
Alternative Structures of ?-Synuclein

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Parameters Used for Structure Description
2.2. Structure of Human Micelle-Bound Alpha-Synuclein (1XQ8)
2.3. Structure of the Amyloid Form of ASyn (2N0A)
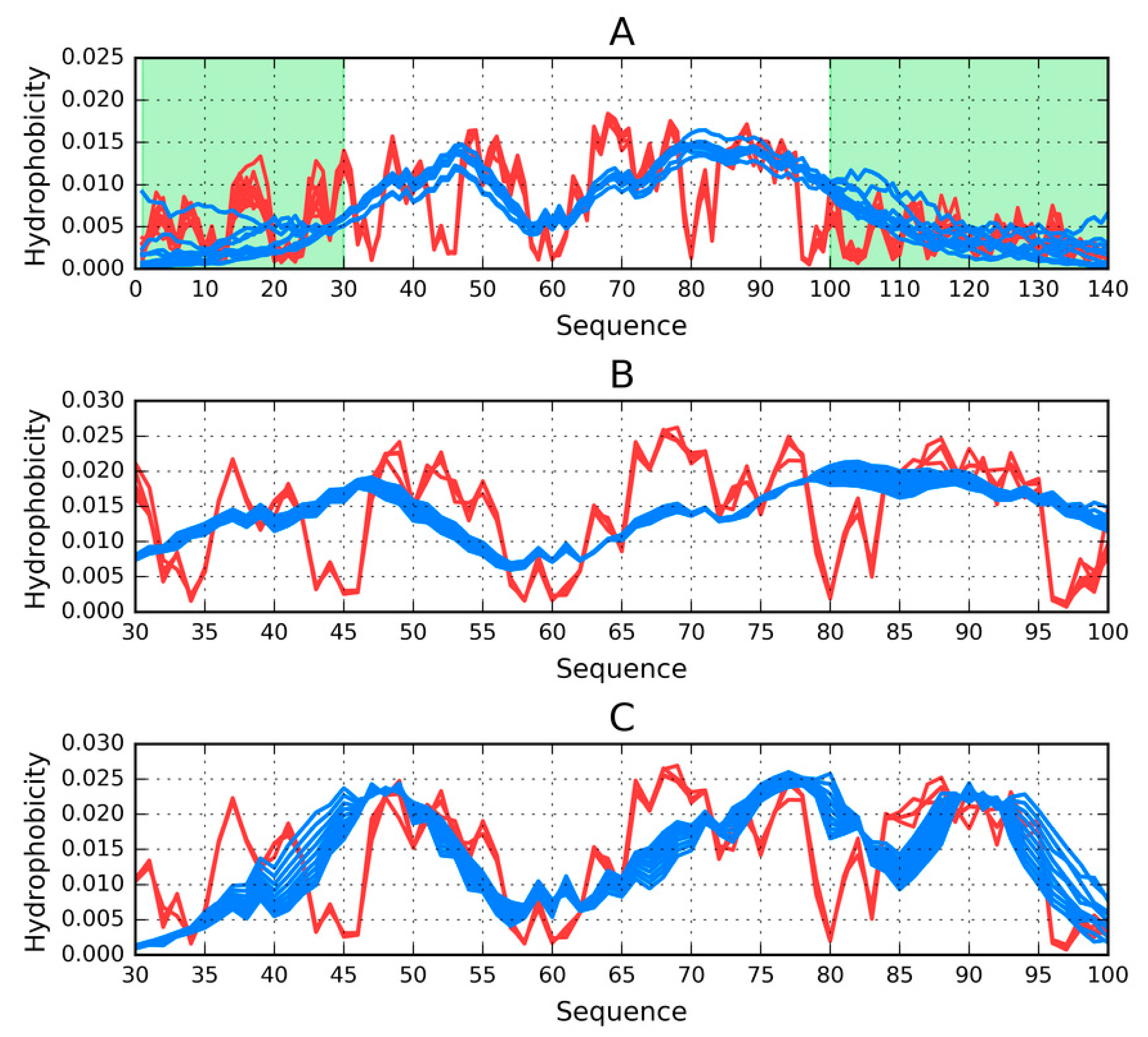
2.4. Models Generated by Specialized Software
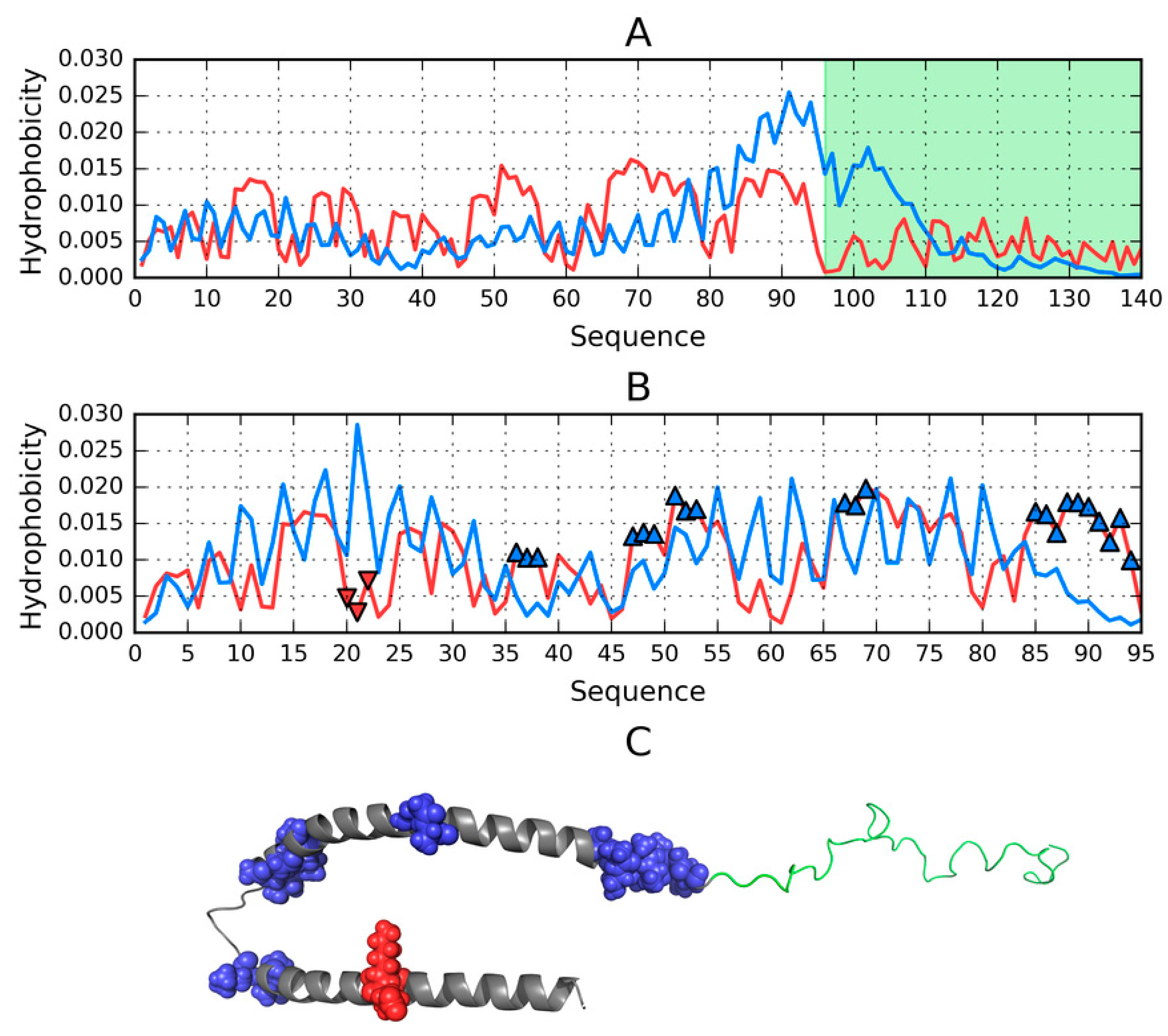
2.5. Analysis of Models for the 1–140 Fragment of ASyn
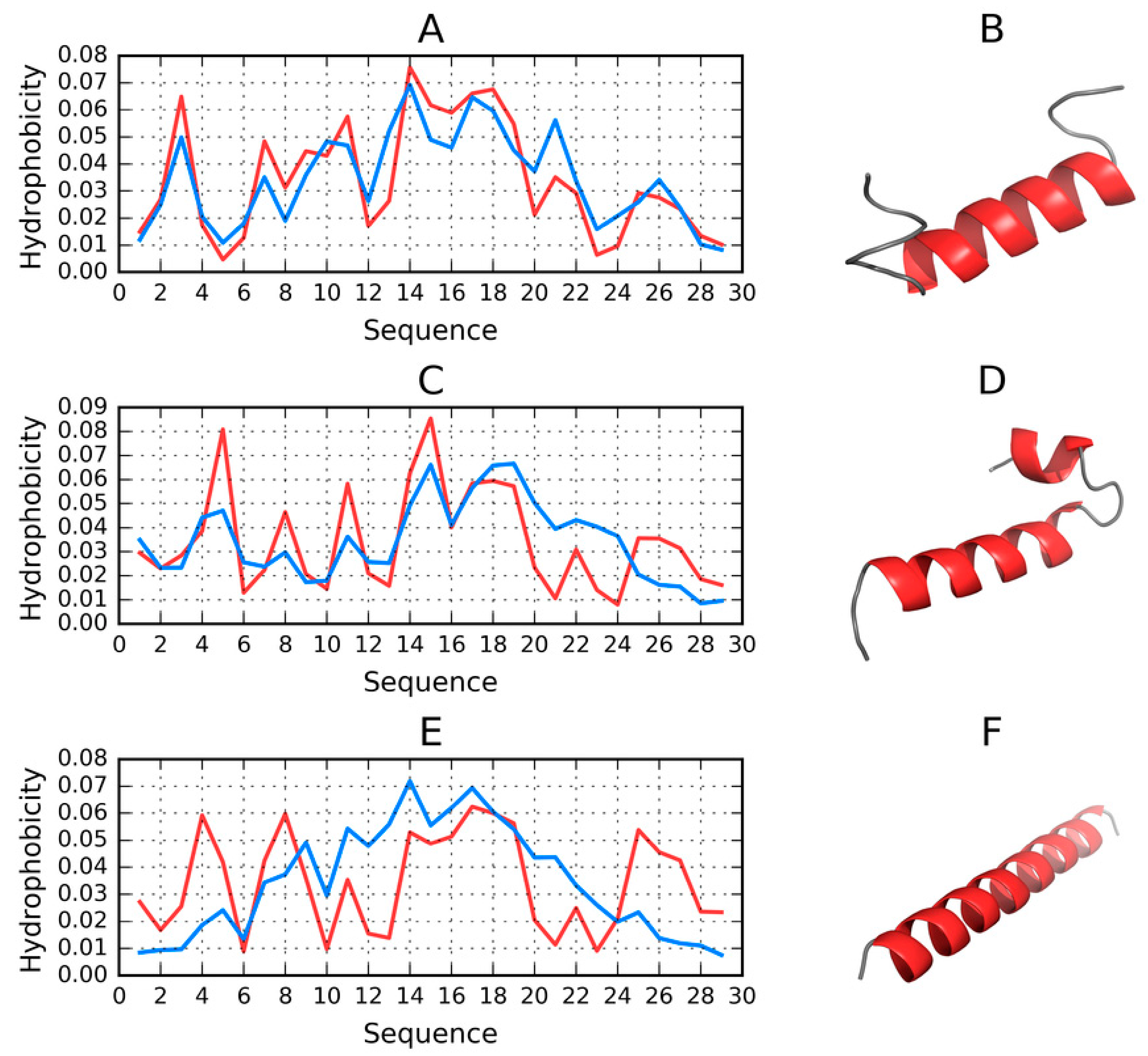
2.6. Structure of the N-Terminal Fragment in FOD, I-Tasser and Robetta Models (1–30 aa)
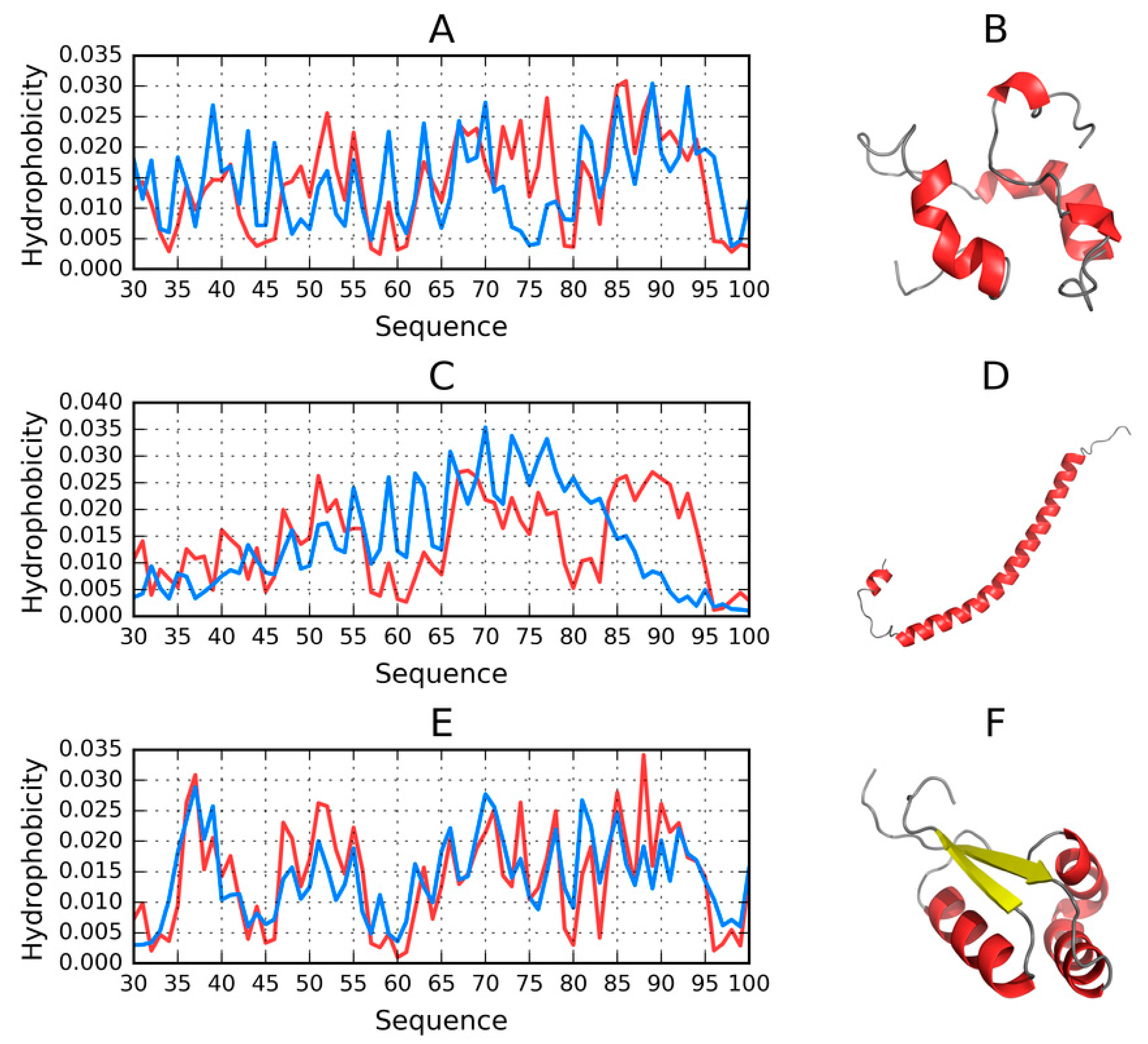
2.7. Structure of the 30–100 Fragment of the ASyn Polypeptide According to FOD, I-Tasser and Robetta
