A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L



Abstract
:1. Introduction
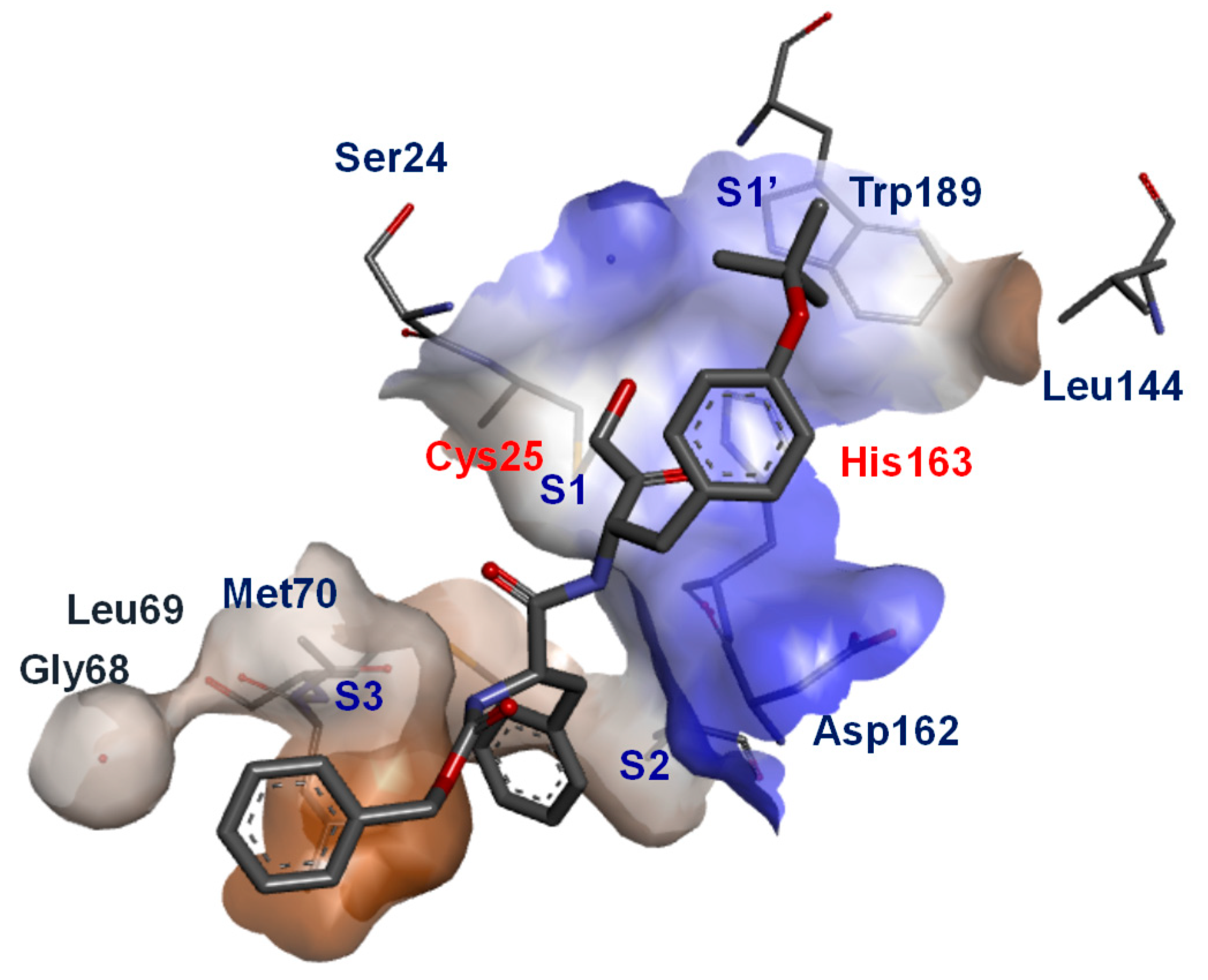
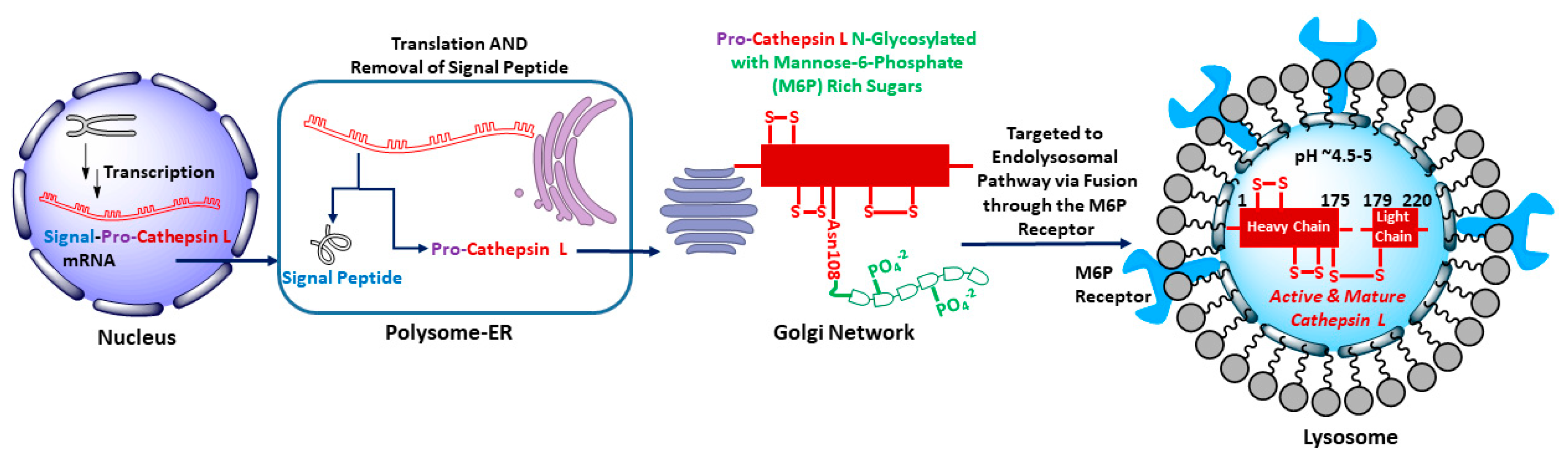
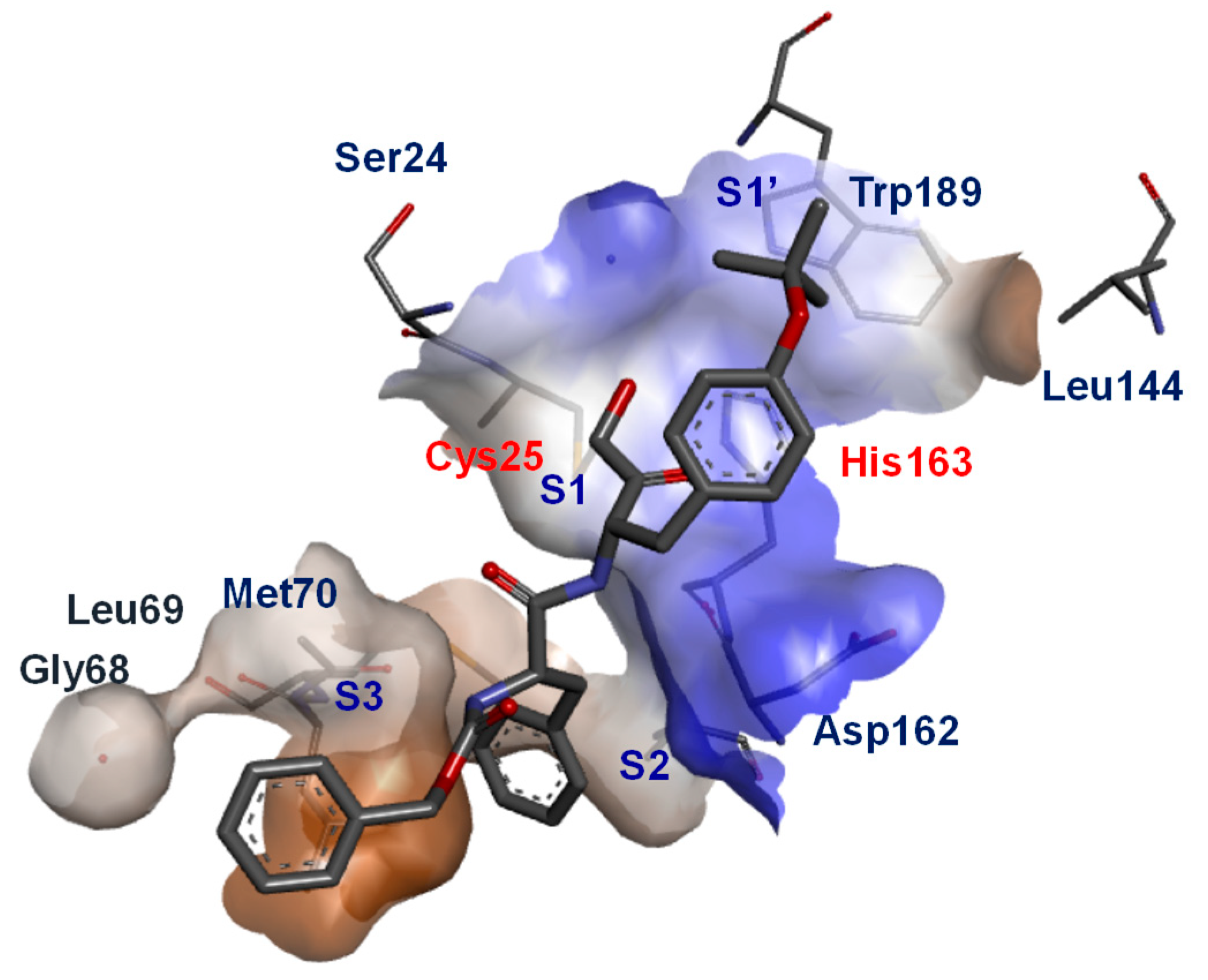
