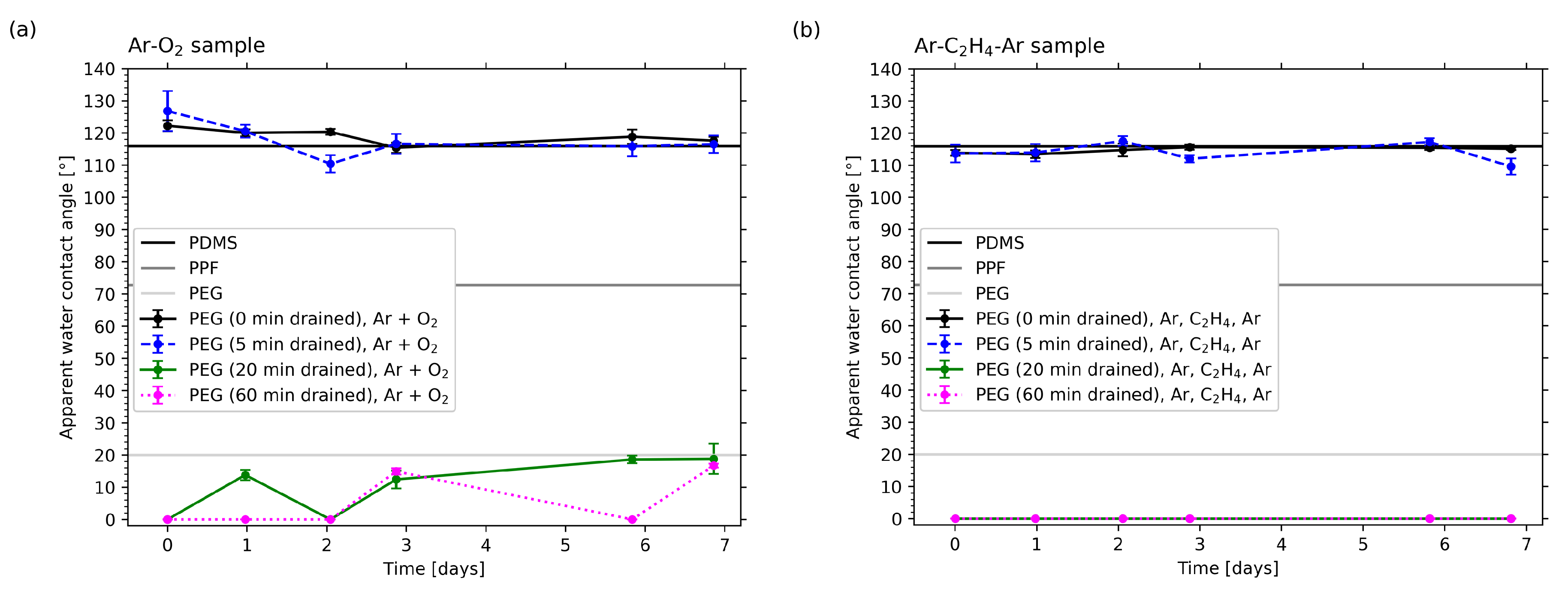
2.5. WCA Measurements
3. Discussion
4. Materials and Methods
4.1. Materials and Sample Preparation
4.2. Analytical Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| a-C:H film | amorphous hydrocarbon film |
| CLSM | confocal laser scanning microscopy |
| FTIR | Fourier transform infrared |
| HMDSO | hexamethyldisiloxane |
| (i)CVD | (initiated) chemical vapor deposition |
| OTS | optical transmission spectroscopy |
| PDMS | polydimethylsiloxane |
| PECVD | plasma-enhanced chemical vapor deposition |
| PEG | polyethylene glycol |
| PHEMA | poly(2-hydroxyethyl methacrylate) |
| PPF | plasma-polymerized film |
| RF | radio frequency |
| SEM | scanning electron microscopy |
| WTA | water contact angle |
| XPS | X-ray photoelectron spectroscopy |
References
- Ye, G.X.; Zhang, Q.R.; Feng, C.M.; Ge, H.L.; Jiao, Z.K. Structural and electrical properties of a metallic rough-thin-film system deposited on liquid substrates. Phys. Rev. B 1996, 54, 14754–14757. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Feng, C.; Ye, G.; Ren, Y.; Jiao, Z. Growth mechanism and electrical properties of metallic films deposited on silicone oil surfaces. J. Appl. Phys. 1997, 82, 5469–5471. [Google Scholar] [CrossRef]
- Ye, G.X.; Michely, T.; Weidenhof, V.; Friedrich, I.; Wuttig, M. Nucleation, Growth, and Aggregation of Ag Clusters on Liquid Surfaces. Phys. Rev. Lett. 1998, 81, 622–625. [Google Scholar] [CrossRef] [Green Version]
- Michely, T.; Ye, G.X.; Weidenhof, V.; Wuttig, M. Silver deposition on oil – cluster nucleation, growth and aggregation on a liquid surface. Surf. Sci. 1999, 432, 228–238. [Google Scholar] [CrossRef]
- Feng, C.M.; Ge, H.L.; Tong, M.R.; Ye, G.X.; Jiao, Z.K. Growth behavior and surface morphology of Ag rough thin films deposited on silicone oil surfaces. Thin Solid Films 1999, 342, 30–34. [Google Scholar] [CrossRef]
- Cai, P.G.; Yu, S.J.; Ye, Q.L.; Jin, J.S.; Ye, G.X. An internal stress pattern in free standing films. Phys. Lett. A 2003, 312, 119–122. [Google Scholar] [CrossRef]
- Ye, Q.L.; Ye, S.J.; Jin, J.S.; Ye, G.X. Formation Mechanism and Orderly Structures of an Iron Film System Deposited on Silicone Oil Surfaces. Chin. Phys. Lett. 2003, 20, 1109–1111. [Google Scholar]
- Yu, S.J.; Zhang, Y.J.; Ye, Q.L.; Cai, P.G.; Tang, X.W.; Ye, G.X. Spontaneous formation of ordered patterns in Al films deposited on silicone oil surfaces. Phys. Rev. B 2003, 68, 193403. [Google Scholar] [CrossRef]
- Yu, S.J.; Ye, Q.L.; Zhang, Y.J.; Cai, P.G.; Xu, X.J.; Chen, J.X.; Ye, G.X. Pattern formation under residual compressive stress in free sustained aluminum films. Thin Solid Films 2005, 491, 311–316. [Google Scholar] [CrossRef]
- Chen, M.G.; Xie, J.P.; Ye, G.X. Formation mechanism and ordered patterns in Cu films deposited on silicone oil surfaces. Phys. Lett. A 2006, 360, 323–326. [Google Scholar] [CrossRef]
- Tao, X.M.; Yang, B.; Ma, H.Z.; Ye, G.X. Expansion effect of liquid substrates on the ordered structures in the Al films. Phys. Lett. A 2006, 351, 350–353. [Google Scholar] [CrossRef]
- Torimoto, T.; Okazaki, K.i.; Kiyama, T.; Hirahara, K.; Tanaka, N.; Kuwabata, S. Sputter deposition onto ionic liquids: Simple and clean synthesis of highly dispersed ultrafine metal nanoparticles. Appl. Phys. Lett. 2006, 89, 243117. [Google Scholar] [CrossRef]