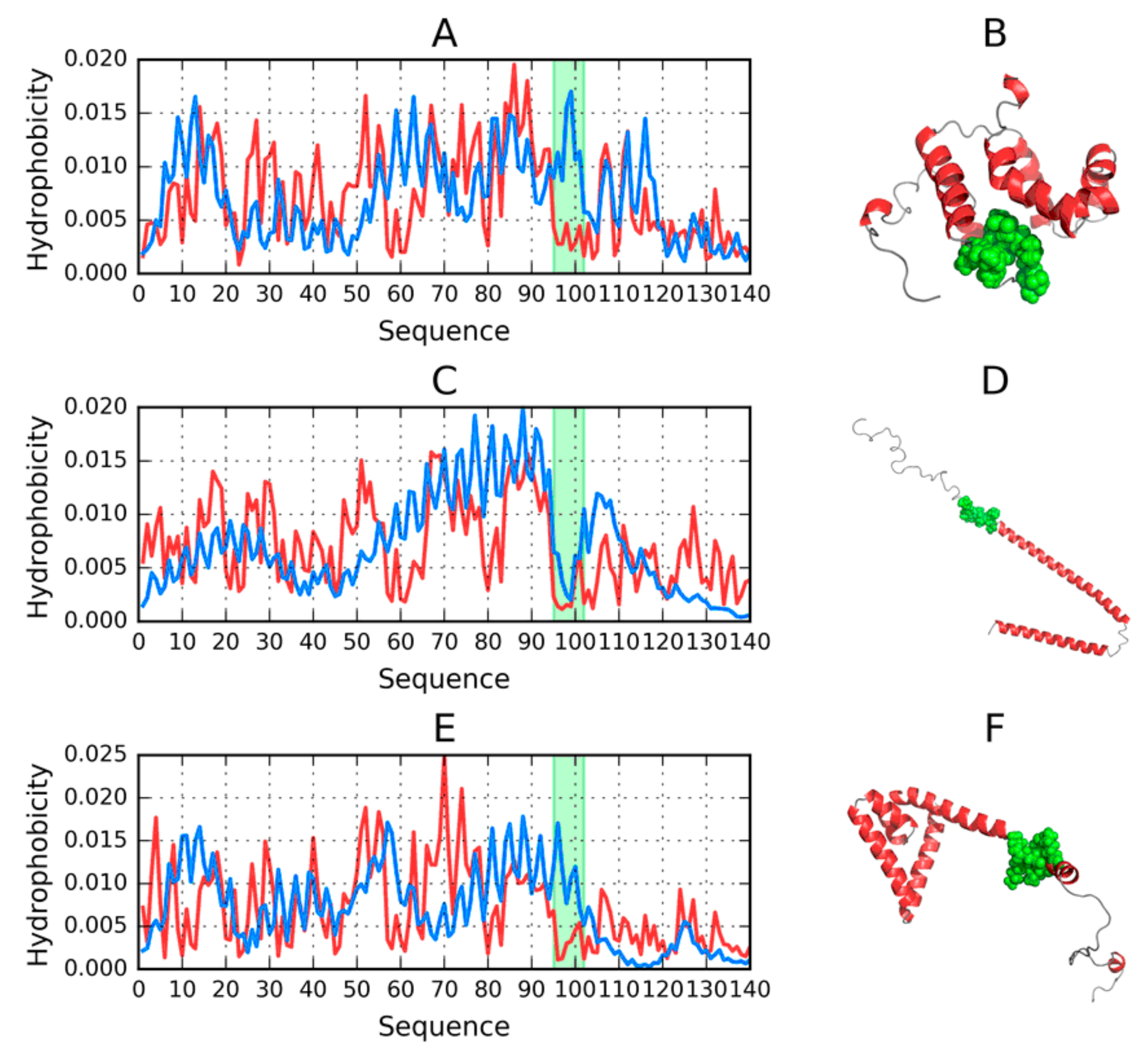
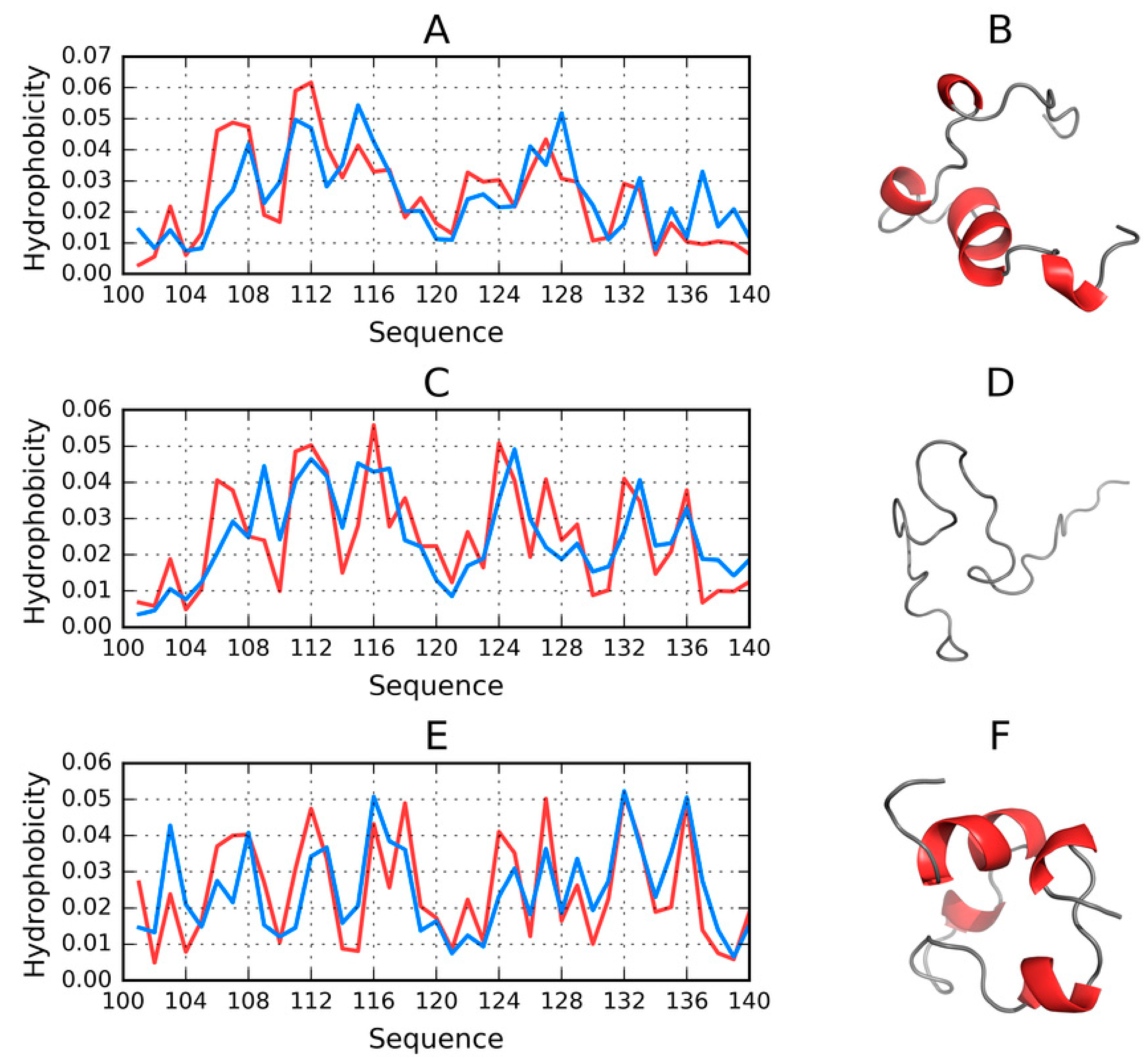
2.8. Comparative Analysis of Models Obtained for the 100–140 Fragment of ASyn
2.9. Effect of Mutations on Amyloid Transformation
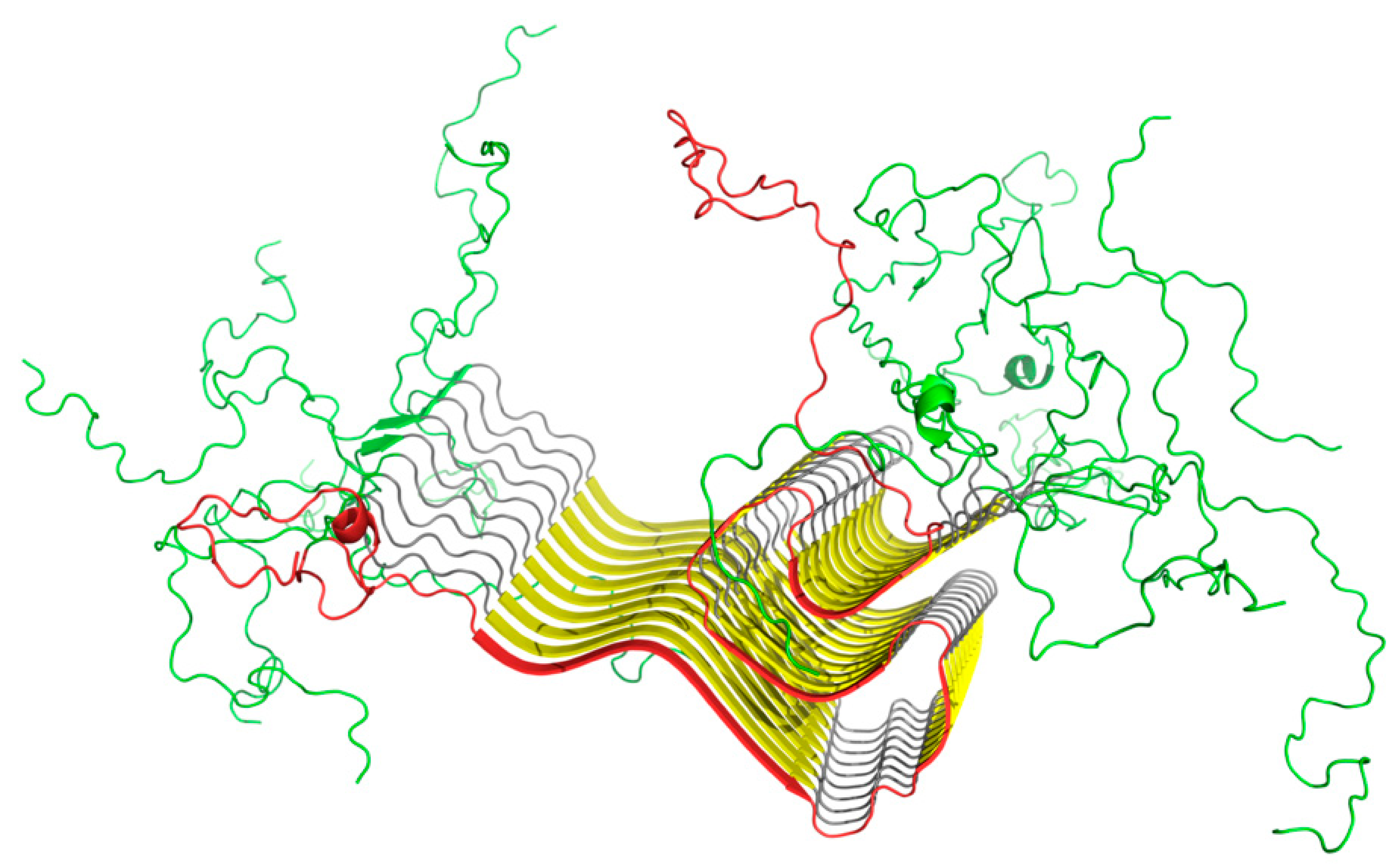
2.10. Status of Selected Fragments Identified in Other Publications as Linked to Amyloid Transformation in Parkinson’s Disease
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Obtaining Alternative ASyn Polypeptide Models
4.3. Fuzzy Oil Drop Model—Protein Folding with Preferential Generation of a Hydrophobic Core
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| ASyn | α-synuclein |
References
