
Development and Validation of Methods for the Determination of Anthocyanins in Physiological Fluids via UHPLC-MSn


Abstract
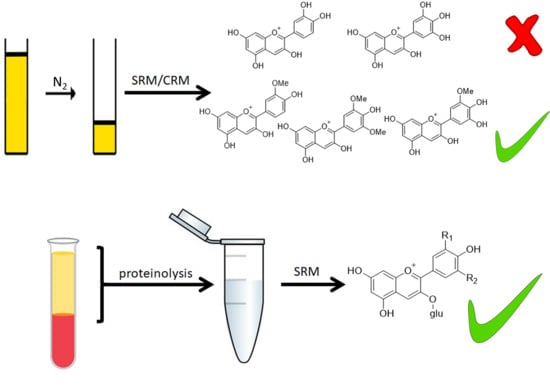
1. Introduction
2. Results and Discussion
2.1. Anthocyanidins in Urine
2.2. Anthocyanins in Plasma
3. Materials and Methods
3.1. Chemicals and Reagents
Stock Solutions
3.2. Sample Preparation
3.2.1. Urine
3.2.2. Plasma
3.3. UHPLC-MS
3.4. Validation
3.5. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smeriglio, A.; Barreca, D.; Bellocco, E.; Trombetta, D. Chemistry, Pharmacology and Health Benefits of Anthocyanins: Anthocyanins and Human Health. Phytother. Res. 2016, 30, 1265–1286. [Google Scholar] [CrossRef]
- Braga, A.R.C.; Murador, D.C.; de Souza Mesquita, L.M.; de Rosso, V.V. Bioavailability of anthocyanins: Gaps in knowledge, challenges and future research. J. Food Compos. Anal. 2018, 68, 31–40. [Google Scholar] [CrossRef]
- Khoo, H.E.; Azlan, A.; Tang, S.T.; Lim, S.M. Anthocyanidins and anthocyanins: Colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. Res. 2017, 61, 1361779. [Google Scholar] [CrossRef]
- Li, D.; Wang, P.; Luo, Y.; Zhao, M.; Chen, F. Health benefits of anthocyanins and molecular mechanisms: Update from recent decade. Crit. Rev. Food Sci. Nutr. 2017, 57, 1729–1741. [Google Scholar] [CrossRef]
- Müller, D.; Schantz, M.; Richling, E. High Performance Liquid Chromatography Analysis of Anthocyanins in Bilberries (Vaccinium myrtillus L.), Blueberries (Vaccinium corymbosum L.), and Corresponding Juices. J. Food Sci. 2012, 77, C340–C345. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, H.; Yi, J.; Yang, B.; Li, M.; He, D.; Yang, W.; Zhang, Y.; Ni, H. Anti-tumor properties of anthocyanins from Lonicera caerulea ‘Beilei’ fruit on human hepatocellular carcinoma: In vitro and in vivo study. Biomed. Pharmacother. 2018, 104, 520–529. [Google Scholar] [CrossRef]
- Shim, S.H.; Kim, J.M.; Choi, C.Y.; Kim, C.Y.; Park, K.H. Ginkgo biloba Extract and Bilberry Anthocyanins Improve Visual Function in Patients with Normal Tension Glaucoma. J. Med. Food 2012, 15, 818–823. [Google Scholar] [CrossRef]
- Miyake, S.; Takahashi, N.; Sasaki, M.; Kobayashi, S.; Tsubota, K.; Ozawa, Y. Vision preservation during retinal inflammation by anthocyanin-rich bilberry extract: Cellular and molecular mechanism. Lab. Invest. 2012, 92, 102–109. [Google Scholar] [CrossRef] [PubMed]
- De Ferrars, R.M.; Czank, C.; Saha, S.; Needs, P.W.; Zhang, Q.; Raheem, K.S.; Botting, N.P.; Kroon, P.A.; Kay, C.D. Methods for Isolating, Identifying, and Quantifying Anthocyanin Metabolites in Clinical Samples. Anal. Chem. 2014, 86, 10052–10058. [Google Scholar] [CrossRef] [PubMed]
