Discovery of Novel Inhibitors Targeting Multi-UDP-hexose Pyrophosphorylases as Anticancer Agents

 and
and
Abstract
:1. Introduction
2. Results
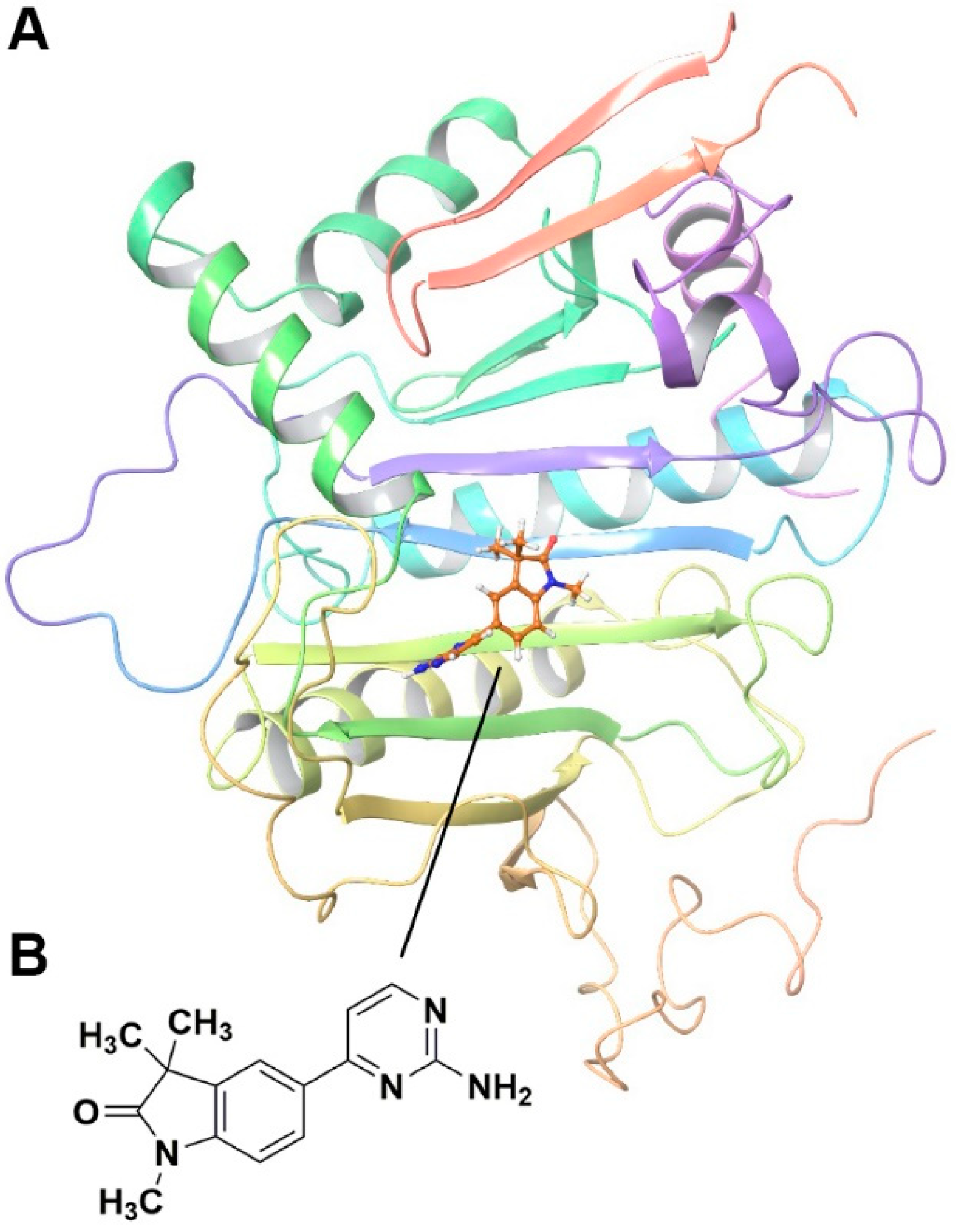
2.1. Fragment-Based Screening for GALT Inhibitors
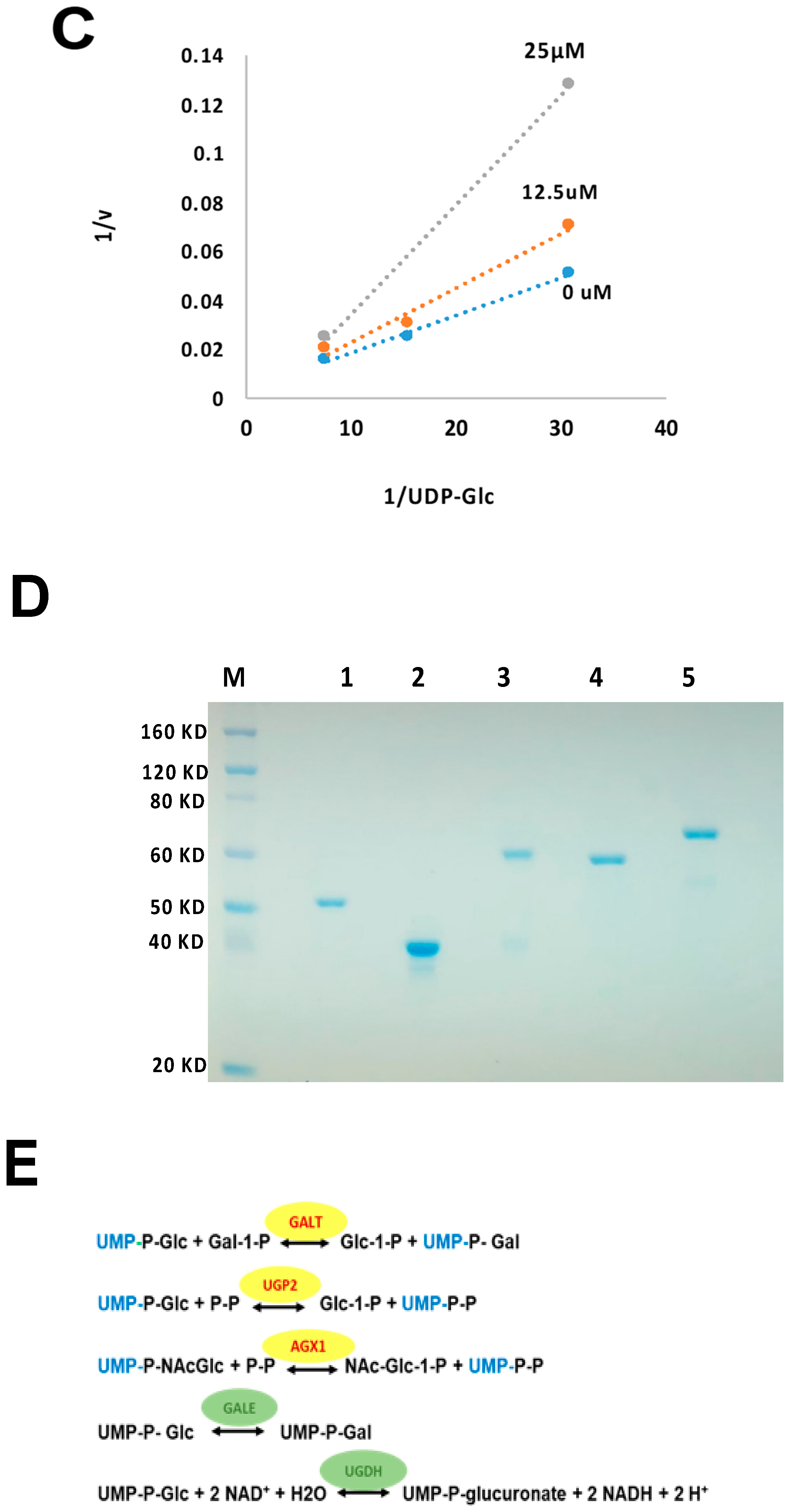
2.2. Fragment GAL-012 Is a Competitive Inhibitor of Recombinant GALT
2.3. Fragment GAL-012 Is a Novel Inhibitor of Multi-UDP-hexose Pyrophosphorylases
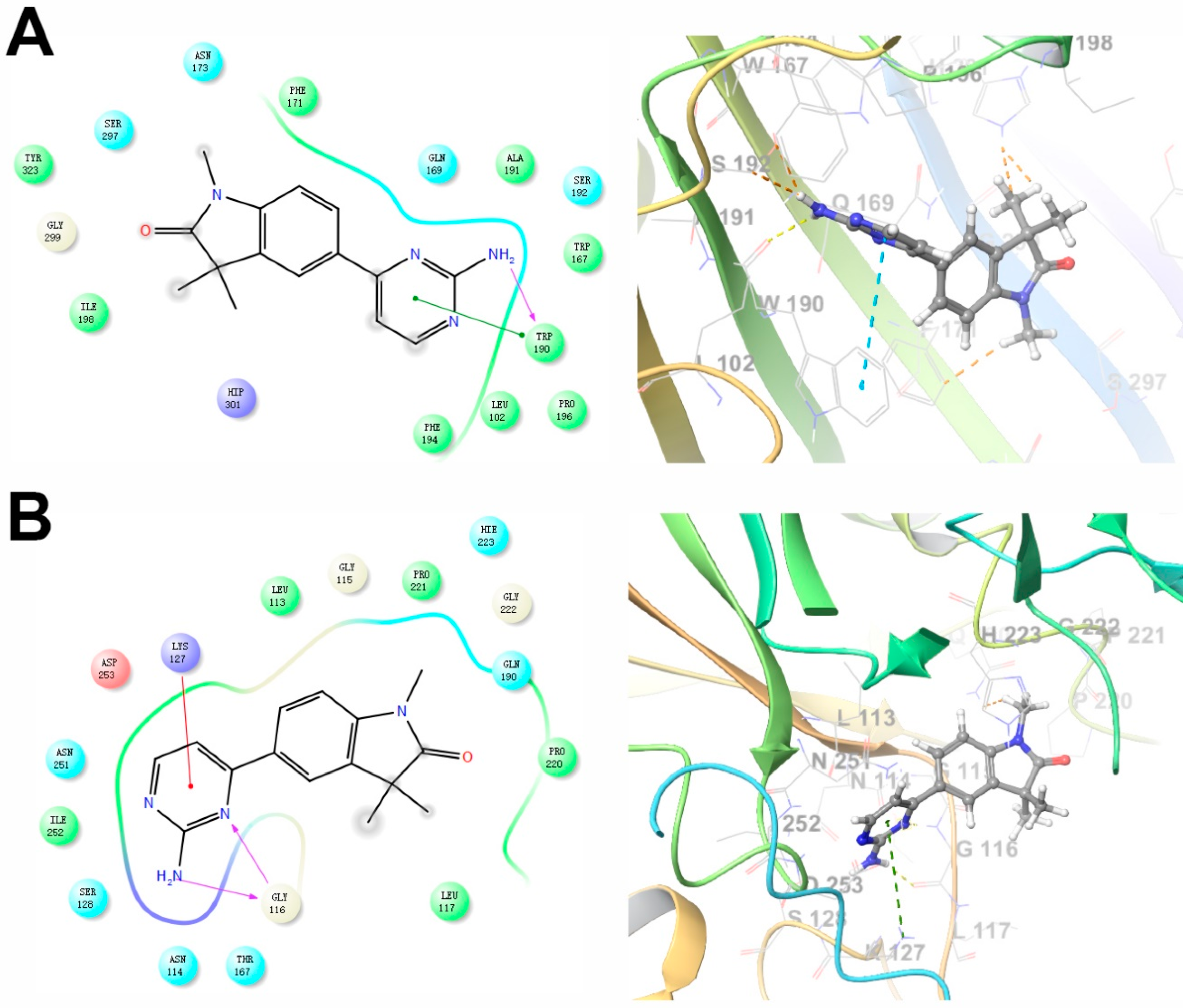
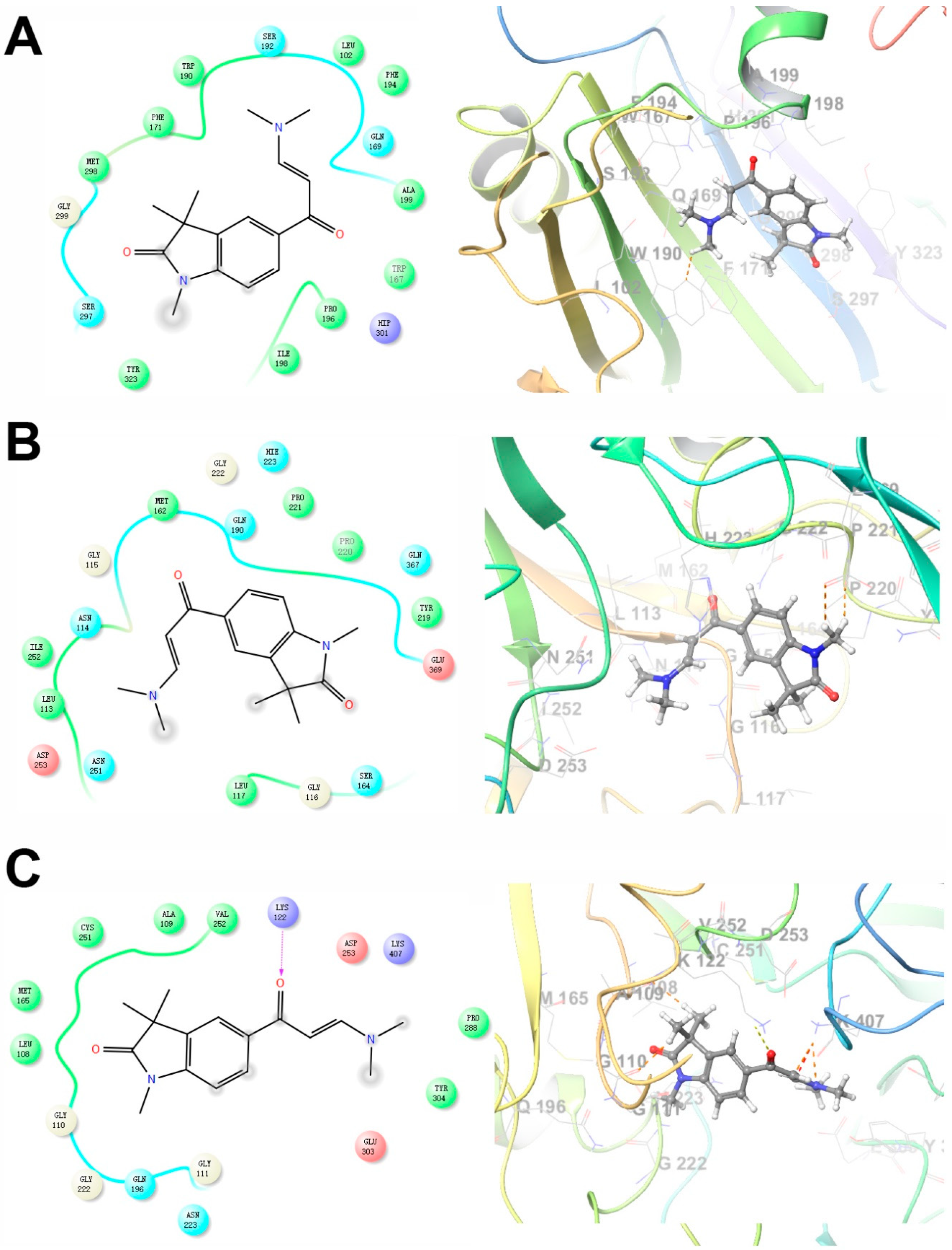
2.4. Predicted Molecular Interactions between GAL-012 and the Respective UDP-hexose Pyrophosphorylases
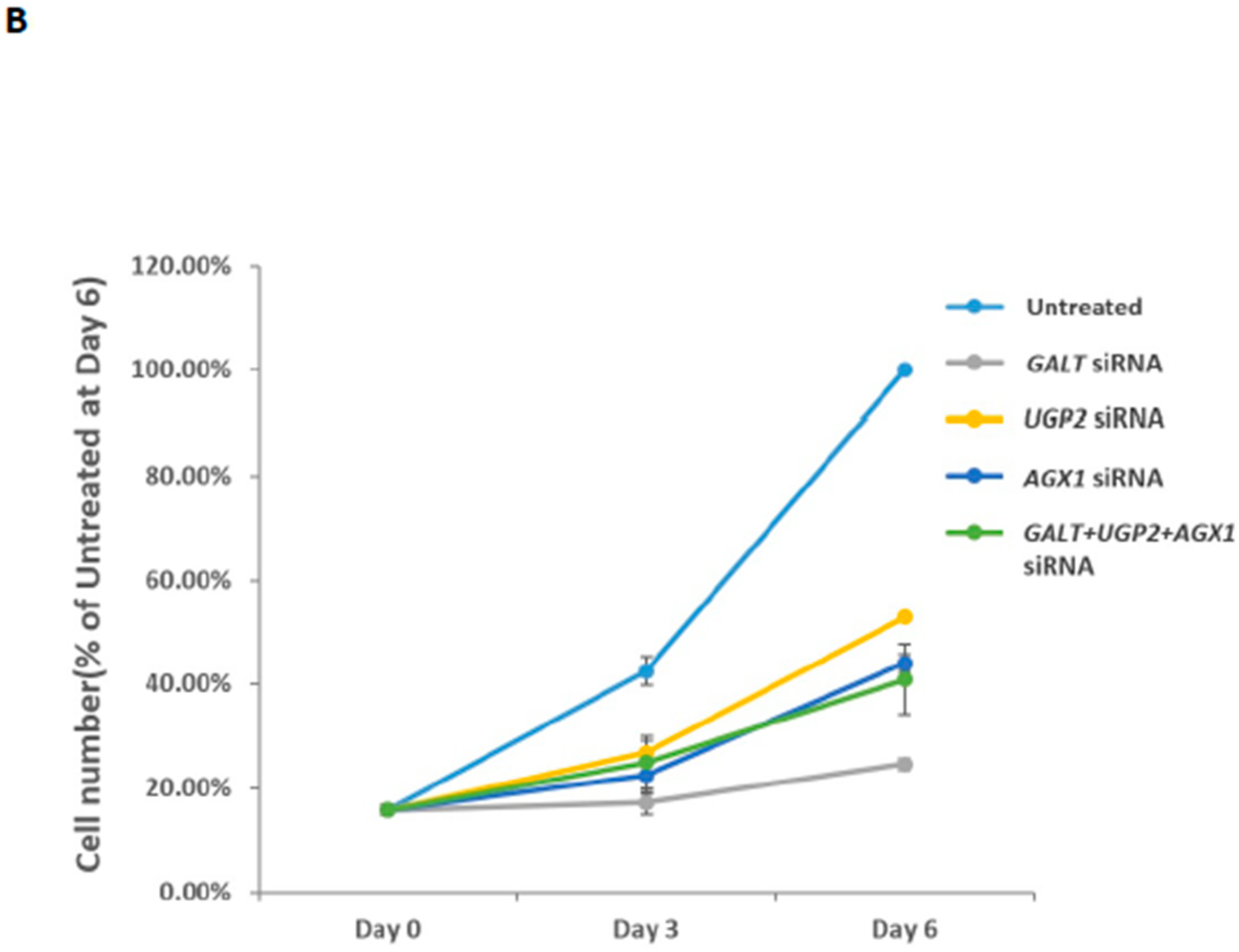
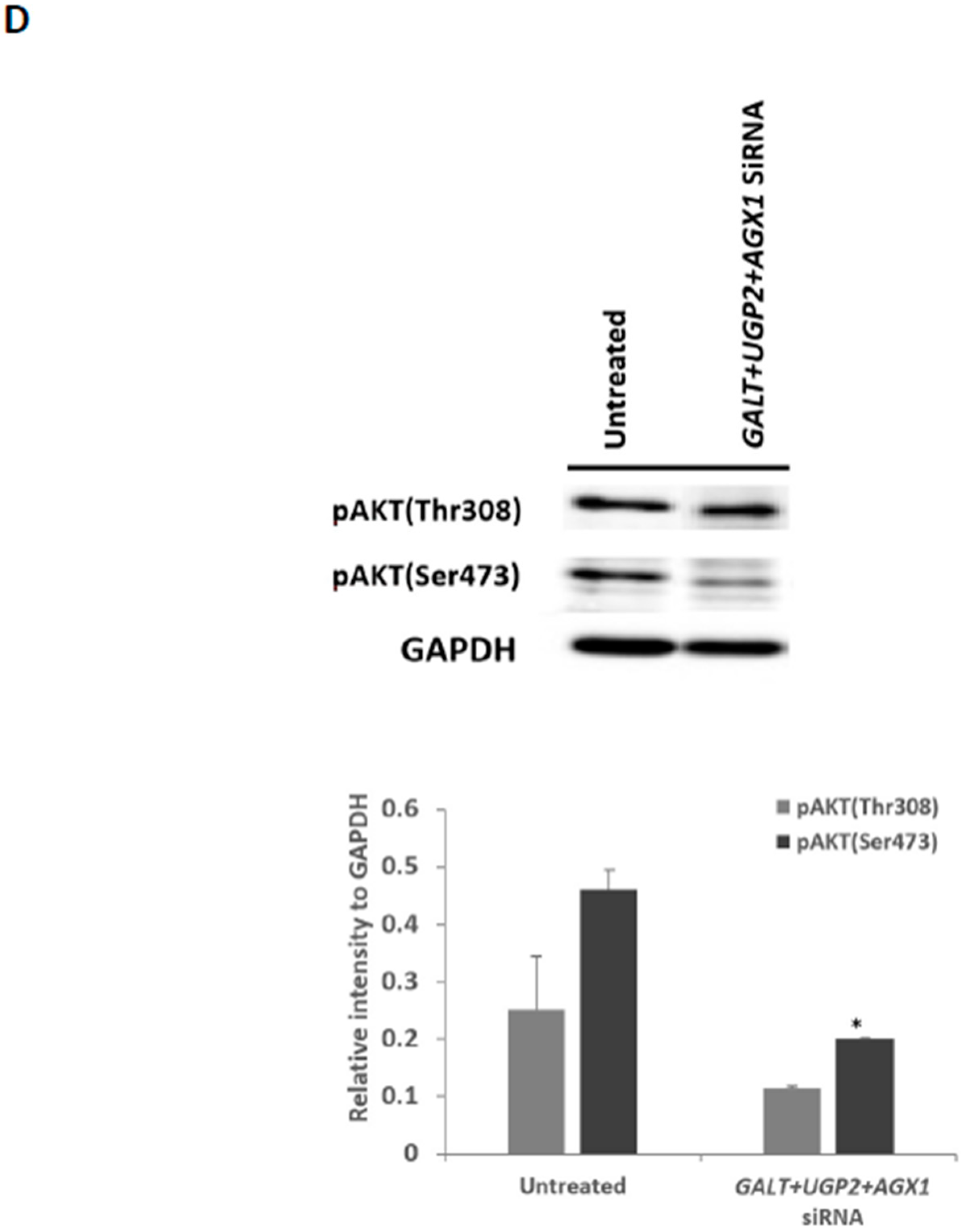
2.5. Effects of UGP2 and AGX1/UAP1 Gene Knockdown in Cultured Cancer Cells
2.6. GAL-012 Derivative GAL-012-2 Inhibits GALT and UGP2
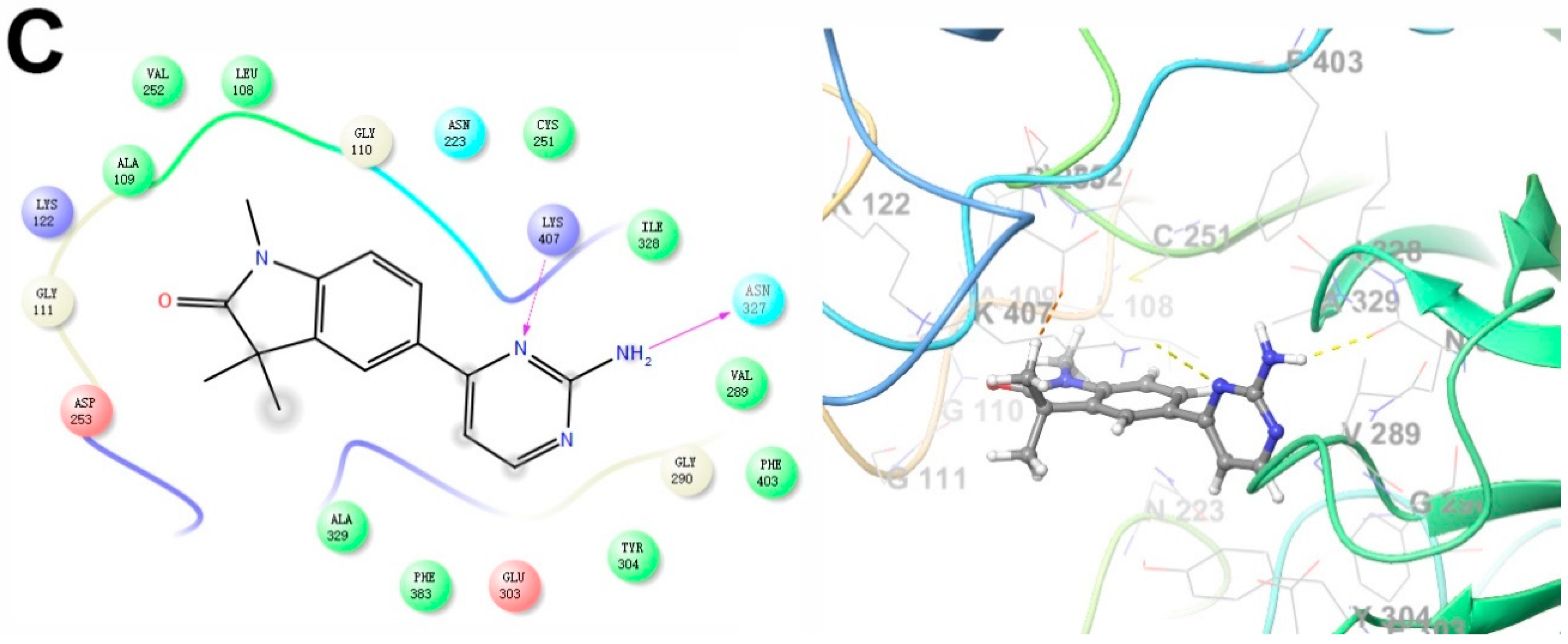
2.7. Docking Study of GAL-012-2 to GALT, UGP2 and AGX1/UAP1
2.8. Preliminary SAR of GAL-012 Series of Compounds
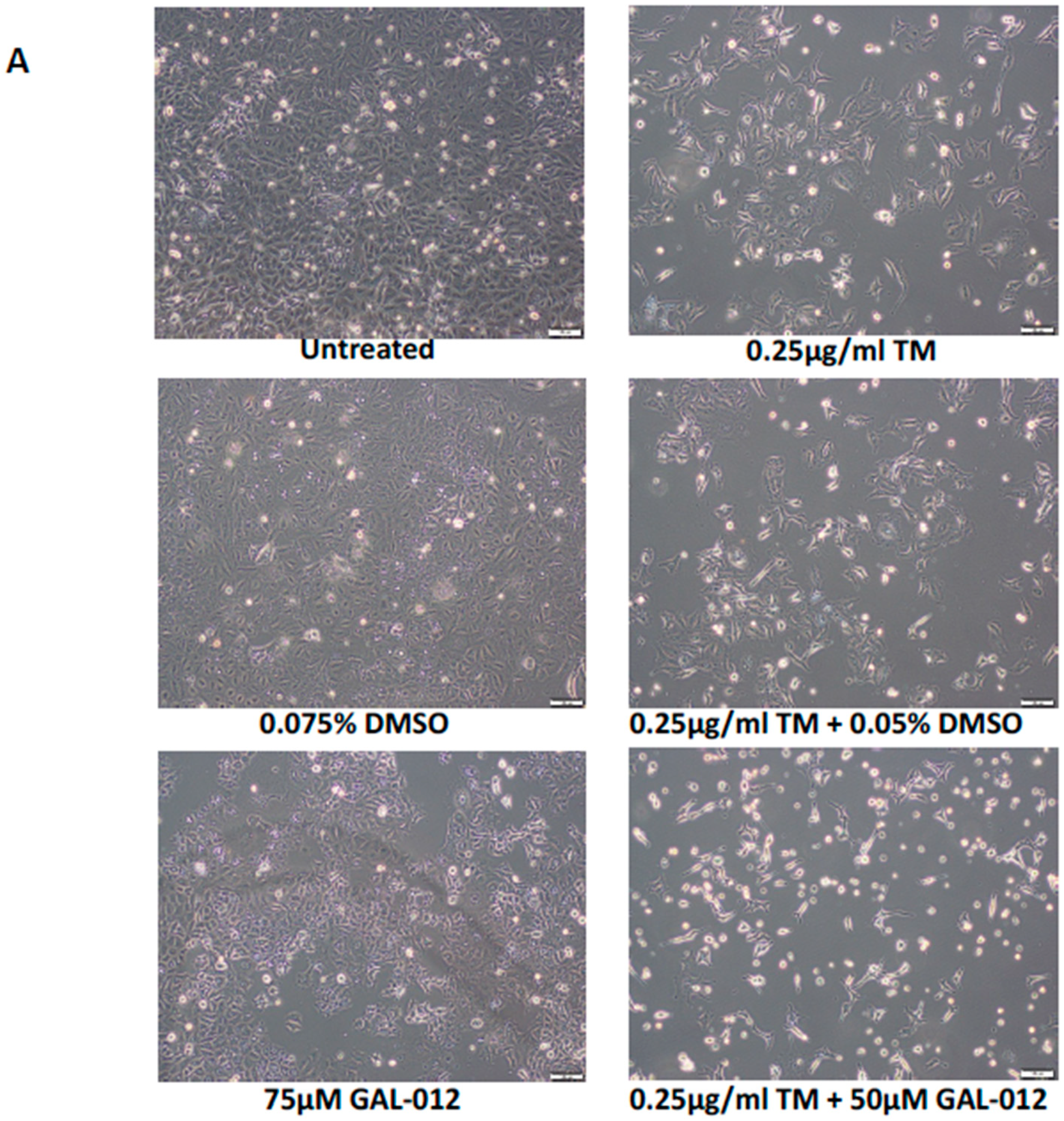
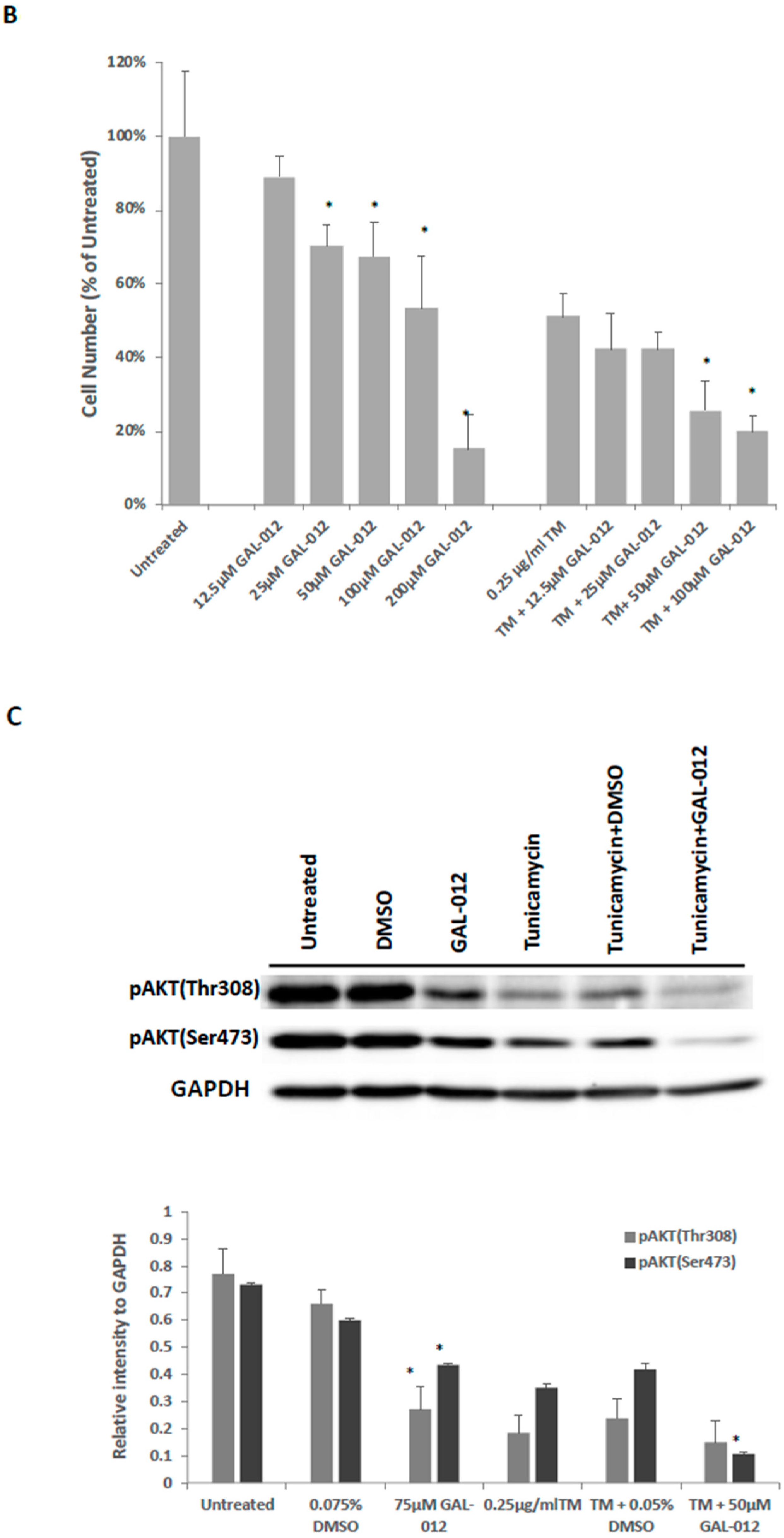
2.9. Fragment GAL-012 Dose Dependently Inhibits PC3 Cell Growth
2.10. GAL-012 Increased Sensitivity to Tunicamycin in PC3 Cells
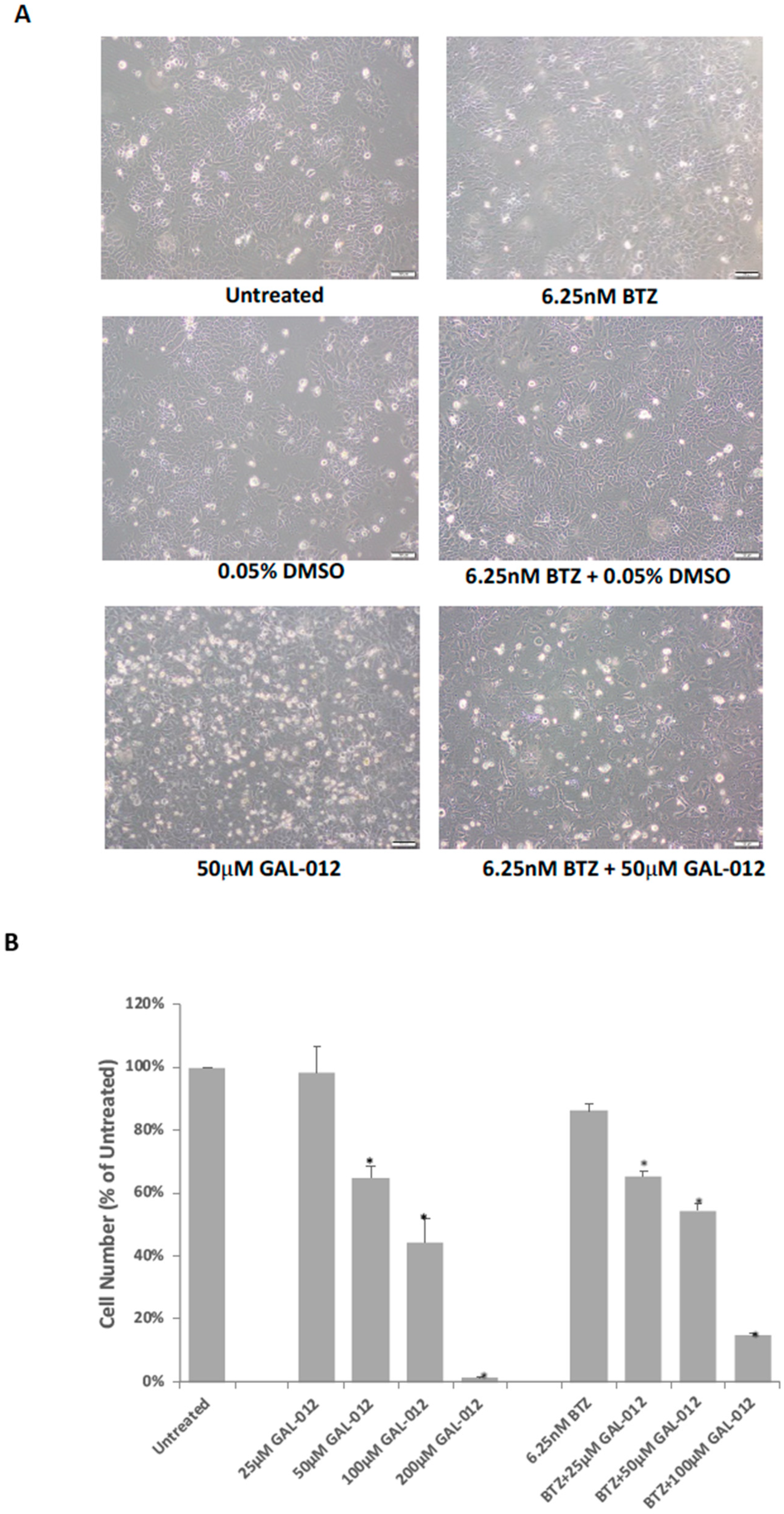
2.11. GAL-012 Increased Sensitivity to Bortezomib (BTZ) in HepG2 Cells
3. Discussion
4. Methods
4.1. In Silico Fragment-Based Screening for GALT Inhibitors
4.2. Expression Cloning of Human UDP-hexose Binding Enzymes: GALT, UDP-galactose 4′ Epimerase (GALE), UDP-Glc Dehydrogenase (UGDH), UGP2, and AGX1/UAP1
4.3. Over-Expression, Affinity Purification, and Quantification of Recombinant GALT, GALE, UGDH, UGP2, and AGX1/UAP1 Enzymes
4.4. Enzyme Activity Assays for Recombinant Human GALT, GALE, UGDH, UGP2 and AGX1/UAP1 Enzymes
4.4.1. GALT, UGP2, AGX1/UAP1 Enzymatic Activities Measurements
4.4.2. GALE Enzyme Activity Measurements
4.4.3. UGDH Enzymatic Activity Measurements
4.5. Inhibition Kinetics of Fragment GAL-012 and Derivatives
4.6. Molecular Docking Experiments of GAL-012 and GAL-012-2 to GALT, UGP2 and AGX1/UAP1
4.7. Cell-Based Studies of siRNA-Treated Culture Cells