- Xie, J.P.; Yu, W.Y.; Zhang, S.L.; Chen, M.G.; Ye, G.X. AFM study on microstructures of metal films deposited on liquid substrates. Phys. Lett. A 2007, 371, 160–164. [Google Scholar] [CrossRef]
- Borra, E.F.; Seddiki, O.; Angel, R.; Eisenstein, D.; Hickson, P.; Seddon, K.R.; Worden, S.P. Deposition of metal films on an ionic liquid as a basis for a lunar telescope. Nature 2007, 447, 979–981. [Google Scholar] [CrossRef]
- Chen, M.; Xie, J.P.; Jin, J.S.; Xia, A.G.; Ye, G.X. Formation mechanism of ordered stress-relief patterns in a free sustained Cu film system. Chin. Phys. B 2008, 17, 669–673. [Google Scholar]
- Yu, S.J.; Zhang, Y.J. Formation Meachanism and Surface Evolution of Silver Films Sputtering Deposited on Silicone Oil Substrates. Surf. Rev. Lett. 2008, 15, 525–530. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Yu, S.J. Experimental Observations of Disk-Shaped Patterns in Fe Films Sputtering Deposited on Silicone Oil Surfaces. Int. J. Mod. Phys. B 2009, 23, 3147–3157. [Google Scholar] [CrossRef]
- Cai, P.; Yu, S.; Xu, X.; Chen, M.; Sui, C.; Ye, G.X. Growth mechanism and stress relief patterns of Ni films deposited on silicone oil surfaces. Appl. Surf. Sci. 2009, 255, 8352–8358. [Google Scholar] [CrossRef]
- Yu, S.J.; Zhang, Y.J.; Chen, M.G. Comparison of Stress Relief Mechanisms of Metal Films Deposited on Liquid Substrates by Thermal Evaporating and Sputtering. Int. J. Mod. Phys. B 2010, 24, 997–1005. [Google Scholar] [CrossRef]
- Yu, S.J. Wrinkle patterns of Fe films deposited on micro-scale silicone oil droplets. Thin Solid Films 2014, 558, 247–251. [Google Scholar] [CrossRef]
- Chen, M.G.; Yu, S.J.; Feng, Y.X.; Jiao, Z.W.; Yu, M.Z.; Yang, B. Microstructure and growth mechanism of Cu ramified aggregates on silicone oil surfaces. Thin Solid Films 2010, 518, 2674–2677. [Google Scholar] [CrossRef]
- Wender, H.; de Oliveira, L.F.; Feil, A.F.; Lissner, E.; Migowski, P.; Meneghetti, M.R.; Teixeira, S.R.; Dupont, J. Synthesis of gold nanoparticles in a biocompatible fluid from sputtering deposition onto castor oil. Chem. Commun. 2010, 46, 7019. [Google Scholar] [CrossRef] [PubMed]
- Wender, H.; Goncalves, R.V.; Feil, A.F.; Migowski, P.; Poletto, F.S.; Pohlmann, A.R.; Dupont, J.; Teixeira, S.R. Sputtering onto Liquids: From Thin Films to Nanoparticles. J. Phys. Chem. C 2011, 115, 16362–16367. [Google Scholar] [CrossRef]
- Wender, H.; Migowski, P.; Feil, A.F.; de Oliveira, L.F.; Prechtl, M.H.G.; Leal, R.; Machado, G.; Teixeira, S.R.; Dupont, J. On the formation of anisotropic gold nanoparticles by sputtering onto a nitrile functionalised ionic liquid. Phys. Chem. Chem. Phys. 2011, 13, 13552. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Ma, R.R.; Li, D.M.; Xia, A.G.; Tao, X.M. Aggregation behavior and microstructure of silver thin films on ionic liquid substrates. Thin Solid Films 2012, 520, 2321–2325. [Google Scholar] [CrossRef]
- Pan, Q.F.; Cheng, Y.; Tao, X.M.; Yang, B.; Li, B.X.; Ye, G.X. Temperature dependence of the aggregation behavior of aluminum nanoparticles on liquid substrate. J. Nanopart. Res. 2015, 17, 161. [Google Scholar] [CrossRef]