- Czank, C.; Cassidy, A.; Zhang, Q.; Morrison, D.J.; Preston, T.; Kroon, P.A.; Botting, N.P.; Kay, C.D. Human metabolism and elimination of the anthocyanin, cyanidin-3-glucoside: A 13C-tracer study. Am. J. Clin. Nutr. 2013, 97, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Maier, C.S. The chemistry of gut microbial metabolism of polyphenols. Phytochem. Rev. 2016, 15, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Tan, Y.; Chen, G.; Wang, G.; Sun, J.; Ou, S.; Chen, W.; Bai, W. Metabolism of anthocyanins and consequent effects on the gut microbiota. Crit. Rev. Food Sci. Nutr. 2019, 59, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Lila, M.A.; Burton-Freeman, B.; Grace, M.; Kalt, W. Unraveling Anthocyanin Bioavailability for Human Health. Annu. Rev. Food Sci. Technol. 2016, 7, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.L.; Wu, X. Anthocyanins: Structural characteristics that result in unique metabolic patterns and biological activities. Free Radic. Res. 2006, 40, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Harel, S.; Akiri, B.; Granit, R.; Kanner, J. pH-Dependent Forms of Red Wine Anthocyanins as Antioxidants. J. Agric. Food Chem. 1999, 47, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H. Chemical Composition of Blood Plasma and Serum. Annu. Rev. Biochem. 1950, 19, 409–430. [Google Scholar] [CrossRef]
- Rose, C.; Parker, A.; Jefferson, B.; Cartmell, E. The Characterization of Feces and Urine: A Review of the Literature to Inform Advanced Treatment Technology. Crit. Rev. Environ. Sci. Technol. 2015, 45, 1827–1879. [Google Scholar] [CrossRef]
- Hribar, U.; Poklar Ulrih, N. The Metabolism of Anthocyanins. Curr. Drug Metab. 2014, 15, 3–13. [Google Scholar] [CrossRef]
- Richter, R.; Schulz-Knappe, P.; Schrader, M.; Ständker, L.; Jürgens, M.; Tammen, H.; Forssmann, W.-G. Composition of the peptide fraction in human blood plasma: Database of circulating human peptides. J. Chromatogr. B. Biomed. Sci. App. 1999, 726, 25–35. [Google Scholar] [CrossRef]
- Kaiser, M.; Lacheta, B.; Passon, M.; Schieber, A. An Innovative Approach to the Preparation of Plasma Samples for UHPLC–MS Analysis. J. Agric. Food Chem. 2019, 67, 6665–6671. [Google Scholar] [CrossRef]
- Nurmi, T.; Mursu, J.; Heinonen, M.; Nurmi, A.; Hiltunen, R.; Voutilainen, S. Metabolism of Berry Anthocyanins to Phenolic Acids in Humans. J. Agric. Food Chem. 2009, 57, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Coletta, W.; Rapisarda, P.; Donati, M.B.; Rotilio, D. Development and validation of an LC-MS/MS analysis for simultaneous determination of delphinidin-3-glucoside, cyanidin-3-glucoside and cyanidin-3-(6-malonylglucoside) in human plasma and urine after blood orange juice administration. J. Sep. Sci. 2007, 30, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Matsumoto, H.; Morifuji, M.; Iida, H.; Takeuchi, Y. Development and Validation of a Liquid Chromatography Tandem Mass Spectrometry Method for Simultaneous Determination of Four Anthocyanins in Human Plasma after Black Currant Anthocyanins Ingestion. J. Agric. Food Chem. 2010, 58, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- De Ferrars, R.M.; Cassidy, A.; Curtis, P.; Kay, C.D. Phenolic metabolites of anthocyanins following a dietary intervention study in post-menopausal women. Mol. Nutr. Food Res. 2014, 58, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, J.; Huang, K.; Michel, D.; Fang, J. HPLC-MS/MS analysis of anthocyanins in human plasma and urine using protein precipitation and dilute-and-shoot sample preparation methods, respectively. Biomed. Chromatogr. 