4.7.1. siRNA Transfection
4.7.2. RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.7.3. Cell Viability Assay
4.7.4. PI3K/AKT Signaling Pathway in siRNA-Treated Cultured Cells
4.8. Cell-Based Characterization of GAL-012-Treated Cultured Cells
4.8.1. Gross Examination of Growth Inhibition
4.8.2. PI3K/AKT Signaling Pathway in GAL-012-Treated Cultured Cells
4.9. Combined GAL-012 and Tunicamycin Treatment of PC3 Cells
4.10. Combined GAL-012 and Bortezomib (BTZ) Treatment HepG2 Cells
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Ward, E.M.; Jemal, A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J. Clin. 2017, 67, 273–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, D.; Chakedis, J.; Sun, S.H.; Spolverato, G.; Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Spartalis, E.; Pawlik, T.M. Management, outcomes, and prognostic factors of ruptured hepatocellular carcinoma: A systematic review. J. Surg. Oncol. 2018, 117, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhu, J.; Lubman, D.M.; Gao, C. Aberrant glycosylation and cancer biomarker discovery: A promising and thorny journey. Clin. Chem. Lab. Med. 2019, 57, 407–416. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Josic, D.; Martinovic, T.; Pavelic, K. Glycosylation and metastases. Electrophoresis 2019, 40, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Very, N.; Lefebvre, T.; El Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2018, 9, 1380–1402. [Google Scholar] [CrossRef] [Green Version]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef]
- Lynch, T.P.; Reginato, M.J. O-GlcNAc transferase: A sweet new cancer target. Cell Cycle 2011, 10, 1712–1713. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.B.; Hou, J.; Ratnam Bandaru, V.V.; Pezhouh, M.K.; Syed Rifat Mannan, A.A.; Sharma, R. Lactosylceramide synthase beta-1,4-GalT-V: A novel target for the diagnosis and therapy of human colorectal cancer. Biochem. Biophys. Res. Commun. 2019, 508, 380–386. [Google Scholar] [CrossRef]