2. Functional Biology of Cathepsin L and Its Role in Human Diseases
3. Small Molecule Inhibitors
3.1. Epoxysuccinates
3.2. Peptidyldiazomethane and Peptidylchloromethane
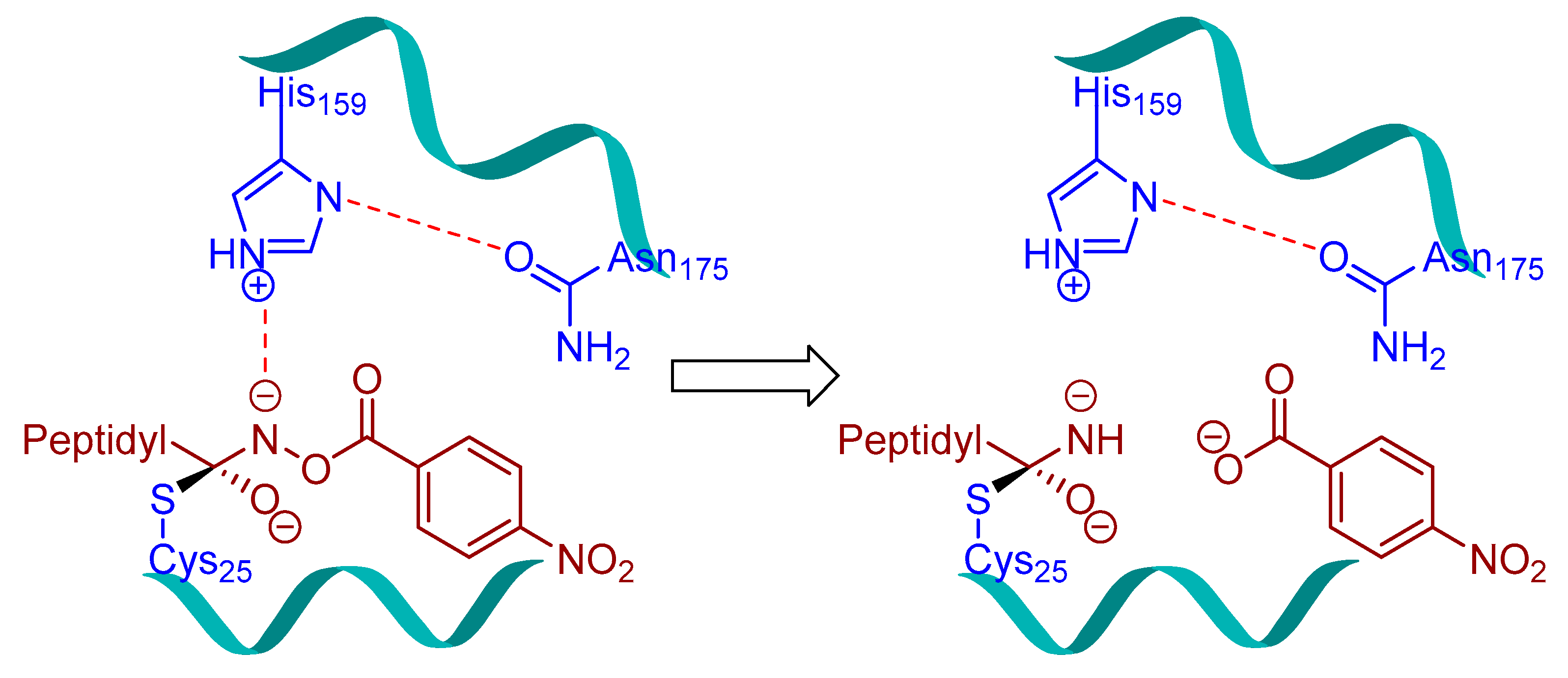
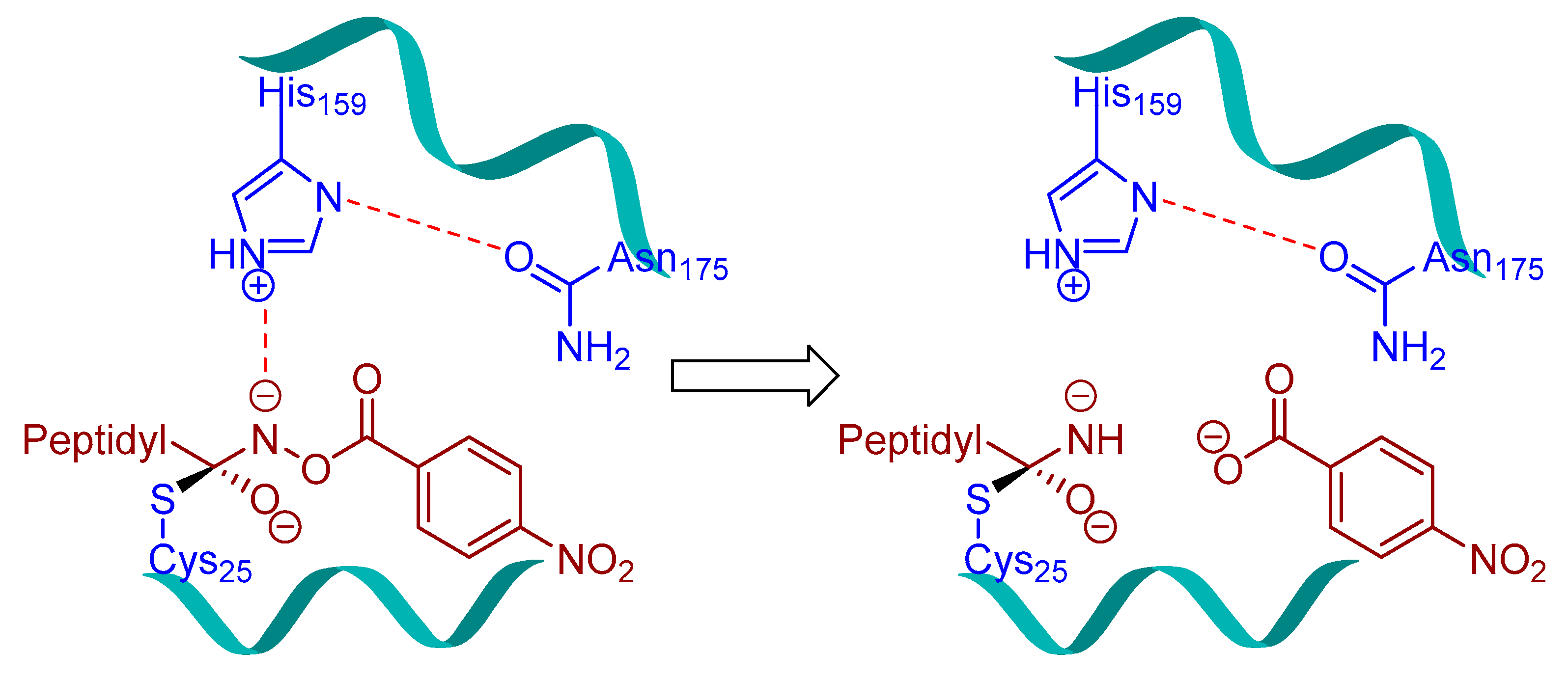
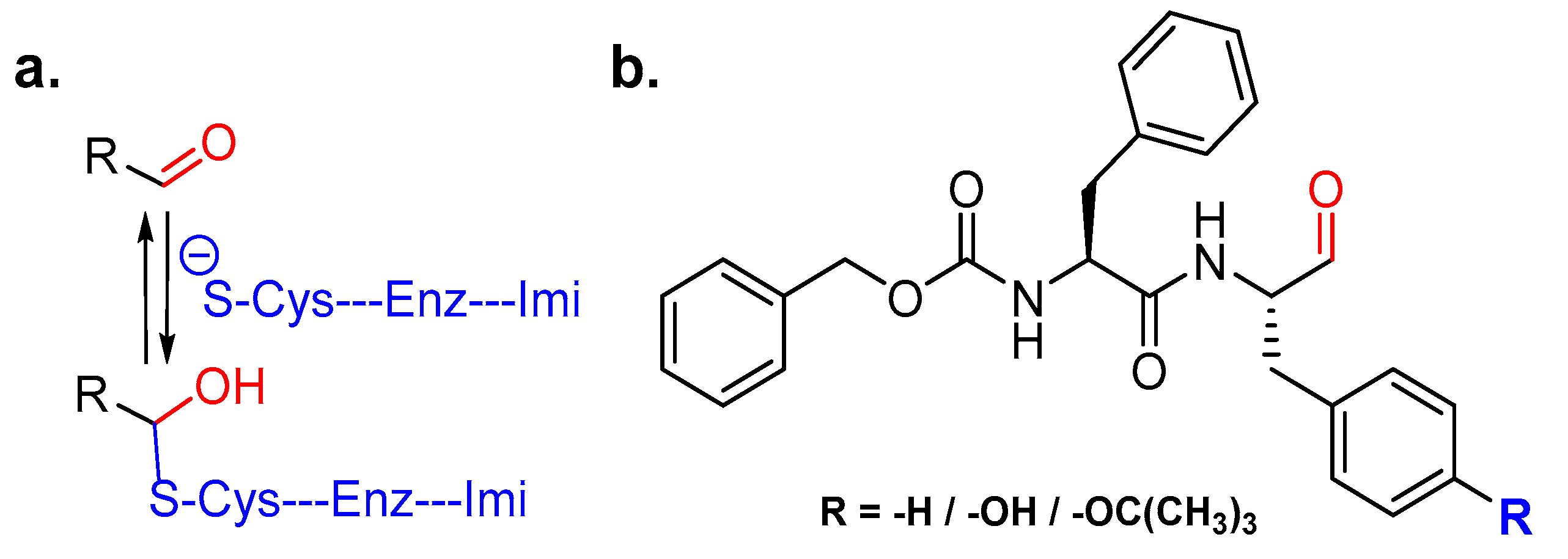