2018, 32, e4177. [Google Scholar] [CrossRef] [PubMed]
- Sperlingova, I.; Dabrowská, L.; Tichý, M.; Kucera, J. Problems of traceability of total protein and catecholamine determinations in human urine. Accreditation Qual. Assur. 2001, 6, 302–305. [Google Scholar] [CrossRef]
- Netzel, M.; Strass, G.; Janssen, M.; Bitsch, I.; Bitsch, R. Bioactive Anthocyanins Detected in Human Urine after Ingestion of Blackcurrant Juice. J. Environ. Pathol. Toxicol. Oncol. 2001, 20, 89–95. [Google Scholar] [CrossRef]
- Mülleder, U.; Murkovic, M.; Pfannhauser, W. Urinary excretion of cyanidin glycosides. J. Biochem. Biophys. Methods 2002, 53, 61–66. [Google Scholar] [CrossRef]
- Behrends, A.; Weber, F. Influence of Different Fermentation Strategies on the Phenolic Profile of Bilberry Wine (Vaccinium myrtillus L.). J. Agric. Food Chem. 2017, 65, 7483–7490. [Google Scholar] [CrossRef]
- Mazza, G.; Kay, C.D.; Cottrell, T.; Holub, B.J. Absorption of Anthocyanins from Blueberries and Serum Antioxidant Status in Human Subjects. J. Agric. Food Chem. 2002, 50, 7731–7737. [Google Scholar] [CrossRef]
- Kuntz, S.; Rudloff, S.; Asseburg, H.; Borsch, C.; Fröhling, B.; Unger, F.; Dold, S.; Spengler, B.; Römpp, A.; Kunz, C. Uptake and bioavailability of anthocyanins and phenolic acids from grape/blueberry juice and smoothie in vitro and in vivo. Br. J. Nutr. 2015, 113, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Vargas, F.; Jiménez, A.R.; Paredes-López, O. Natural Pigments: Carotenoids, Anthocyanins, and Betalains—Characteristics, Biosynthesis, Processing, and Stability. Crit. Rev. Food Sci. Nutr. 2000, 40, 173–289. [Google Scholar] [CrossRef] [PubMed]
- Gleichenhagen, M.; Schieber, A. Current challenges in polyphenol analytical chemistry. Curr. Opin. Food Sci. 2016, 7, 43–49. [Google Scholar] [CrossRef]
- Schmitt, S.; Tratzka, S.; Schieber, A.; Passon, M. Hemisynthesis of Anthocyanin Phase II Metabolites by Porcine Liver Enzymes. J. Agric. Food Chem. 2019, 67, 6177–6189. [Google Scholar] [CrossRef]
- Wellmitz, J.; Gluschke, M. Leitlinie zur Methodenvalidierung im BLMP 2005; Umweltbundesamt: Berlin, Germany, 2005. [Google Scholar]
- Furey, A.; Moriarty, M.; Bane, V.; Kinsella, B.; Lehane, M. Ion suppression: A critical review on causes, evaluation, prevention and applications. Talanta 2013, 115, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Booth, B.; Kadavil, J. Guidance for Industry—Bioanalytical Method Validation—Biopharmaceutics, Revision 1; Food and Drug Administration: Silver Spring, MD, USA, 2013.