- Homsy, A.; Laux, E.; Jeandupeux, L.; Charmet, J.; Bitterli, R.; Botta, C.; Rebetez, Y.; Banakh, O.; Keppner, H. Solid on liquid deposition, a review of technological solutions. Microelectron. Eng. 2015, 141, 267–279. [Google Scholar]
- Ye, G.X.; Xia, A.G.; Gao, G.L.; Lao, Y.F.; Tao, X.M. Experimental observation of large ramified Au aggregates on melting glass surfaces. Phys. Rev. B 2001, 63, 125405. [Google Scholar] [CrossRef]
- Yang, B.; Scheidtmann, J.; Mayer, J.; Wuttig, M.; Michely, T. Fragmentation, rings and coarsening: Structure and transformations of nanocrystal aggregate networks on a liquid surface. Surf. Sci. 2002, 497, 100–112. [Google Scholar] [CrossRef]
- Voigt, M.; Dorsfeld, S.; Volz, A.; Sokolowski, M. Nucleation and Growth of Molecular Organic Crystals in a Liquid Film under Vapor Deposition. Phys. Rev. Lett. 2003, 91, 026103. [Google Scholar]
- Liu, X.; Kaiser, V.; Wuttig, M.; Michely, T. Unidirectional anisotropies in perylene crystal growth on a liquid surface. J. Cryst. Growth 2004, 269, 542–549. [Google Scholar] [CrossRef]
- Liu, X.; Wuttig, M. Inherent features in the growth of perylene crystals on an oil substrate. Phys. Rev. B 2006, 73, 033405. [Google Scholar] [CrossRef]
- Lu, C.; Cheng, Y.; Pan, Q.; Tao, X.; Yang, B.; Ye, G. One-dimensional Growth of Zinc Crystals on a Liquid Surface. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.W.; Ye, X.H.; Bao, Z.L.; Li, L.; Yang, B.; Tao, X.M.; Ye, G.X. Growth and aggregation of Cu nanocrystals on ionic liquid surfaces. Chin. Phys. B 2020, 29, 066801. [Google Scholar] [CrossRef]
- Li, L.; Bao, Z.L.; Ye, X.H.; Shen, J.W.; Yang, B.; Ye, G.X.; Tao, X.M. Nucleation, Growth, and Aggregation of Au Nanocrystals on Liquid Surfaces. Chin. Phys. Lett. 2020, 37, 028102. [Google Scholar] [CrossRef]
- Keppner, H.; Benkhaïra, M. Method for producing a plastic membrane device and the thus obtained device. Patent WO/2006/063955, 16 December 2004. [Google Scholar]
- Haller, P.D.; Frank-Finney, R.J.; Gupta, M. Vapor-Phase Free Radical Polymerization in the Presence of an Ionic Liquid. Macromolecules 2011, 44, 2653–2659. [Google Scholar] [CrossRef]
- Bradley, L.C.; Gupta, M. Encapsulation of Ionic Liquids within Polymer Shells via Vapor Phase Deposition. Langmuir 2012, 28, 10276–10280. [Google Scholar] [CrossRef]
- Frank-Finney, R.J.; Haller, P.D.; Gupta, M. Ultrathin Free-Standing Polymer Films Deposited onto Patterned Ionic Liquids and Silicone Oil. Macromolecules 2012, 45, 165–170. [Google Scholar] [CrossRef]
- Haller, P.D.; Bradley, L.C.; Gupta, M. Effect of Surface Tension, Viscosity, and Process Conditions on Polymer Morphology Deposited at the Liquid-Vapor Interface. Langmuir 2013, 29, 11640–11645. [Google Scholar] [CrossRef]
- Haller, P.D.; Gupta, M. Synthesis of Polymer Nanoparticles via Vapor Phase Deposition onto Liquid Substrates. Macromol. Rapid Comm. 2014, 35, 2000–2004. [Google Scholar] [CrossRef]
- Frank-Finney, R.J.; Gupta, M. Two-Stage Growth of Polymer Nanoparticles at the Liquid–Vapor Interface by Vapor-Phase Polymerization. Langmuir 2016, 32, 11014–11020. [Google Scholar] [CrossRef] [PubMed]