- Bonini, N.M.; Giasson, B.I. Snaring the Function of α-Synuclein. Cell 2005, 123, 359–361. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; De Silva, H.R.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Visanji, N.P.; Wislet-Gendebien, S.; Oschipok, L.W.; Zhang, G.; Aubert, I.; Fraser, P.E.; Tandon, A. Effect of Ser-129 Phosphorylation on Interaction of α-Synuclein with Synaptic and Cellular Membranes*. J. Boil. Chem. 2011, 286, 35863–35873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lykkebo, S.; Jensen, P.H. Alpha-Synuclein and Presynaptic Function: Implications for Parkinson’s Disease. NeuroMol. Med. 2002, 2, 115–130. [Google Scholar] [CrossRef]
- Cremades, N.; Chen, S.; Dobson, C. Structural Characteristics of α-Synuclein Oligomers. Int. Rev. Cell. Mol. Biol. 2017, 329, 79–143. [Google Scholar] [CrossRef]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G.; Tata, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Boil. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Sgourakis, N.G.; Yau, W.-M.; Qiang, W. Modeling an In-Register, Parallel “Iowa” Aβ Fibril Structure Using Solid-State NMR Data from Labeled Samples with Rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Boil. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-Resolution Three-Dimensional Structure of Amyloid β Fibrils Bearing the Osaka Mutation**. Angew. Chem. Int. Ed. 2014, 54, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.V.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.S.