- González, O.; Blanco, M.E.; Iriarte, G.; Bartolomé, L.; Maguregui, M.I.; Alonso, R.M. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. J. Chromatogr. A 2014, 1353, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Kromidas, S. Handbuch Validierung in der Analytik; 2; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32938-0. [Google Scholar]
- Hibbert, D.B. Systematic errors in analytical measurement results. J. Chromatogr. A 2007, 1158, 25–32. [Google Scholar] [CrossRef][Green Version]
- Ohnishi, R.; Ito, H.; Kasajima, N.; Kaneda, M.; Kariyama, R.; Kumon, H.; Hatano, T.; Yoshida, T. Urinary Excretion of Anthocyanins in Humans after Cranberry Juice Ingestion. Biosci. Biotechnol. Biochem. 2006, 70, 1681–1687. [Google Scholar] [CrossRef]
- Kalt, W.; Liu, Y.; McDonald, J.E.; Vinqvist-Tymchuk, M.R.; Fillmore, S.A.E. Anthocyanin Metabolites Are Abundant and Persistent in Human Urine. J. Agric. Food Chem. 2014, 62, 3926–3934. [Google Scholar] [CrossRef]
- Milbury, P.E.; Vita, J.A.; Blumberg, J.B. Anthocyanins are Bioavailable in Humans following an Acute Dose of Cranberry Juice. J. Nutr. 2010, 140, 1099–1104. [Google Scholar] [CrossRef]
- Felgines, C.; Talavéra, S.; Gonthier, M.-P.; Texier, O.; Scalbert, A.; Lamaison, J.-L.; Rémésy, C. Strawberry anthocyanins are recovered in urine as glucuro-and sulfoconjugates in humans. J. Nutr. 2003, 133, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Ovando, A.; de Lourdes Pacheco-Hernández, M.; Páez-Hernández, M.E.; Rodríguez, J.A.; Galán-Vidal, C.A. Chemical studies of anthocyanins: A review. Food Chem. 2009, 113, 859–871. [Google Scholar] [CrossRef]
- Cao, H.; Liu, X.; Ulrih, N.P.; Sengupta, P.K.; Xiao, J. Plasma protein binding of dietary polyphenols to human serum albumin: A high performance affinity chromatography approach. Food Chem. 2019, 270, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, B.F.; Papagiannopoulos, M.; Brachmann, S.; Lorenz, M.; Stangl, V.; Galensa, R. A shortcut from plasma to chromatographic analysis: Straightforward and fast sample preparation for analysis of green tea catechins in human plasma. J. Chromatogr. B 2009, 877, 823–826. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |



{kind=link}
{kind=link}
{kind=link}
| Linearity | Analytical Limits | Precision | Recovery | Repeatability | Robustness | ||||
|---|---|---|---|---|---|---|---|---|---|
| Range (µg/mL) | LOD (ng/mL) | LOQ (LLOQ) (ng/mL) | System CV (%) | Method CV (%) | (%) | (%) | Process Stability CV (%) | ||
| Dp | 0.125–67 | 58.1 | 205.3 (189.7) | 9.4 | 38.0 | 46.7 | 51.5 | 6.7 | |
| Cy | 0.116–71 | 124.1 | 427.2 (353.5) | 6.8 | 26.7 | 78.7 | 60.5 | 7.1 | |
| Pt | 0.006–11 | 12.7 | 46.0 (15.7) | 2.9 | 15.3 | 96.5 | 42.5 | 7.4 | |
| Pn | 0.005–38 | 9.6 | 35.3 (25.6) | 2.2 | 19.1 | 64.3 | 31.7 | 3.7 | |
| Mn | 0.005–15 | 5.1 | 18.8 (15.1) | 3.7 | 18.7 | 65.8 | 30.9 | 5.6 | |
| Pg | 0.106–54 | 120.3 | 416.2 (412.2) | 7.4 | -- | -- | -- | -- | |
| Analyte | MS (M)+ | MS2 (M)+ | MS3 (M)+ | Collision Energies MS2/MS3 (%) |
|---|---|---|---|---|
| Dp | 303 | 257 | 229 | 60/45 |