3.3. Peptidylhydroxylamine and Peptidylhydroxamates
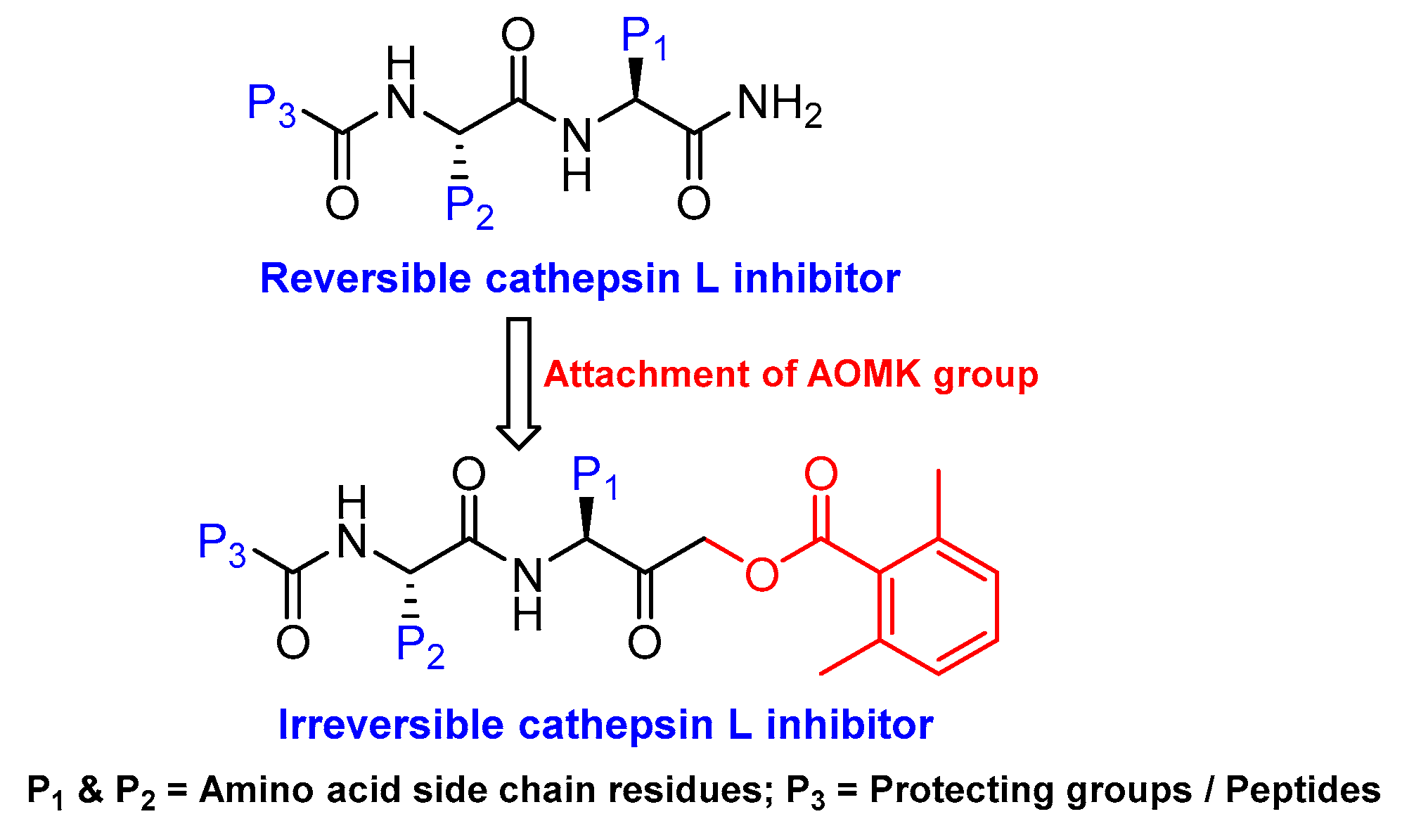