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, N.; Roterman, I. Amyloid as a ribbon-like micelle. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020; pp. 177–192. [Google Scholar]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-bound Human -Synuclein. J. Boil. Chem. 2004, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)-round x. Proteins: Struct. Funct. Bioinform. 2013, 82, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, L.; Brylinski, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. Silico Boil. 2006, 6, 15–22. [Google Scholar]
- Gadzała, M.; Dułak, D.; Kalinowska, B.; Baster, Z.; Bryliński, M.; Konieczny, L.; Banach, M.; Roterman, I. The aqueous environment as an active participant in the protein folding process. J. Mol. Graph. Model. 2019, 87, 227–239. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Dułak, D.; Banach, M.; Gadzała, M.; Konieczny, L.; Roterman, I. Structural analysis of the Aβ(15-40) amyloid fibril based on hydrophobicity distribution. Acta Biochim. Pol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dułak, D.; Gadzała, M.; Banach, M.; Ptak, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Filamentous Aggregates of Tau Proteins Fulfil Standard Amyloid Criteria Provided by the Fuzzy Oil Drop (FOD) Model. Int. J. Mol. Sci. 2018, 19, 2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dułak, D.; Gadzała, M.; Stapor, K.; Fabian, P.; Konieczny, L.; Roterman, I. Holding with active paticipation of water. In From Globular Proteins to Amyloids; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Banach, M.; Konieczny, L.; Roterman, I. Fuzzy Oil Drop Model Application—From Globular Proteins to Amyloids. In Springer Series on Bio- and Neurosystems; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2018; pp. 639–658. [Google Scholar]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Boil. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Available online: https://zhanglab.ccmb.med.umich.edu/I-TASSER/ (accessed on 15 December 2018).
- Available online: http://robetta.org/submit.jsp (accessed on 15 December 2018).