- Laux, E.; Charmet, J.; Haquette, H.; Banakh, O.; Jeandupeux, L.; Graf, B.; Keppner, H. General aspects of solid on liquid growth mechanisms. J. Phys. Conf. Ser. 2009, 182, 012029. [Google Scholar] [CrossRef]
- Binh-Khiem Nguyen, N.; Eiji Iwase, N.; Kiyoshi Matsumoto, N.; Isao Shimoyama, N. Electrically driven varifocal micro lens fabricated by depositing parylene directly on liquid. In Proceedings of the 2007 IEEE 20th International Conference on Micro Electro Mechanical Systems (MEMS), Hyogo, Japan, 21–25 January 2007. [Google Scholar]
- Binh-Khiem, N.; Matsumoto, K.; Shimoyama, I. Polymer thin film deposited on liquid for varifocal encapsulated liquid lenses. Appl. Phys. Lett. 2008, 93, 124101. [Google Scholar]
- Binh-Khiem, N.; Matsumoto, K.; Shimoyama, I. Tensile Film Stress of Parylene Deposited on Liquid. Langmuir 2010, 26, 18771–18775. [Google Scholar] [CrossRef] [PubMed]
- Charmet, J.; Banakh, O.; Laux, E.; Graf, B.; Dias, F.; Dunand, A.; Keppner, H.; Gorodyska, G.; Textor, M.; Noell, W.; et al. Solid on liquid deposition. Thin Solid Films 2010, 518, 5061–5065. [Google Scholar] [CrossRef]
- Thangawng, A.L.; Ruoff, R.S.; Swartz, M.A.; Glucksberg, M.R. An ultra-thin PDMS membrane as a bio/micro–nano interface: Fabrication and characterization. Biomed. Microdevices 2007, 9, 587–595. [Google Scholar] [CrossRef]
- Lötters, J.C.; Olthuis, W.; Veltink, P.H.; Bergveld, P. The mechanical properties of the rubber elastic polymer polydimethylsiloxane for sensor applications. J. Micromech. Microeng. 1997, 7, 145–147. [Google Scholar] [CrossRef]
- Gaiser, S.; Schütz, U.; Hegemann, D. Top-down approach to attach liquid polyethylene glycol to solid surfaces by plasma interaction. Plasma Process. Polym. 2020, 17, e1900211. [Google Scholar] [CrossRef]
- Mahato, R.I. Biomaterials for Delivery and Targeting of Proteins and Nucleic Acids; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Dalsin, J.L.; Lin, L.; Tosatti, S.; Vörös, J.; Textor, M.; Messersmith, P.B. Protein Resistance of Titanium Oxide Surfaces Modified by Biologically Inspired mPEG-DOPA. Langmuir 2005, 21, 640–646. [Google Scholar] [CrossRef]
- Wyman, P. Coatings for Biomedical Applications; Woodhead Publishing: Cambridge, UK, 2012; pp. 3–42. [Google Scholar]
- Lee, J.; Lee, H.; Andrade, J. Blood compatibility of polyethylene oxide surfaces. Prog. Polym. Sci. 1995, 20, 1043–1079. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeong, B.J.; Lee, H.B. Plasma protein adsorption and platelet adhesion onto comb-like PEO gradient surfaces. J. Biomed. Mater. Res. 1997, 34, 105–114. [Google Scholar] [CrossRef]
- Wang, R.L.C.; Kreuzer, H.J.; Grunze, M. Molecular Conformation and Solvation of Oligo(ethylene glycol)-Terminated Self-Assembled Monolayers and Their Resistance to Protein Adsorption. J. Phys. Chem. B 1997, 101, 9767–9773. [Google Scholar] [CrossRef]
- Kingshott, P.; Wei, J.; Bagge-Ravn, D.; Gadegaard, N.; Gram, L. Covalent Attachment of Poly(ethylene glycol) to Surfaces, Critical for Reducing Bacterial Adhesion. Langmuir 2003, 19, 6912–6921. [Google Scholar] [CrossRef]
