1. Introduction
Lewis acid–Lewis base interactions continue to be a heavily explored area of modern chemistry. The wide diversity of Lewis acids and bases involves them in numerous different chemical and biological processes [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12]. Within this general topic, an enormous amount of research currently centers on the topic of σ-hole/π-hole interactions [
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24]. One aspect which makes these noncovalent interactions both surprising and unique is the attractive force between electronegative atoms, which simple chemical intuition would have guessed would be repulsive. The resolution of this paradox, initially explained for halogen atom–nucleophile contacts [
25,
26,
27,
28] is the anisotropy of the electron density around the halogen X atom. This density is thinned in the vicinity of the outer lobe of the
p orbital engaged in the R-X covalent bond, which is commonly denoted as a σ-hole. There are also planar molecules in which an electron deficiency occurs above the plane, referred to as a π-hole [
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42]. Following the initial introduction of the σ-hole concept to rationalize the halogen bond, [
43,
44] the same idea has been expanded to various other groups of the periodic table [
45], now denoted as chalcogen, pnicogen, tetrel, triel, and even aerogen bonds [
46,
47,
48,
49,
50,
51,
52,
53,
54,
55,
56].
As triel (Tr) atoms (B, Al, etc) typically find themselves in planar TrR
3 molecular arrangements, a π-hole can be found above and below this atom, which facilitates the formation of a triel bond (TrB) with an approaching nucleophile. There are also exceptional cases wherein a tetravalent Tr atom can generate a σ-hole [
57]. This TrB has generated sufficient interest so as to be the focus of a number of prior quantum chemical studies [
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73]. Our own earlier study of the TrB [
73] in complexes of TrR
3 (Tr = B, Al, Ga; R = H, F, Cl, Br, CH
3) with pyrazine provides some information about the influence of various substituents on the energy, geometry, and properties of this interaction. Complexation occurred in one of two ways, either through a Tr π-hole to the N-lone pair of pyrazine or via a stacked arrangement. The former was many times stronger than the latter. The two geometries also differed significantly in their fundamental nature: the stronger π-hole complex relied primarily on Coulombic forces and orbital interactions, whereas dispersion was the chief contributor to the stacked dimers. Despite their reliance on electrostatic attraction, the binding energies of the π-hole complexes did not correlate well with the intensity of this hole. A similar contradiction was noted also by Xu and Li [
67], in their calculations of RTrH
2···NH
3. These authors explained these inconsistencies in terms of the crucial role of orbital interaction and polarization energy.
Recently, the prevalence of the triel bond has been expanded [
61] to encompass carbenes and silylenes as electron donors to TrR
3. The interaction energies of these complexes were surprisingly large, reaching 90 kcal/mol. There were also quite substantial geometric deformations in the monomers caused by the interaction, which lowered the binding energies, which appears to be a common feature of TrBs. Consistant with the aforementioned findings of a poor relationship between binding energy and π-hole intensity, there was little correlation observed here between the electron-withdrawing power of the substituent attached to the Lewis acid and the binding energy. The cooperativity involved in TrBs has been explored, combined with the halogen bond [
74], anion–π interactions [
58] or the regium bond [
68].
Not all noncovalent interactions involve separate molecules. Just as in the case of H-bonds, intramolecular interactions are important in terms of establishing the structure and function of essential molecules such as proteins [
75,
76]. These internal noncovalent bonds are also involved in supramolecular recognition [
77,
78]. There has been a respectable amount of study of intramolecular contacts steered by pnicogen [
79], tetrel [
80,
81,
82] or chalcogen bonds [
78,
83,
84]. In contrast, however, there is far less information available regarding the intramolecular triel bond (IMTrB). As one of only a few examples, Pla et al. [
85] examined a series of naphthyl-bridged amino-borane derivatives and concluded that the naphthyl scaffold exhibits flexibility as a response to B···N interactions. Their NBO analysis revealed a B···N dative bond between an N lone pair and a vacant virtual B orbital. Very recently, the bifurcated TrB in hydrides, fluorides, and chlorides of 1,8-bis(dichloroboryl)naphthalene and 1,2-bis(dichloroboryl)benzene was examined by Grabowski [
64]. The anionic structures linked by a BXB bridge (X = H, F, Cl) were characterized as partly covalent. A CSD survey confirmed the presence of similar crystal structures with not only boron but other triel atoms: Al and Ga.
