
Validation of a LLME/GC-MS Methodology for Quantification of Volatile Compounds in Fermented Beverages

 , , ,
, , ,  and
and
Abstract
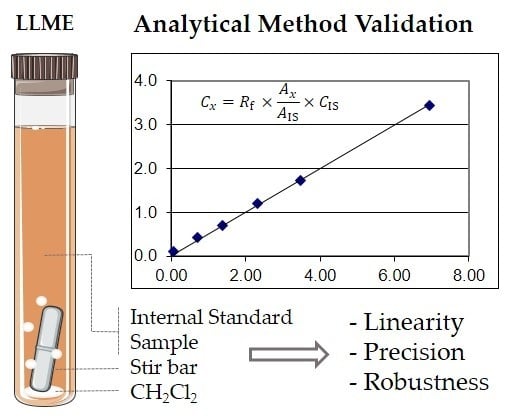
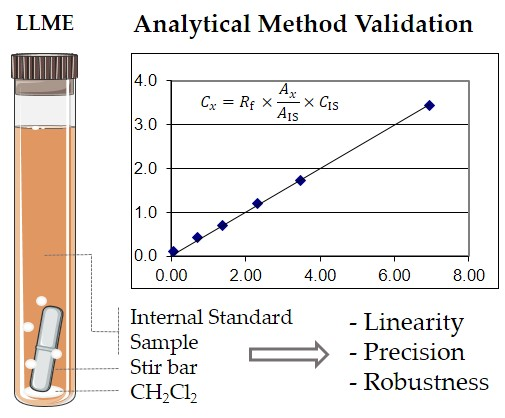
:
1. Introduction
2. Results and Discussion
2.1. Linearity and Sensitivity
2.2. Limits of Detection and Quantification
2.3. Precision
2.4. Accuracy
2.5. Robustness
3. Materials and Methods
3.1. LLME-GC/MS Method
3.1.1. Liquid-Liquid Microextraction of Volatile Compounds
3.1.2. Chromatographic Analysis
3.1.3. Identification of Volatile Compounds
3.2. Method Validation
3.2.1. Base Standard Solution
3.2.2. Linearity
3.2.3. Precision
3.2.4. Accuracy
3.2.5. Robustness
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Briggs, D.E.; Boulton, C.A.; Brookes, P.A.; Stevens, R. Brewing Science and Practice; Woodhead Publishing Limited: Cambridge, UK; CRC Press LLC: Boca Raton, FL, USA, 2004; p. 9062. ISBN 978185573. [Google Scholar]
- Robinson, A.L.; Boss, P.K.; Solomon, P.S.; Trengove, R.D.; Heymann, H.; Ebeler, S.E. Origins of Grape and Wine Aroma. Part 1. Chemical Components and Viticultural Impacts. Am. J. Enol. Vitic. 2013, 65, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Hirst, M.B.; Richter, C.L. Review of aroma formation through metabolic pathways of saccharomyces cerevisiae in beverage fermentations. Am. J. Enol. Vitic. 2016, 67, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Le Floch, A.; Jourdes, M.; Teissedre, P.-L. Polysaccharides and lignin from oak wood used in cooperage: Composition, interest, assays: A review. Carbohydr. Res. 2015, 417, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Ugliano, M.; Henschke, P.A. Yeasts and wine flavour. In Wine Chemistry and Biochemistry; Moreno-Arribas, M.V., Polo, M.C., Eds.; Springer: New York, NY, USA, 2009; pp. 313–392. ISBN 978-0-387-74116-1. [Google Scholar]
- Coelho, E.; Vilanova, M.; Genisheva, Z.; Oliveira, J.M.; Teixeira, J.A.; Domingues, L. Systematic approach for the development of fruit wines from industrially processed fruit concentrates, including optimization of fermentation parameters, chemical characterization and sensory evaluation. LWT 2015, 62, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Coelho, E.; Genisheva, Z.; Oliveira, J.M.; Teixeira, J.A.; Domingues, L. Vinegar production from fruit concentrates: Effect on volatile composition and antioxidant activity. J. Food Sci. Technol. 2017, 54, 4112–4122. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.; Boss, P.K.; Solomon, P.S.; Trengove, R.D.; Heymann, H.; Ebeler, S.E. Origins of Grape and Wine Aroma. Part 2. Chemical and Sensory Analysis. Am. J. Enol. Vitic. 2014, 65, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Dambergs, R.; Gishen, M.; Cozzolino, D. A review of the state of the art, limitations, and perspectives of infrared spectroscopy for the analysis of wine grapes, must, and grapevine tissue. Appl. Spectrosc. Rev. 2015, 50, 261–278. [Google Scholar] [CrossRef]
- Genisheva, Z.; Quintelas, C.; Mesquita, D.; Ferreira, E.; Oliveira, J.; Amaral, A. New PLS analysis approach to wine volatile compounds characterization by near infrared spectroscopy (NIR). Food Chem. 2018, 246, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Perestrelo, R.; Fernandes, A.; Albuquerque, F.; Marques, J.C.; Câmara, J. Analytical characterization of the aroma of Tinta Negra Mole red wine: Identification of the main odorants compounds. Anal. Chim. Acta 2006, 563, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Andújar-Ortiz, I.; Moreno-Arribas, M.; Martin-Alvarez, P.; Pozo-Bayon, M.A. Analytical performance of three commonly used extraction methods for the gas chromatography–mass spectrometry analysis of wine volatile compounds. J. Chromatogr. A 2009, 1216, 7351–7357. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.; Lopez, R.; Escudero, A.; Cacho, J.F. Quantitative determination of trace and ultratrace flavour active compounds in red wines through gas chromatographic–ion trap mass spectrometric analysis of microextracts. J. Chromatogr. A 1998, 806, 349–354. [Google Scholar] [CrossRef]