- Brockhausen, I.; Benn, M.; Bhat, S.; Marone, S.; Riley, J.G.; Montoya-Peleaz, P.; Vlahakis, J.Z.; Paulsen, H.; Schutzbach, J.S.; Szarek, W.A. UDP-Gal: GlcNAc-R beta1,4-galactosyltransferase—A target enzyme for drug design. Acceptor specificity and inhibition of the enzyme. Glycoconj. J. 2006, 23, 525–541. [Google Scholar] [CrossRef]
- Tang, M.; Etokidem, E.; Lai, K. The Leloir Pathway of Galactose Metabolism-A Novel Therapeutic Target for Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 6265–6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isselbacher, K.J.; Anderson, E.P.; Kurahashi, K.; Kalckar, H.M. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science 1956, 123, 635–636. [Google Scholar] [CrossRef] [PubMed]
- Maratha, A.; Stockmann, H.; Coss, K.P.; Gozalbo, M.E.R.; Knerr, I.; Fitzgibbon, M.; McVeigh, T.P.; Foley, P.; Moss, C.; Colhoun, H.O.; et al. Classical galactosemia: Novel insights in IgG N-glycosylation and N-glycan biosynthesis. Eur. J. Hum. Genet. 2016, 24, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Waisbren, S.E.; Potter, N.L.; Gordon, C.M.; Green, R.C.; Greenstein, P.; Gubbels, C.S.; Rubio-Gozalbo, E.; Schomer, D.; Welt, C.; Anastasoaie, V.; et al. The adult galactosemic phenotype. J. Inherit. Metab. Dis. 2012, 35, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.; Langley, S.D.; Singh, R.H.; Dembure, P.P.; Hjelm, L.N.; Elsas, L.J. A prevalent mutation for galactosemia among black Americans. J. Pediatr. 1996, 128, 89–95. [Google Scholar] [CrossRef]
- Yuzyuk, T.; Balakrishnan, B.; Schwarz, E.L.; De Biase, I.; Hobert, J.; Longo, N.; Mao, R.; Lai, K.; Pasquali, M. Effect of genotype on galactose-1-phosphate in classic galactosemia patients. Mol. Genet. Metab. 2018, 125, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.; Engedal, N.; Babaie, E.; Luhr, M.; Guldvik, I.J.; Minner, S.; Hohloch, J.; Tsourlakis, M.C.; Schlomm, T.; Mills, I.G. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene 2015, 34, 3744–3750. [Google Scholar] [CrossRef]
- Yarema, K.I.; Saeui, C.T.; Quinones-Hinojosa, A.; Shan, S.R. Use of UAP Inhibitors to Inhibit Flux through the Hexosamine Biosynthetic Pathway. U.S. Patent 20180148468A1, 31 May 2018. [Google Scholar]