- Banach, M.; Konieczny, L.; Roterman, I. The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules 2019, 24, 4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, L.; Roterman, I. Protein is ane intelligent micelle. In From Globular Proteins to Amyloids; Roterman-Konieczna, I., Ed.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Cambridge, MA, USA, 2020; pp. 241–252. [Google Scholar]
- Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation E46K increases phospholipids binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar]
- Xu, L.; Bhattacharya, S.; Thompson, D. The fold preference and thermodynamic stability of-synuclein fibrils is encoded in the non-amyloid-component region. Phys. Chem. Chem. Phys. 2018, 20, 4502–4512. [Google Scholar] [CrossRef]
- Schwalbe, M.; Ozenne, V.; Bibow, S.; Jaremko, M.; Jaremko, L.; Gajda, M.; Jensen, M.R.; Biernat, J.; Becker, S.; Mandelkow, E.; et al. Predictive Atomic Resolution Descriptions of Intrinsically Disordered hTau40 and α-Synuclein in Solution from NMR and Small Angle Scattering. Structure 2014, 22, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Sałapa, K.; Kalinowska, B.; Jadczyk, T.; Roterman, I. Measurement of Hydrophobicity Distribution in Proteins (2012) Non-redundant Protein Data Bank. BAMS 2012, 8, 327–334. [Google Scholar]
- Pech, U.; Verstreken, P. α-Synuclein and Tau: Mitochondrial Kill Switches. Neuron 2018, 97, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terakawa, M.S.; Lee, Y.-H.; Kinoshita, M.; Lin, Y.; Sugiki, T.; Fukui, N.; Ikenoue, T.; Kawata, Y.; Goto, Y. Membrane-induced initial structure of α-synuclein control its amyloidogenesis on model membranes. BBA-Biomembranes. 2018, 1860, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupyshev, A.B.; Korolenko, T.A.; Akopyan, A.A.; Amstislavskaya, T.G.; Tikhonova, M.A. Suppression of autophagy in the brain of transgenic mice with overexpression of A53T-mutant α-synuclein as an early event at synucleinopathy progression. Neurosci. Lett. 2018, 672, 140–144. [Google Scholar] [CrossRef]
- Flynn, J.D.; McGlinchey, R.P.; Walker, R.L.; Lee, J.C. Structural features of α-synuclein amyloid fibrils revealed by Raman spectroscopy. J. Boil. Chem. 2017, 293, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Tamgüney, G.; Korczyn, A.D. A critical review of the prion hypothesis of human synucleinopathies. Cell Tissue Res. 2017, 373, 213–220. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Jha, N.N.; Kumar, R.; Panigrahi, R.; Navalkar, A.; Ghosh, D.; Sahay, S.; Mondal, M.; Kumar, A.; Maji, S.K. Comparison of α-Synuclein Fibril Inhibition by Four Different Amyloid Inhibitors. ACS Chem. Neurosci. 2017, 8, 2722–2733. [Google Scholar] [CrossRef]
- Avila, C.L.; Chaves, S.; Socias, S.B.; Vera-Pingitore, E.; González-Lizárraga, F.; Vera, C.; Ploper, D.; Chehín, R. Lessons learned from protein aggregation: Toward technological and biomedical applications. Biophys. Rev. 2017, 9, 501–515. [Google Scholar] [CrossRef]
- Lek, S.; Vargas-Medrano, J.; Villanueva, E.; Marcus, B.S.; Godfrey, W.; Perez, R.G. Recombinant α- β- and γ-Synucleins Stimulate Protein Phosphatase 2A Catalytic Subunit Activity in Cell Free Assays. J. Vis. Exp. 2017, 126, e55361. [Google Scholar]
- Ono, K. The Oligomer Hypothesis in α-Synucleinopathy. Neurochem. Res. 2017, 42, 3362–3371. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2017, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- PLGrid User Manual. Available online: https://docs.cyfronet.pl/display/PLGDoc/User+manual (accessed on 5 July 2018).
- Ptak-Kaczor, M.; Banach, M.; Konieczny, L.; Roterman, I. Internal force field in selected proteins. Acta Biochim. Pol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. BBA-Biomembranes 2000, 1502, 16–30. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dułak, D.; Gadzała, M.; Banach, M.; Konieczny, L.; Roterman, I. Alternative Structures of ?-Synuclein. Molecules 2020, 25, 600. https://doi.org/10.3390/molecules25030600
Dułak D, Gadzała M, Banach M, Konieczny L, Roterman I. Alternative Structures of ?-Synuclein. Molecules. 2020; 25(3):600. https://doi.org/10.3390/molecules25030600
Chicago/Turabian StyleDułak, Dawid, Małgorzata Gadzała, Mateusz Banach, Leszek Konieczny, and Irena Roterman. 2020. "Alternative Structures of ?-Synuclein" Molecules 25, no. 3: 600. https://doi.org/10.3390/molecules25030600
APA StyleDułak, D., Gadzała, M., Banach, M., Konieczny, L., & Roterman, I. (2020). Alternative Structures of ?-Synuclein. Molecules, 25(3), 600. https://doi.org/10.3390/molecules25030600