- Heuberger, M.; Drobek, T.; Vörös, J. About the Role of Water in Surface-Grafted Poly(ethylene glycol) Layers. Langmuir 2004, 20, 9445–9448. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Jiang, H.; Manolache, S.; Wong, A.C.L.; Denes, F.S. Plasma-Mediated Grafting of Poly(ethylene glycol) on Polyamide and Polyester Surfaces and Evaluation of Antifouling Ability of Modified Substrates. Langmuir 2007, 23, 7306–7313. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; Ashurst, W.R. Interfacial Water Structure on a Highly Hydroxylated Silica Film. Langmuir 2009, 25, 11549–11554. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Yoon, J.Y. Reusable, polyethylene glycol-structured microfluidic channel for particle immunoassays. J. Biol. Eng. 2009, 3, 6. [Google Scholar] [PubMed] [Green Version]
- Zhang, H.; Chiao, M. Anti-fouling Coatings of Poly(dimethylsiloxane) Devices for Biological and Biomedical Applications. J. Med. Biol. Eng. 2015, 35, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, A.S. Non-Fouling Surface Technologies. J. Biomat. Sci. Polym. E 1999, 10, 1011–1014. [Google Scholar] [CrossRef]
- Wang, P.; Tan, K.; Kang, E.; Neoh, K. Plasma-induced immobilization of poly(ethylene glycol) onto poly(vinylidene fluoride) microporous membrane. J. Membr. Sci. 2002, 195, 103–114. [Google Scholar] [CrossRef]
- Herrmann, M.; Roy, E.; Veres, T.; Tabrizian, M. Microfluidic ELISA on non-passivated PDMS chip using magnetic bead transfer inside dual networks of channels. Lab Chip 2007, 7, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbers, G.M.; Emoto, K.; Greef, C.; Metzger, S.W.; Woodward, H.N.; Mascali, J.J.; Grainger, D.W.; Lochhead, M.J. Functionalized Poly(ethylene glycol)-Based Bioassay Surface Chemistry That Facilitates Bio-Immobilization and Inhibits Nonspecific Protein, Bacterial, and Mammalian Cell Adhesion. Chem. Mater. 2007, 19, 4405–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, K.; Inoue, H.; Ikada, Y. Protein adsorption and platelet adhesion onto polyurethane grafted with methoxy-poly(ethylene glycol) methacrylate by plasma technique. J. Biomed. Mater. Res. 1993, 27, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Choukourov, A.; Gordeev, I.; Polonskyi, O.; Artemenko, A.; Hanyková, L.; Krakovský, I.; Kylián, O.; Slavínská, D.; Biederman, H. Poly(ethylene oxide)-like Plasma Polymers Produced by Plasma-Assisted Vacuum Evaporation. Plasma Process. Polym. 2010, 7, 445–458. [Google Scholar] [CrossRef]
- Kang, E.; Neoh, K. Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Nisol, B.; Oldenhove, G.; Preyat, N.; Monteyne, D.; Moser, M.; Perez-Morga, D.; Reniers, F. Atmospheric plasma synthesized PEG coatings: Non-fouling biomaterials showing protein and cell repulsion. Surf. Coat. Technol. 2014, 252, 126–133. [Google Scholar] [CrossRef]
- Ottani, S.; Vitalini, D.; Comelli, F.; Castellari, C. Densities, Viscosities, and Refractive Indices of Poly(ethylene glycol) 200 and 400 + Cyclic Ethers at 303.15 K. J. Chem. Eng. Data 2002, 47, 1197–1204. [Google Scholar]
- Emoto, K.; Van Alstine, J.M.; Harris, J.M. Stability of Poly(ethylene glycol) Graft Coatings. Langmuir 1998, 14, 2722–2729. [Google Scholar] [CrossRef]
- Dong, B.; Manolache, S.; Somers, E.B.; Lee Wong, A.C.; Denes, F.S. Generation of antifouling layers on stainless steel surfaces by plasma-enhanced crosslinking of polyethylene glycol. J. Appl. Polym. Sci. 2005, 97, 485–497. [Google Scholar] [CrossRef]