- McCorvie, T.J.; Kopec, J.; Pey, A.L.; Fitzpatrick, F.; Patel, D.; Chalk, R.; Shrestha, L.; Yue, W.W. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum. Mol. Genet. 2016, 25, 2234–2244. [Google Scholar] [CrossRef]
- Ye, J.; Yang, X.; Xu, M.; Chan, P.K.; Ma, C. Novel N-Substituted oseltamivir derivatives as potent influenza neuraminidase inhibitors: Design, synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2019, 182, 111635. [Google Scholar] [CrossRef] [PubMed]
- Surani, M.A. Glycoprotein synthesis and inhibition of glycosylation by tunicamycin in preimplantation mouse embryos: Compaction and trophoblast adhesion. Cell 1979, 18, 217–227. [Google Scholar] [PubMed]
- Leavitt, R.; Schlesinger, S.; Kornfeld, S. Tunicamycin inhibits glycosylation and multiplication of Sindbis and vesicular stomatitis viruses. J. Virol. 1977, 21, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities (review). Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.P.; Mahoney, M.R.; Szydlo, D.; Mok, T.S.; Marshke, R.; Holen, K.; Picus, J.; Boyer, M.; Pitot, H.C.; Rubin, J.; et al. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma. Investig. New Drugs 2012, 30, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Lafaro, K.J.; Demirjian, A.N.; Pawlik, T.M. Epidemiology of hepatocellular carcinoma. Surg. Oncol. Clin. N. Am. 2015, 24, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyronnet, B.; Brucker, B.M. Management of Overactive Bladder Symptoms after Radical Prostatectomy. Curr. Urol. Rep. 2018, 19, 95. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Baquero, R.; Fernandez-Avila, C.M.; Alvarez-Ossorio, J.L. Functional results in the treatment of localized prostate cancer. An updated literature review. Rev. Int. Androl. 