- Ortega, C.; López, R.; Cacho, J.; Ferreira, V. Fast analysis of important wine volatile compounds development and validation of a new method based on gas chromatographic-flame ionisation detection analysis of dichloromethane microextracts. J. Chromatogr. A 2001, 923, 205–214. [Google Scholar] [CrossRef]
- Jain, A.; Verma, K.K. Recent advances in applications of single-drop microextraction: A review. Anal. Chim. Acta 2011, 706, 37–65. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.M.; Oliveira, P.; Baumes, R.L.; Maia, O. Changes in aromatic characteristics of Loureiro and Alvarinho wines during maturation. J. Food Compos. Anal. 2008, 21, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhu, J.; Wang, L.; Li, Z. Development of a SPME-GC-MS method for the determination of volatile compounds in Shanxi aged vinegar and its analytical characterization by aroma wheel. J. Food Sci. Technol. 2016, 53, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Souza-Silva, É.A.; Gionfriddo, E.; Pawliszyn, J. A critical review of the state of the art of solid-phase microextraction of complex matrices II. Food analysis. TrAC Trends Anal. Chem. 2015, 71, 236–248. [Google Scholar] [CrossRef]
- Cirlini, M.; Caligiani, A.; Palla, L.; Palla, G. HS-SPME/GC–MS and chemometrics for the classification of Balsamic Vinegars of Modena of different maturation and ageing. Food Chem. 2011, 124, 1678–1683. [Google Scholar] [CrossRef]
- Martín, S.G.; Herrero, C.; Peña, R.; Barciela, J.; Crecente, R.M.P. Solid-phase microextraction gas chromatography–mass spectrometry (HS-SPME-GC–MS) determination of volatile compounds in orujo spirits: Multivariate chemometric characterisation. Food Chem. 2010, 118, 456–461. [Google Scholar] [CrossRef]
- Castro, L.F.; Ross, C.F. Determination of flavour compounds in beer using stir-bar sorptive extraction and solid-phase microextraction. J. Inst. Brew. 2015, 121, 197–203. [Google Scholar] [CrossRef]
- Horak, T.; Čulík, J.; Kellner, V.; Jurková, M.; Čejka, P.; Hašková, D.; Dvořák, J. Analysis of Selected Esters in Beer: Comparison of Solid-Phase Microextraction and Stir Bar Sorptive Extraction. J. Inst. Brew. 2010, 116, 81–85. [Google Scholar] [CrossRef]
- Rodríguez-Bencomo, J.J.; Schneider, R.; Lepoutre, J.P.; Rigou, P. Improved method to quantitatively determine powerful odorant volatile thiols in wine by headspace solid-phase microextraction after derivatization. J. Chromatogr. A 2009, 1216, 5640–5646. [Google Scholar] [CrossRef]
- Hartmann, C.; Smeyers-Verbeke, J.; Massart, D.L.; McDowall, R.D. Validation of bioanalytical chromatographic methods. J. Pharm. Biomed. Anal. 1998, 17, 193–218. [Google Scholar] [CrossRef]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part I. Anal. Chim. Acta 2015, 870, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part II. Anal. Chim. Acta 2015, 870, 8–28. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Faria, M.; Sá, F.; Barros, F.; Araújo, I.M. C6-alcohols as varietal markers for assessment of wine origin. Anal. Chim. Acta 2006, 563, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Araujo, P. Key aspects of analytical method validation and linearity evaluation. J. Chromatogr. B 2009, 877, 2224–2234. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Eurachem: Torino, Italy, 2014; Available online: http://eurachem.org (accessed on 2 January 2020).