| Dp-glc | 465 | 303 | - | 10 |
| Cy | 287 | 213 | - | 65 |
| Cy-glc | 449 | 287 | - | 10 |
| Pt | 317 | 302 | 274 | 40/25 |
| Pt-glc | 479 | 317 | - | 10 |
| Pn | 301 | 286 | 258 | 40/40 |
| Pn-glc | 463 | 301 | - | 10 |
| Mn | 331 | 316 | 299 | 50/20 |
| Mn-glc | 493 | 331 | - | 10 |
| Pg | 271 | 149215 | - | 67 |
| Pg-glc | 433 | 271 | - | 10 |
| Urine Sample | c(Dp) (ng/mL) | c(Cy) (ng/mL) | c(Pt) (ng/mL) | Øc(Pt) (ng/mL) | CV (%) | c(Pn) (ng/mL) | Øc(Pn) (ng/mL) | CV (%) | c(Mn) (ng/mL) | Øc(Mn) (ng/mL) | CV (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | n.d. n.d. n.d. n.d. | n.d. n.q. n.d. n.q. | 19.4 20.8 17.5 20.5 | 19.5 | 7.6 | 27.6 34.7 30.3 34.8 | 31.8 | 11.1 | 16.6 22.1 18.8 21.9 | 19.8 | 13.3 |
| #2 | n.d. n.d. n.d. n.d. | n.q. n.q. n.q. n.q. | 22.1 18.8 19.2 22.6 | 20.7 | 9.4 | 37.2 38.4 35.8 38.1 | 37.4 | 3.1 | 26.6 28.7 25.1 22.8 | 25.8 | 9.6 |
| #3 | n.d. n.d. n.d. n.d. | n.d. n.q. n.d. n.d. | 29.7 27.3 25.2 27.2 | 27.3 | 6.7 | 43.2 41.1 40.7 40.1 | 41.3 | 3.3 | 21.6 21.4 19.5 17.4 | 20.0 | 9.8 |
| #4 | n.d. n.d. n.d. n.d. | n.q. n.q. n.q. n.q. | 32.6 30.0 38.0 43.0 | 35.9 | 16.1 | 32.5 29.6 37.0 40.9 | 35.0 | 14.2 | 18.9 19.5 20.0 25.3 | 21.0 | 14.1 |
| #5 | n.d. n.d. n.d. n.d. | n.d. n.d. n.d. n.d. | 26.3 29.8 22.7 26.7 | 26.4 | 11.0 | 45.9 43.9 43.6 44.8 | 44.5 | 2.3 | 29.1 29.2 29.0 29.1 | 29.1 | 0.3 |
| Linearity | Analytical Limits | Precision | Recovery | Repeat-Abilityr | Robust-Ness | |||
|---|---|---|---|---|---|---|---|---|
| Range (µg/mL) | LOD (ng/mL) | LOQ (LLOQ)(ng/mL) | System CV (%) | Method CV (%) | (%) | (%) | Process Stability CV (%) | |
| Dp-glc | 0.002–24 | 2.3 | 8.1 (4.2) | 1.9 | 15.6 | 53.8 | 20.7 | 5.0 |
| Cy-glc | 0.003–23 | 2.0 | 7.3 (6.9) | 2.6 | 7.7 | 84.6 | 10.8 | 5.7 |
| Pt-glc | 0.004–25 | 2.1 | 7.7 (6.1) | 2.8 | 14.5 | 68.6 | 23.2 | 3.1 |
| Pn-glc | 0.002–24 | 1.2 | 4.1 (3.7) | 3.3 | 4.9 | 98.8 | 13.2 | 5.4 |
| Mn-glc | 0.002–25 | 2.2 | 7.9 (3.3) | 5.1 | 12.2 | 107.6 | 20.7 | 5.5 |
| Pg-glc | 0.003–25 | 2.2 | 8.0 (4.8) | 3.0 | -- | -- | -- | -- |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, M.; Müller-Ehl, L.; Passon, M.; Schieber, A. Development and Validation of Methods for the Determination of Anthocyanins in Physiological Fluids via UHPLC-MSn. Molecules 2020, 25, 518. https://doi.org/10.3390/molecules25030518
Kaiser M, Müller-Ehl L, Passon M, Schieber A. Development and Validation of Methods for the Determination of Anthocyanins in Physiological Fluids via UHPLC-MSn. Molecules. 2020; 25(3):518. https://doi.org/10.3390/molecules25030518
Chicago/Turabian StyleKaiser, Michael, Lisa Müller-Ehl, Maike Passon, and Andreas Schieber. 2020. "Development and Validation of Methods for the Determination of Anthocyanins in Physiological Fluids via UHPLC-MSn" Molecules 25, no. 3: 518. https://doi.org/10.3390/molecules25030518
APA StyleKaiser, M., Müller-Ehl, L., Passon, M., & Schieber, A. (2020). Development and Validation of Methods for the Determination of Anthocyanins in Physiological Fluids via UHPLC-MSn. Molecules, 25(3), 518. https://doi.org/10.3390/molecules25030518