2018, 17, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Gaither, T.W.; Awad, M.A.; Osterberg, E.C.; Murphy, G.P.; Allen, I.E.; Chang, A.; Rosen, R.C.; Breyer, B.N. The Natural History of Erectile Dysfunction after Prostatic Radiotherapy: A Systematic Review and Meta-Analysis. J. Sex. Med. 2017, 14, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, J.J.; Yin, P.H.; Xu, K.; Wang, Y.Z.; Shi, F.; Gao, J.; Fu, X.L. Strategies to target energy metabolism in consensus molecular subtype 3 along with Kirsten rat sarcoma viral oncogene homolog mutations for colorectal cancer therapy. J. Cell Physiol. 2019, 234, 5601–5612. [Google Scholar] [CrossRef]
- Pope, E.D., III; Kimbrough, E.O.; Vemireddy, L.P.; Surapaneni, P.K.; Copland, J.A., III; Mody, K. Aberrant lipid metabolism as a therapeutic target in liver cancer. Expert Opin. Ther. Targets 2019, 23, 473–483. [Google Scholar] [CrossRef]
- Ocana, M.C.; Martinez-Poveda, B.; Quesada, A.R.; Medina, M.A. Metabolism within the tumor microenvironment and its implication on cancer progression: An ongoing therapeutic target. Med. Res. Rev. 2019, 39, 70–113. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Ko, H.; Yun, M. Cancer Metabolism as a Mechanism of Treatment Resistance and Potential Therapeutic Target in Hepatocellular Carcinoma. Yonsei. Med. J. 2018, 59, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.S.; Reith, M.E.; Dutta, A.K. Three-dimensional quantitative structure-activity relationship (3D QSAR) and pharmacophore elucidation of tetrahydropyran derivatives as serotonin and norepinephrine transporter inhibitors. J. Comput. Aided Mol. Des. 2008, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiong, L.; Wu, Z.; Miao, X.; Liu, Z.; Li, D.; Zou, Q.; Yang, Z. Expression of UGP2 and CFL1 expression levels in benign and malignant pancreatic lesions and their clinicopathological significance. World J. Surg. Oncol. 2018, 16, 11. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.S.; Lim, K.H.; Tan, H.T.; Khoo, M.L.; Tan, S.H.; Toh, H.C.; Ching Ming Chung, M. Novel proteomic biomarker panel for prediction of aggressive metastatic hepatocellular carcinoma relapse in surgically resectable patients. J. Proteome Res. 2014, 13, 4833–4846. [Google Scholar] [CrossRef]