3.4. Peptidyl Acyloxymethanes/Acyloxymethyl Ketones
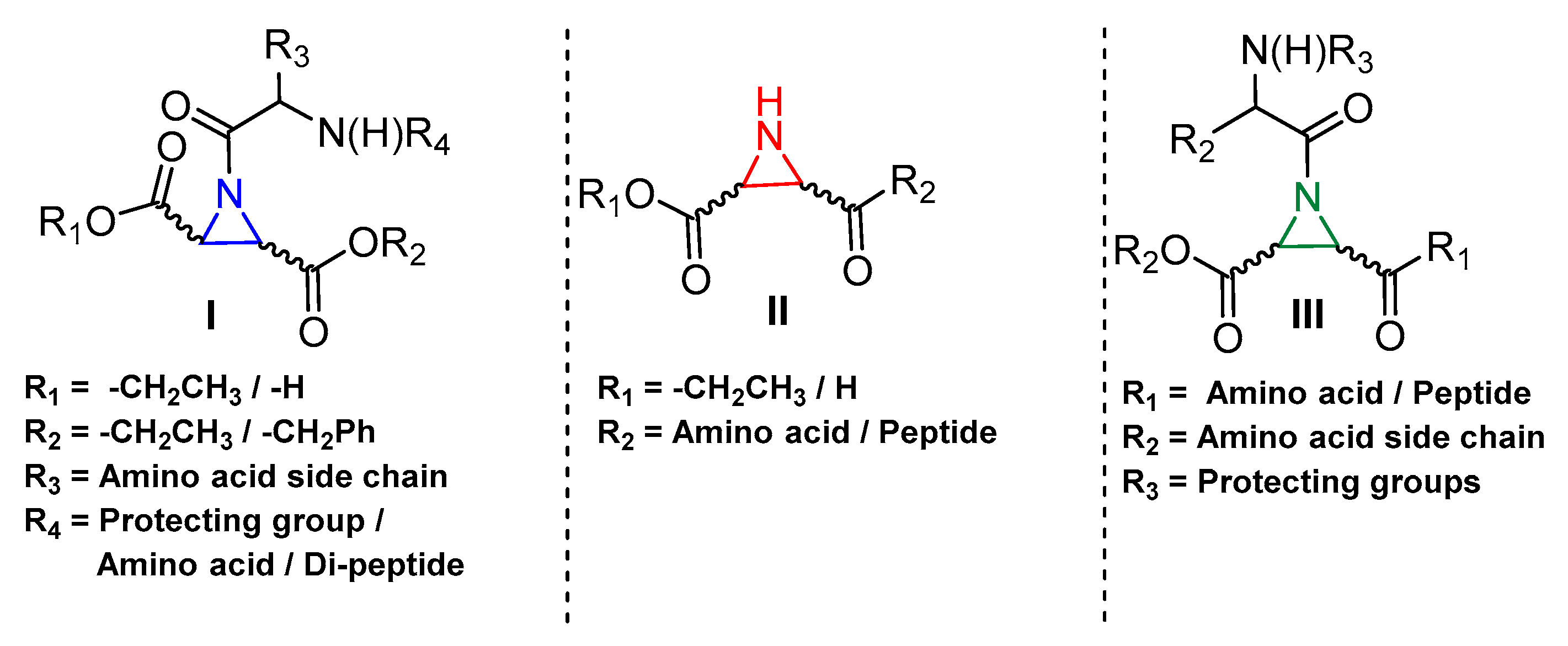