- Ulbricht, J.; Jordan, R.; Luxenhofer, R. On the biodegradability of polyethylene glycol, polypeptoids and poly(2-oxazoline)s. Biomaterials 2014, 35, 4848–4861. [Google Scholar]
- Morgese, G.; Verbraeken, B.; Ramakrishna, S.N.; Gombert, Y.; Cavalli, E.; Rosenboom, J.G.; Zenobi-Wong, M.; Spencer, N.D.; Hoogenboom, R.; Benetti, E.M. Chemical Design of Non-Ionic Polymer Brushes as Biointerfaces: Poly(2-oxazine)s Outperform Both Poly(2-oxazoline)s and PEG. Angew. Chem. Int. Ed. 2018, 57, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers; John Wiley & Sons: Chichester, UK, 1992. [Google Scholar]
- Girard-Lauriault, P.L.; Unger, W.E.S.; Dietrich, P.M.; Holländer, A. Innovative and Established Strategies for the Surface Analysis of Nitrogen and Oxygen-Rich Plasma Polymer Films by XPS: An Introductory Guide. Plasma Process. Polym. 2015, 12, 953–967. [Google Scholar] [CrossRef]
- Homma, H.; Kuroyagi, T.; Izumi, K.; Mirley, C.; Ronzello, J.; Boggs, S. Diffusion of low molecular weight siloxane from bulk to surface. IEEE Trans. Dielectr. Electr. Insul. 1999, 6, 370–375. [Google Scholar] [CrossRef]
- Pan, F.; Altenried, S.; Liu, M.; Hegemann, D.; Bülbül, E.; Moeller, J.; Schmahl, W.W.; Maniura-Weber, K.; Ren, Q. A nanolayer coating on polydimethylsiloxane surfaces enables a mechanistic study of bacterial adhesion influenced by material surface physicochemistry. Mater. Horiz. 2020, 7, 93–103. [Google Scholar] [CrossRef]
- Mata, A.; Fleischman, A.J.; Roy, S. Characterization of Polydimethylsiloxane (PDMS) Properties for Biomedical Micro/Nanosystems. Biomed. Microdevices 2005, 7, 281–293. [Google Scholar] [CrossRef]
- Hegemann, D.; Lorusso, E.; Butron-Garcia, M.I.; Blanchard, N.E.; Rupper, P.; Favia, P.; Heuberger, M.; Vandenbossche, M. Suppression of Hydrophobic Recovery by Plasma Polymer Films with Vertical Chemical Gradients. Langmuir 2016, 32, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Bacharouche, J.; Haidara, H.; Kunemann, P.; Vallat, M.F.; Roucoules, V. Singularities in hydrophobic recovery of plasma treated polydimethylsiloxane surfaces under non-contaminant atmosphere. Sens. Actuators A Phys. 2013, 197, 25–29. [Google Scholar] [CrossRef]
- Zuri, L.; Narkis, M.; Silverstein, M.S. Film formation and crack development in plasma polymerized hexamethyldisiloxane. Polym. Eng. Sci. 1997, 37, 1188–1194. [Google Scholar]
- Sato, K. The internal stress of coating films. Prog. Org. Coat. 1980, 8, 143–160. [Google Scholar]
- Matuda, N.; Baba, S.; Kinbara, A. Internal stress, young’s modulus and adhesion energy of carbon films on glass substrates. Thin Solid Films 1981, 81, 301–305. [Google Scholar] [CrossRef]
- Yasuda, H. Plasma Polymerization; Academic Press: Orlando, FA, USA, 1985. [Google Scholar]
- Yu, S.J.; Zhou, H.; Zhang, Y.J.; Chen, M.G.; Jiao, Z.W.; Si, P.Z. Stress relief patterns of Co films deposited on circular silicone oil substrates. Thin Solid Films 2012, 520, 5683–5690. [Google Scholar] [CrossRef]
- Hegemann, D.; Indutnyi, I.; Zajickova, L.; Makhneva, E.; Farka, Z.; Ushenin, Y.; Vandenbossche, M. Stable, nanometer-thick oxygen-containing plasma polymer films suited for enhanced biosensing. Plasma Process. Polym. 2018, 15, e1800090. [Google Scholar] [CrossRef]