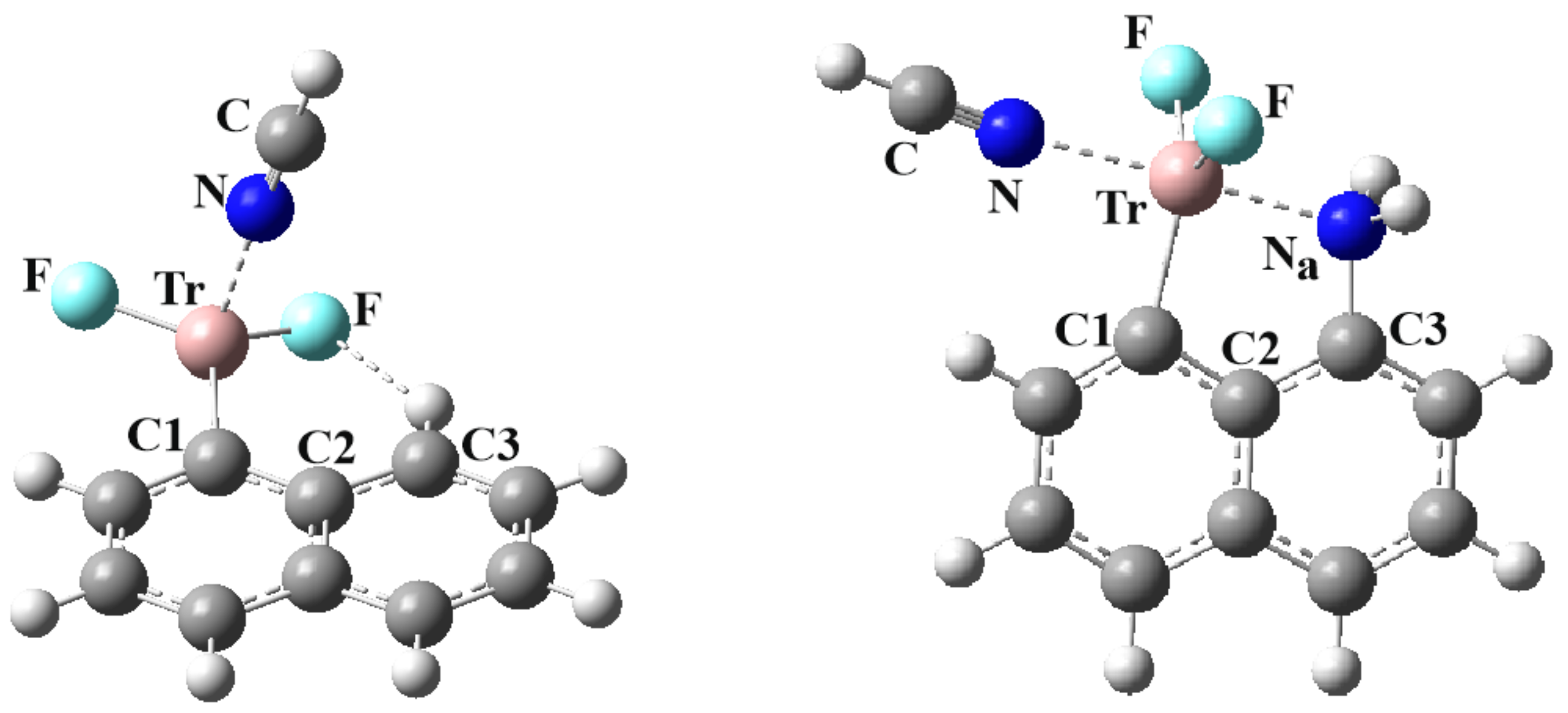
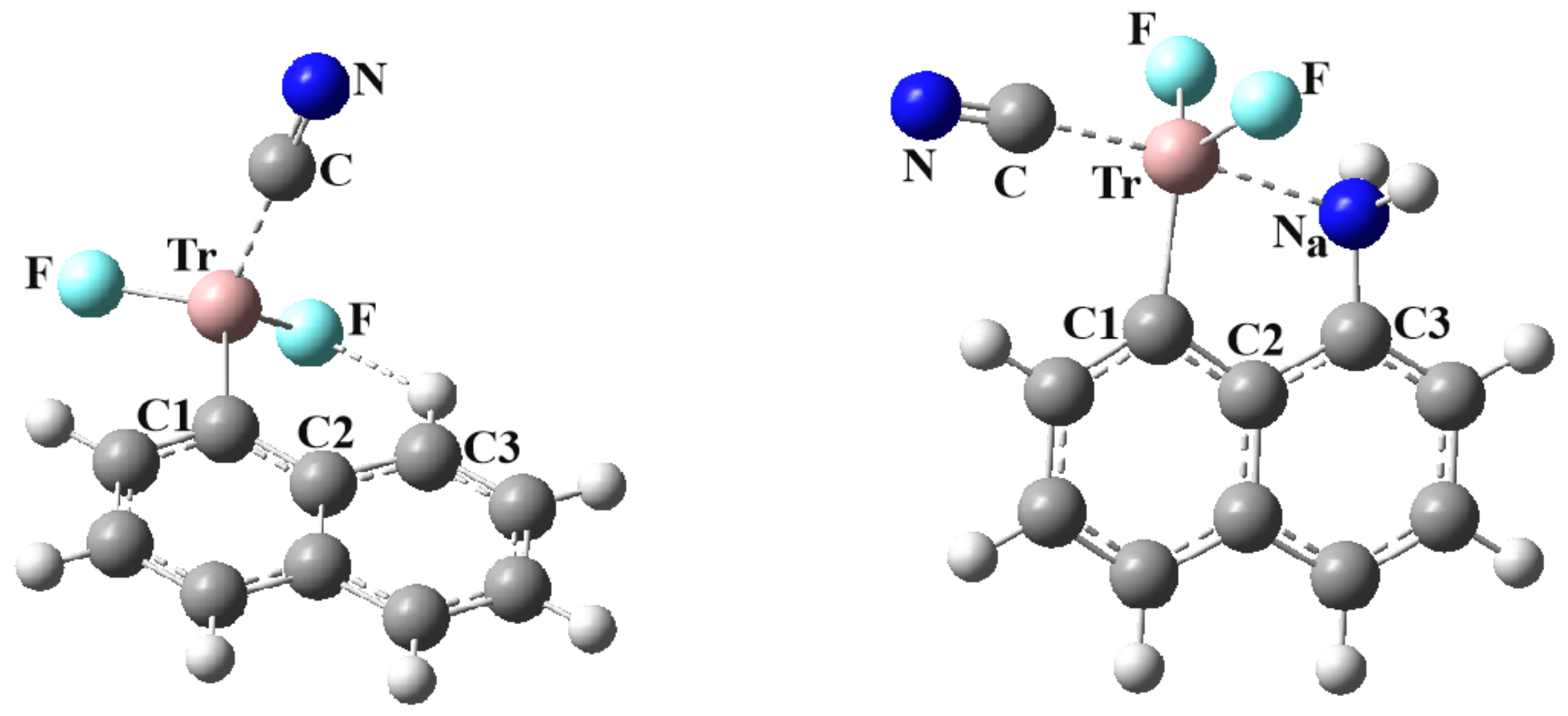
The current work examines the issue of both inter and intramolecular TrBs, and the competition between them. The naphthalene system offers a convenient and well-structured skeleton on which to base this work. A TrF2 group is placed on one of the α-positions. On the neighboring Cα of the other ring is positioned a NH2 nucleophile. These two groups are thus well oriented to engage in an internal Tr···B interaction. Due to its geometry, the TrF2 group ought to have a second π-hole that can engage in a second TrB with an approaching nucleophile. The central question is how these two TrBs, one internal and the other external, affect one another. Does the presence of one inhibit the formation of the second, or might the two reinforce one another? Does the intermolecular TrB, free to adopt its optimal geometry with no internal structural restraints, cause the internal TrB to break? How do the two TrBs, either separately or cumulatively, affect the geometry of the TrF2 unit, and how does its deformation play into the properties of the two TrBs? In order to address this problem in a broad sense, the Tr atom was varied, from the smallest B, all the way up to Tl. In terms of the approaching external nucleophile, NCH was considered as a weak base with sp hybridation of the N. On the other end of the spectrum, the CN− anion, with its full negative charge, ought to represent a very strong base.
3. Discussion
The Cambridge Structural Database (CSD) [
86] provides some experimental context for the bonding schemes studied here. First is the question of systems containing an internal triel bond of the sort depicted in