- Li, Y.; Zhuang, H.; Zhang, X.; Li, Y.; Liu, Y.; Yi, X.; Qin, G.; Wei, W.; Chen, R. Multiomics Integration Reveals the Landscape of Prometastasis Metabolism in Hepatocellular Carcinoma. Mol. Cell. Proteom. 2018, 17, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Chaudhai, R.; Zhang, S. Polypharmacology in Drug Development: A Minireview of Current Technologies. ChemMedChem 2016, 11, 1211–1218. [Google Scholar] [CrossRef]
- Saenz-Mendez, P.; Eriksson, L.A. Exploring Polypharmacology in Drug Design. Methods Mol. Biol. 2018, 1824, 229–243. [Google Scholar]
- Rosini, M. Polypharmacology: The rise of multitarget drugs over combination therapies. Future Med. Chem. 2014, 6, 485–487. [Google Scholar] [CrossRef]
- Antolin, A.A.; Workman, P.; Mestres, J.; Al-Lazikani, B. Polypharmacology in Precision Oncology: Current Applications and Future Prospects. Curr. Pharm. Des. 2016, 22, 6935–6945. [Google Scholar] [CrossRef] [Green Version]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharm. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Zhang, H.; Lu, P.; Liu, X.; Cao, H. Synergy evaluation by a pathway-pathway interaction network: A new way to predict drug combination. Mol. Biosyst. 2016, 12, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Coss, K.P.; Colin, P.H.; Barnara, A.; Stockmamm, H.; Crushell, E.; Saldova, R.; Knerr, I.; Gozalbo, M.E.R.; Monavari, A.; Rudd, P.M.; et al. N-Glycan abnormalities in children with Galactosemia. J. Proteome Res. 2014, 13, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xia, B.; Gleason, T.J.; Castaneda, U.; He, M.; Berry, G.T.; Fridovich-Keil, J.L. N- and O-linked glycosylation of total plasma glycoproteins in galactosemia. Mol. Genet. Metab. 2012, 106, 442–454. [Google Scholar] [CrossRef] [Green Version]
- Maratha, A.; Colhoun, H.O.; Knerr, I.; Coss, K.P.; Doran, P.; Treacy, E.P. Classical Galactosaemia and CDG, the N-Glycosylation Interface. A Review. JIMD Rep. 2016, 34, 33–42. [Google Scholar]
- Charlwood, J.; Clayton, P.; Keir, G.; Mian, N.; Winchester, B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology 1998, 8, 351–357. [Google Scholar] [CrossRef]