- Long, H.; Lai, C.; Chung, C. Polyethylene glycol coating for hydrophilicity enhancement of polydimethylsiloxane self-driven microfluidic chip. Surf. Coat. Technol. 2017, 320, 315–319. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Tu, Q.; Nie, N.; Sha, J.; Liu, W.; Liu, R.; Zhang, Y.; Wang, J. Surface modification of PDMS by surface-initiated atom transfer radical polymerization of water-soluble dendronized PEG methacrylate. Colloid Surf. B 2011, 88, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Efimenko, K.; Finlay, J.; Callow, M.E.; Callow, J.A.; Genzer, J. Development and Testing of Hierarchically Wrinkled Coatings for Marine Antifouling. ACS Appl. Mater. Interfaces 2009, 1, 1031–1040. [Google Scholar] [CrossRef]
- Khang, D.Y.; Rogers, J.A.; Lee, H.H. Mechanical Buckling: Mechanics, Metrology, and Stretchable Electronics. Adv. Funct. Mater. 2009, 19, 1526–1536. [Google Scholar] [CrossRef]
- Greco, F.; Ventrelli, L.; Dario, P.; Mazzolai, B.; Mattoli, V. Micro-wrinkled palladium surface for hydrogen sensing and switched detection of lower flammability limit. Int. J. Hydrogen Energy 2012, 37, 17529–17539. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, J. Wrinkled interfaces: Taking advantage of surface instabilities to pattern polymer surfaces. Prog. Polym. Sci. 2015, 42, 1–41. [Google Scholar] [CrossRef] [Green Version]
- Thiry, D.; Vinx, N.; Damman, P.; Aparicio, F.J.; Tessier, P.Y.; Moerman, D.; LeclAre, P.; Godfroid, T.; Desprez, S.; Snyders, R. The wrinkling concept applied to plasma-deposited polymer-like thin films: A promising method for the fabrication of flexible electrodes. Plasma Process. Polym. 2020, 17, 2000119. [Google Scholar] [CrossRef]
- Cutolo, A.; Pagliarulo, V.; Merola, F.; Coppola, S.; Ferraro, P.; Fraldi, M. Wrinkling prediction, formation and evolution in thin films adhering on polymeric substrata. Mater. Des. 2020, 187, 108314. [Google Scholar] [CrossRef]
- Yan, X.; Perry, S.S.; Spencer, N.D.; Pasche, S.; De Paul, S.M.; Textor, M.; Lim, M.S. Reduction of Friction at Oxide Interfaces upon Polymer Adsorption from Aqueous Solutions. Langmuir 2004, 20, 423–428. [Google Scholar] [CrossRef]
- Lee, S.; Spencer, N.D. Superlubricity; Elsevier Science B.V.: Amsterdam, The Netherlands, 2007; pp. 365–396. [Google Scholar]
- Drobek, T.; Spencer, N.D. Nanotribology of Surface-Grafted PEG Layers in an Aqueous Environment. Langmuir 2008, 24, 1484–1488. [Google Scholar] [CrossRef]
- Yoon, J.Y.; Garrell, R.L. Preventing Biomolecular Adsorption in Electrowetting-Based Biofluidic Chips. Anal. Chem. 2003, 75, 5097–5102. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R. When Microfluidic Devices Go Bad. Anal. Chem. 2005, 77, 429A–432A. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegemann, D.; Bülbül, E.; Hanselmann, B.; Schütz, U.; Amberg, M.; Gaiser, S. Plasma polymerization of hexamethyldisiloxane: Revisited. Plasma Process. Polym. 2020, e2000176. [Google Scholar]
- Sigma-Aldrich. Data Sheet: Polyethylene Glycol 400. Available online: https://www.sigmaaldrich.com/catalog/product/mm/807485 (accessed on 16 June 2020).