Figure 1. Taking the naphthalene unit as a building block, the CSD was searched for systems wherein a Tr atom was located on one C
α atom and an N on the neighboring C
α, as in
Figure 1. The definition of an internal triel bond is based on the R(Tr··N) distance being less than the sum of vdW radii of the Tr and N atoms. The minimum for this range of distances is an arbitrary one, but ought to avoid a purely covalent bond. Each row of
Table S4 refers to a particular percentage of the sum of covalent atomic radii, varying from 110% to 140%. It may be seen that the bulk of observations arise for Tr = B where there are between nine and 13 cases, depending upon the chosen minimum distance. There are fewer observations for the heavier Tr atoms, most of which are only slightly longer than the covalent bond length. This finding is consistent with the computed data in
Table 1 that suggest the internal B···N bond is the shortest and strongest of those considered, hovering around the range of a covalent bond.
Table 1 documents the manner in which the Tr and N atoms bend toward one another as a result of the internal triel bond. This sort of bending is plainly seen in a number of crystal structures, examples of which are provided in
Table S5. The θ(C2C1Tr) and θ(C2C3H) angles are 120°–125° and 120°, respectively, when the N atom is absent. But these angles are quite a bit smaller in those cases where a N substituent is present on the naphthalene system, particularly when the R(Tr···N) distance is short. It might be noted, finally, that the CSD provides some evidence in
Table S5 wherein the Tr atom participates not only in this internal Tr···N bond, but also in a second Tr bond of the sort depicted in
Figure 3 and
Figure 4. It is not only B for which this is true but also Al, In, and Tl.