- Dobbie, J.A.; Holton, J.B.; Clamp, J.R. Defective galactosylation of proteins in cultured skin fibroblasts from galactosaemic patients. Ann. Clin. Biochem. 1990, 27, 274–275. [Google Scholar] [CrossRef]
- Ornstein, K.S.; McGuire, E.J.; Berry, G.T.; Roth, S.; Segal, S. Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose-1-phosphate uridyltransferase deficiency. Pediatric Res. 1992, 31, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Sturiale, L.; Barone, R.; Fiumara, A.; Perez, M.; Zaffanello, M. Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology 2005, 15, 1268–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.; Langley, S.D.; Khwaja, F.W.; Schmitt, E.W.; Elsas, L.J. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology 2003, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.G.; Xu, Y.K.; Kaufman, F.R.; Donnell, G.N.; Wolff, J.; Allen, R.J.; Koritala, S.; Reichardt, J.K. Biochemical and molecular studies of 132 patients with galactosemia. Hum. Genet. 1994, 94, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.G.; Xu, Y.K.; Kaufman, F.R.; Donnell, G.N. Measurements of uridine diphosphate hexoses in galactosemia. J. Pediatr. 1993, 123, 1015–1016. [Google Scholar] [CrossRef]
- Wang, B.B.; Xu, Y.K.; Ng, W.G.; Wong, L.J. Molecular and biochemical basis of galactosemia. Mol. Genet. Metab. 1998, 63, 263–269. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. Medicinal Chemistry of Anticancer Drugs, 2008; Elsevier Inc.: Amsterdam, The Netherlands, 2008; Chapter 6; pp. 177–198. [Google Scholar]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The ‘rule of three’ for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644–645. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.; Willis, A.C.; Elsas, L.J. The biochemical role of glutamine 188 in human galactose-1-phosphate uridyltransferase. J. Biol. Chem. 1999, 274, 6559–6566. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.; Elsas, L.J. Overexpression of human UDP-glucose pyrophosphorylase rescues galactose-1-phosphate uridyltransferase-deficient yeast. Biochem. Biophys. Res. Commun. 2000, 271, 392–400. [Google Scholar] [CrossRef]