- Brettell, T.A.; Lester, R.E. Validation and QA/QC of Gas Chromatographic Methods. Mod. Pract. Gas Chromatogr. 2004, 969–988. [Google Scholar]
- Gonzalez, O.; Blanco, M.E.; Iriarte, G.; Bartolomé, L.; Maguregui, M.I.; Alonso, R.M.; Rojas, R.M.A. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. J. Chromatogr. A 2014, 1353, 10–27. [Google Scholar] [CrossRef]
- Huber, L. Validation of analytical methods: review and strategy. LC-CG Int. 1998, 11, 96–105. [Google Scholar]
- AOAC International AOAC Official Methods of Analysis—Appendix K: Guidelines for Dietary Supplements and Botanicals. Available online: http://www.eoma.aoac.org/app_k.pdf (accessed on 2 January 2020).
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
| Compound | Reference | P/% | Range C/(µg/L) | R2 | LOQ/(µg/L) | |
|---|---|---|---|---|---|---|
| 4-methyl-2-pentanone | Fluka 02474 | ≥ 99.7 | 24.8 to 248 | 0.9991 | 6.9 | 1.32 ± 0.05 |
| Ethyl butyrate | Aldrich E15701 | 99 | 5.76 to 576 | 0.9995 | 4.7 | 1.58 ± 0.04 |
| Ethyl 2-methylbutyrate | Aldrich 306886 | 99 | 2.48 to 248 | 0.9997 | 1.8 | 0.87 ± 0.02 |
| Ethyl 3-methylbutyrate | Aldrich 112283 | 98 | 3.12 to 312 | 0.9993 | 2.2 | 0.91 ± 0.03 |
| 3-methyl-1-butyl acetate | Aldrich 306967 | ≥ 99 | 21.32 to 2132 | 0.9990 | 3.9 | 2.00 ± 0.07 |
| Ethyl hexanoate | Aldrich 148962 | ≥ 99 | 9.64 to 964 | 0.9978 | 2.2 | 1.32 ± 0.07 |
| Hexyl acetate | Aldrich 108154 | 99 | 2.76 to 276 | 0.9983 | 2.9 | 1.57 ± 0.08 |
| 3-methyl-1-pentanol | Aldrich 111112 | 99 | 25.6 to 256 | 0.9968 | 14.2 | 4.63 ± 0.30 |
| Ethyl lactate | Aldrich E34102 | 98 | 113.2 to 1132 | 0.9978 | 107.4 | 44.90 ± 2.45 |
| 1-hexanol | Fluka 73117 | > 99.9 | 14.72 to 1472 | 0.9976 | 6.7 | 3.63 ± 0.20 |
| E-3-hexen-1-ol | Aldrich 224715 | 97 | 6.32 to 632 | 0.9971 | 5.1 | 5.11 ± 0.32 |
| Z-3-hexen-1-ol | Fluka 53056 | ≥ 98 | 7.20 to 720 | 0.9968 | 5.9 | 5.23 ± 0.34 |
| Linalool | Aldrich L2602 | 97 | 4.76 to 476 | 0.9998 | 3.2 | 1.71 ± 0.03 |
| Diethyl succinate | Aldrich 112402 | 99 | 6.12 to 612 | 0.9977 | 2.4 | 1.25 ± 0.07 |
| α-terpineol | Merck 8.21078 | ≥ 98 | 2.60 to 260 | 0.9979 | 2.6 | 1.37 ± 0.07 |
| Citronellol | Aldrich C83201 | 95 | 2.72 to 272 | 0.9999 | 2.2 | 1.43 ± 0.02 |
| Nerol | Aldrich 268909 | 97 | 3.04 to 304 | 0.9988 | 3.1 | 1.83 ± 0.07 |
| 2-phenylethyl acetate | Fluka 46030 | > 99 | 10.32 to 1032 | 0.9995 | 2.6 | 1.39 ± 0.03 |
| Geraniol | Aldrich 163333 | 98 | 3.08 to 308 | 0.9994 | 2.4 | 1.26 ± 0.04 |
| Guaiacol | Aldrich G10903 | 98 | 2.92 to 292 | 0.9984 | 5.1 | 2.65 ± 0.12 |
| 4-ethylphenol | Aldrich E44205 | 99 | 4.88 to 488 | 0.9983 | 4.2 | 2.03 ± 0.10 |