The findings presented above can be placed in the context of several earlier sets of calculations. For example, an earlier study [
87] of an intramolecular B···N interaction in various naphthyl-bridged amino–borane compounds calculated NBO E(2) energies for LP(N)→LV(B) transfer that reached 164.2 kcal/mol, although this quantity is much smaller, in the range of 4–10 kcal/mol, for the other systems. The first large value is consistent with our own finding by NBO of a covalent B···N bond, with LP(N)→LP*(Tr) values between 39 and 97 kcal/mol.
Our own earlier study of a triel bond pairing TrR
3 with pyrazine [
73] applied the same level of theory as here, but involved a different base. In order to facilitate some comparison, it could be considered that the GaH
3 molecule has a π-hole very similar in magnitude to that of C
10H
7GaF
2. Conflating the data for the different bases, the order of interaction energy diminishes as CN
− > pyrazine > NCH. As in the current work, the earlier calculations also indicated a non-negligible role of deformation energy, which was largest for B. In keeping with its placement between NCH and CN
− in terms of binding energy, pyrazine also is associated with deformation energies between these two extremes. Energy decompositions are also similar for the various bases, in that the contribution of electrostatic and orbital contributions are nearly equal for B, while the percentage contribution of electrostatics grows along with a decrease in the orbital interaction component for the other Tr atoms.
Grabowski’s [
64] systems encompassed dichloroboryl derivatives of naphthalene and benzene, wherein the Tr atoms situated on the same molecule were linked by H, F or Cl anions, somewhat akin to our own CN
− nucleophile. As such, each anion was held by what might be considered a pair of Tr bonds to B, which might explain the large interaction energies between 104 and 161 kcal/mol. But nonetheless, these systems also showed a large influence of deformation energies, as high as 50 kcal/mol. Despite some difference in structural form, these systems displayed a pattern of energy decomposition components similar to those observed here. It might also be noted that the AIM BCP densities for some of the stronger complexes exceeded 0.1 au, suggesting covalency. Similarly, large ρ
BCP values were observed here for complexes of the CN
− anion with B-derivatives. Earlier calculations [
86] verify the ability of a Tr atom to engage in a pair of triel bonds simultaneously, one on each side of a planar TrF
3 molecule, and echo the weakening the second bond causes to the first, as was observed here.
4. Computational Methods
The geometries of the naphthalene derivatives C
10H
7TrF
2 and C
10H
6NH
2TrF
2 (Tr = B, Al, Ga, In, Tl) and their complexes with HCN and CN
− were all optimized at the MP2 level of theory in combination with the aug-cc-pVDZ basis set [
88,
89]. For the purpose of including relativistic effects for heavy In and Tl atoms, aug-cc-pVDZ-PP pseudopotentials were incorporated [
90,
91,
92,
93,
94,
95]. Frequency calculations were carried out at the same level to verify that the obtained geometries are true minima with no imaginary frequencies. The energies were recalculated at the CCSD(T)/aug-cc-pVDZ level (making use of MP2 geometries) to verify their accuracy [
96,
97,
98]. The interaction energy (E
int) is defined here as the difference between the energy of the complex and the sum of monomers, with the latter in the geometry they adopt within the complex. The binding energy (E
b) takes as its reference the optimized pre-deformed isolated monomers. Thus, the difference between interaction and binding energy is the deformation energy E
def. All energies were corrected for basis set superposition error (BSSE) via the counterpoise protocol [
99]. Computations were performed using the Gaussian 16 suite of codes [
100].
The molecular electrostatic potentials of isolated monomers were analyzed through the MultiWFN and WFA-SAS programs in order to obtain V
s,max and V
s,min values as well as electrostatic potential maps on the 0.001 au electronic isodensity surface [
101,
102,
103,
104]. The quantum theory of atoms in molecules (QTAIM) was employed to locate and characterize bond critical points (BCPs) in interacting systems using AIMAll software [
105]. The NBO method was applied (GenNBO 6.0 program) to compute the orbital–orbital interactions using the wavefunction generated at the DFT level for the MP2 optimized geometries [
106,
107,
108,
109]. The decomposition of interaction energies was applied to partition E
int into the following components: electrostatic, orbital interaction, dispersion and repulsive forces. The EDA scheme embedded in the ADF software at the BLYP/ZORA/TZ2P level was used for this purpose [
110,
111,
112]. Finally, the CSD survey [
86] with specified bond distances criteria was performed using the ConQuest program [
113] so as to identify experimental crystal structures with certain bonding patterns related to those found via calculations.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}