- Hegemann, D.; Körner, E.; Albrecht, K.; Schütz, U.; Guimond, S. Growth Mechanism of Oxygen-Containing Functional Plasma Polymers. Plasma Process. Polym. 2010, 7, 889–898. [Google Scholar] [CrossRef]
- Hegemann, D.; Michlicek, M.; Blanchard, N.E.; Schütz, U.; Lohmann, D.; Vandenbossche, M.; Zajickova, L.; Drabik, M. Deposition of Functional Plasma Polymers Influenced by Reactor Geometry in Capacitively Coupled Discharges. Plasma Process. Polym. 2016, 13, 279–286. [Google Scholar]
- Hegemann, D.; Nisol, B.; Gaiser, S.; Watson, S.; Wertheimer, M.R. Energy conversion efficiency in low- and atmospheric-pressure plasma polymerization processes with hydrocarbons. Phys. Chem. Chem. Phys. 2019, 21, 8698–8708. [Google Scholar] [CrossRef]
- Mundry, T.; Surmann, P.; Schurreit, T. Surface characterization of polydimethylsiloxane treated pharmaceutical glass containers by X-ray-excited photo- and Auger electron spectroscopy. Fresen. J. Anal. Chem. 2000, 368, 820–831. [Google Scholar] [CrossRef]
- Hillborg, H.; Gedde, U. Hydrophobicity recovery of polydimethylsiloxane after exposure to corona discharges. Polymer 1998, 39, 1991–1998. [Google Scholar]
- Morra, M.; Occhiello, E.; Marola, R.; Garbassi, F.; Humphrey, P.; Johnson, D. On the aging of oxygen plasma-treated polydimethylsiloxane surfaces. J. Colloid Interfaces Sci. 1990, 137, 11–24. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Physical Electronics Inc.: Eden Prairie, MN, USA, 1995. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Gas (Gas Flow Rate [sccm]) | Pressure [mbar] | Power [W] | Duration [min] | |
|---|---|---|---|---|---|
| Process 1 | 1 | Ar (40) + O (10) | 0.1 | 50 | 20 |
| Process 2 | 1 | Ar (40) | 0.1 | 50 | 10 |
| 2 | CH (16) | 0.2 → 0.1 | 30 | 1 + 9 | |
| 3 | Ar (40) | 0.1 | 50 | 15 |
Sample Availability: Samples of the type Ar-O2 and Ar-C2H4-Ar are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaiser, S.; Schütz, U.; Rupper, P.; Hegemann, D. Plasma Processing of Low Vapor Pressure Liquids to Generate Functional Surfaces. Molecules 2020, 25, 6024. https://doi.org/10.3390/molecules25246024
Gaiser S, Schütz U, Rupper P, Hegemann D. Plasma Processing of Low Vapor Pressure Liquids to Generate Functional Surfaces. Molecules. 2020; 25(24):6024. https://doi.org/10.3390/molecules25246024
Chicago/Turabian StyleGaiser, Sandra, Urs Schütz, Patrick Rupper, and Dirk Hegemann. 2020. "Plasma Processing of Low Vapor Pressure Liquids to Generate Functional Surfaces" Molecules 25, no. 24: 6024. https://doi.org/10.3390/molecules25246024
APA StyleGaiser, S., Schütz, U., Rupper, P., & Hegemann, D. (2020). Plasma Processing of Low Vapor Pressure Liquids to Generate Functional Surfaces. Molecules, 25(24), 6024. https://doi.org/10.3390/molecules25246024