- Fuhring, J.I.; Cramer, J.T.; Schneider, J.; Baruch, P.; Gerardy-Schahn, R.; Fedorov, R. A quaternary mechanism enables the complex biological functions of octameric human UDP-glucose pyrophosphorylase, a key enzyme in cell metabolism. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Peneff, C.; Ferrari, P.; Charrier, V.; Taburet, Y.; Monnier, C.; Zamboni, V.; Winter, J.; Harnois, M.; Fassy, F.; Bourne, Y. Crystal structures of two human pyrophosphorylase isoforms in complexes with UDPGlc(Gal)NAc: Role of the alternatively spliced insert in the enzyme oligomeric assembly and active site architecture. EMBO J. 2001, 20, 6191–6202. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Enzyme Inhibitory Activity | ||
|---|---|---|---|---|
| GALT | UGP2 | AGX1/UAP1 | ||
| GAL-012 | 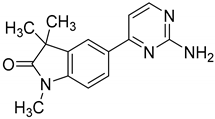 | 63.2 ± 2.6% | 58.5 ± 6.5% | 56.4 ± 5.3% |
| GAL-012-1 | 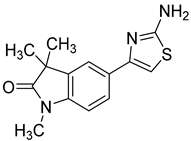 | 113.4 ± 0.8% | 111.9 ± 2.1% | 119 ± 1.8% |
| GAL-012-2 | 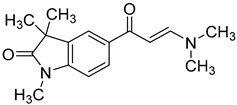 | 63.4 ± 2.3% | 63.8 ± 4% | 107.9 ± 4.7% |
| GAL-012-3 | 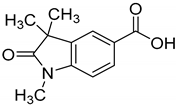 | 121.2 ± 14.3% | 106.1 ± 4% | 104 ± 8.9% |
| GAL-012-4 | 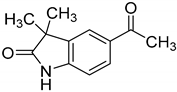 | 125.8 ± 7.9% | 103.2 ± 4.5% | 107.7 ± 5.3% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Vankayalapati, H.; Tang, M.; Zheng, Y.; Li, Y.; Ma, C.; Lai, K. Discovery of Novel Inhibitors Targeting Multi-UDP-hexose Pyrophosphorylases as Anticancer Agents. Molecules 2020, 25, 645. https://doi.org/10.3390/molecules25030645
Yang Y, Vankayalapati H, Tang M, Zheng Y, Li Y, Ma C, Lai K. Discovery of Novel Inhibitors Targeting Multi-UDP-hexose Pyrophosphorylases as Anticancer Agents. Molecules. 2020; 25(3):645. https://doi.org/10.3390/molecules25030645
Chicago/Turabian StyleYang, Yueqin, Hariprasad Vankayalapati, Manshu Tang, Yingbo Zheng, Yingri Li, Cong Ma, and Kent Lai. 2020. "Discovery of Novel Inhibitors Targeting Multi-UDP-hexose Pyrophosphorylases as Anticancer Agents" Molecules 25, no. 3: 645. https://doi.org/10.3390/molecules25030645