| Compound | Repeatability | Intermediate Precision | Accuracy | Robustness | ||
|---|---|---|---|---|---|---|
| RSD/% | RSD/% | RE/% | Rec/% (t = 30 min) | Rec/% (Synthetic Wine) | Rec/% (Synthetic Vinegar) | |
| 4-methyl-2-pentanone | 6.5 | 9.3 | 11.2 | 103.2 | 100.6 | 103.1 |
| Ethyl butyrate | 5.3 | 7.4 | 10.8 | 99.9 | 91.0 | 95.2 |
| Ethyl 2-methylbutyrate | 9.0 | 9.3 | 13.2 | 96.6 | 86.0 | 83.0 |
| Ethyl 3-methylbutyrate | 5.3 | 6.7 | 20.5 | 98.0 | 88.8 | 91.8 |
| 3-methyl-1-butyl acetate | 4.5 | 5.7 | 1.6 | 104.7 | 93.9 | 96.2 |
| Ethyl hexanoate | 3.3 | 6.0 | 2.9 | 100.6 | 101.9 | 100.3 |
| Hexyl acetate | 3.8 | 11.2 | 15.5 | 98.0 | 92.3 | 97.8 |
| 3-methyl-1-pentanol | 6.3 | 18.0 | 2.3 | 96.1 | 148.6 | 113.6 |
| Ethyl lactate | 8.4 | 18.9 | 2.8 | 88.5 | 161.6 | 106.7 |
| 1-hexanol | 5.0 | 12.5 | 15.8 | 91.3 | 115.3 | 95.9 |
| E-3-hexen-1-ol | 7.0 | 18.0 | 14.2 | 86.0 | 116.3 | 96.2 |
| Z-3-hexen-1-ol | 6.8 | 19.7 | 15.9 | 85.8 | 116.4 | 90.5 |
| Linalool | 4.0 | 10.8 | 9.5 | 103.8 | 98.6 | 93.4 |
| Diethyl succinate | 3.3 | 10.3 | 13.3 | 113.0 | 117.7 | 113.4 |
| α-terpineol | 4.9 | 10.2 | 10.8 | 109.7 | 112.7 | 105.6 |
| Citronellol | 4.4 | 12.5 | 6.6 | 87.8 | 90.6 | 86.1 |
| Nerol | 5.9 | 13.8 | 13.7 | 108.6 | 100.0 | 93.2 |
| 2-phenylethyl acetate | 3.4 | 7.9 | 0.7 | 108.6 | 109.6 | 109.9 |
| Geraniol | 2.6 | 9.6 | 16.7 | 107.2 | 97.3 | 93.9 |
| Guaiacol | 6.3 | 14.0 | 1.6 | 97.3 | 120.5 | 105.5 |
| 4-ethylphenol | 4.5 | 9.9 | 5.0 | 72.9 | 112.5 | 97.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, E.; Lemos, M.; Genisheva, Z.; Domingues, L.; Vilanova, M.; Oliveira, J.M. Validation of a LLME/GC-MS Methodology for Quantification of Volatile Compounds in Fermented Beverages. Molecules 2020, 25, 621. https://doi.org/10.3390/molecules25030621
Coelho E, Lemos M, Genisheva Z, Domingues L, Vilanova M, Oliveira JM. Validation of a LLME/GC-MS Methodology for Quantification of Volatile Compounds in Fermented Beverages. Molecules. 2020; 25(3):621. https://doi.org/10.3390/molecules25030621
Chicago/Turabian StyleCoelho, Eduardo, Margarida Lemos, Zlatina Genisheva, Lucília Domingues, Mar Vilanova, and José M. Oliveira. 2020. "Validation of a LLME/GC-MS Methodology for Quantification of Volatile Compounds in Fermented Beverages" Molecules 25, no. 3: 621. https://doi.org/10.3390/molecules25030621
APA StyleCoelho, E., Lemos, M., Genisheva, Z., Domingues, L., Vilanova, M., & Oliveira, J. M. (2020). Validation of a LLME/GC-MS Methodology for Quantification of Volatile Compounds in Fermented Beverages. Molecules, 25(3), 621. https://doi.org/10.3390/molecules25030621