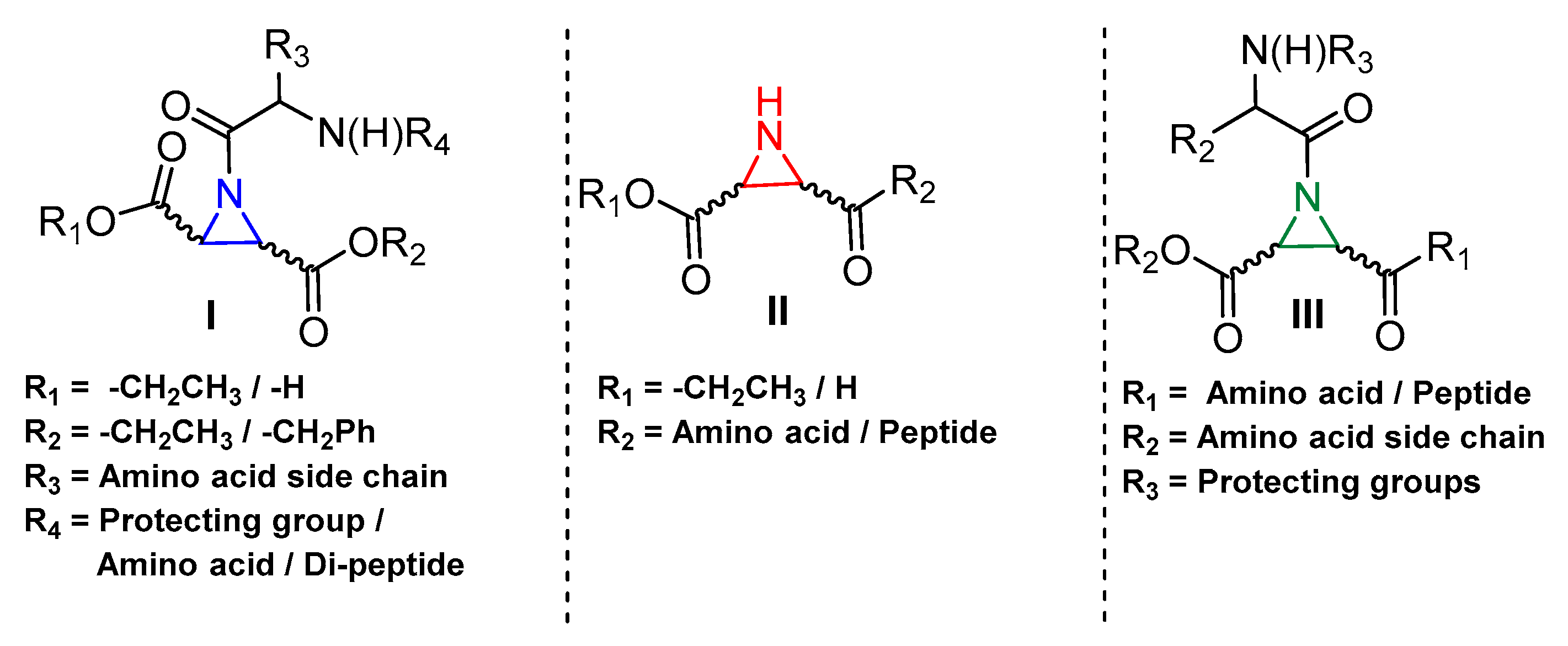
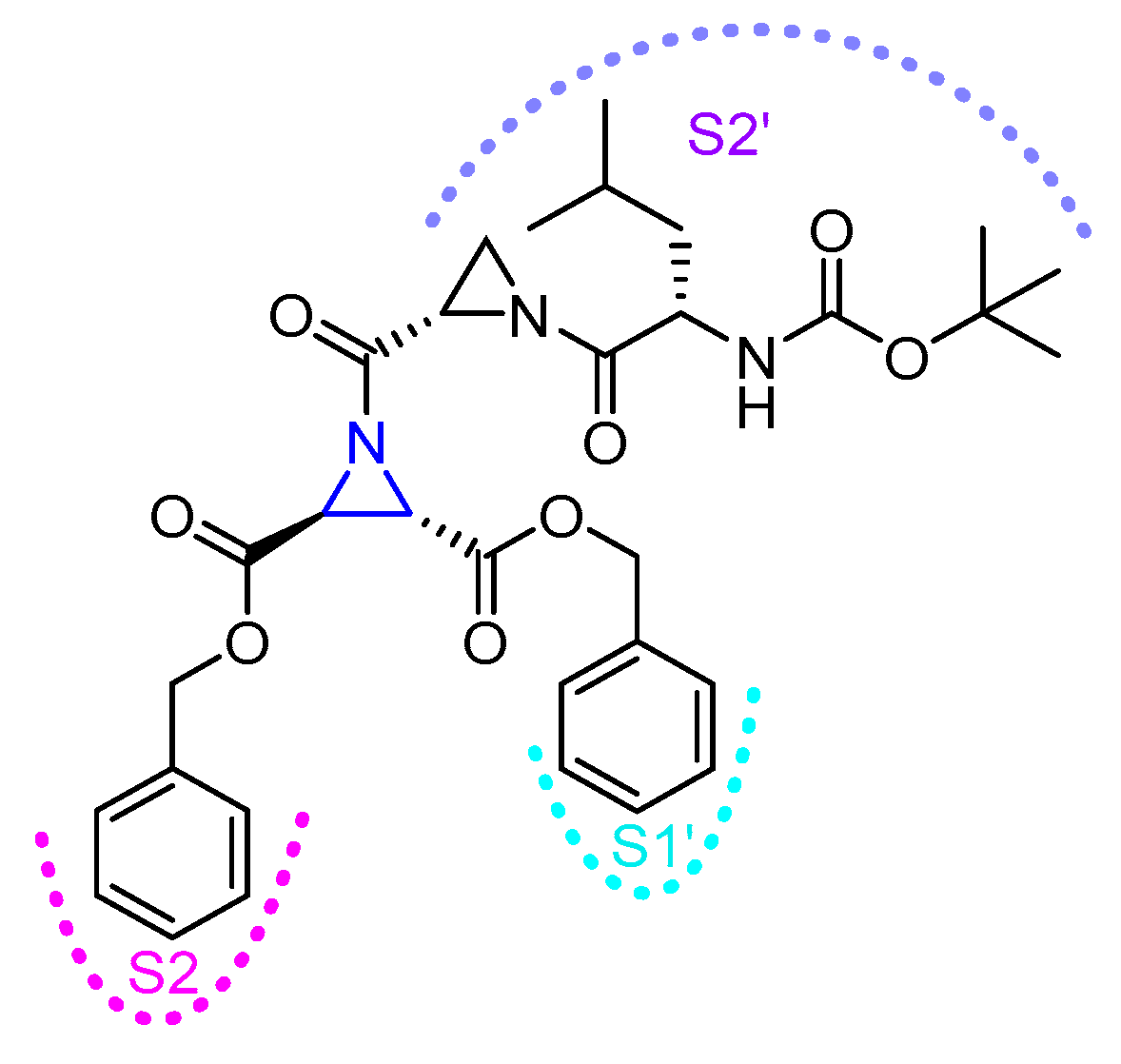
3.5. Peptidyl Aziridine
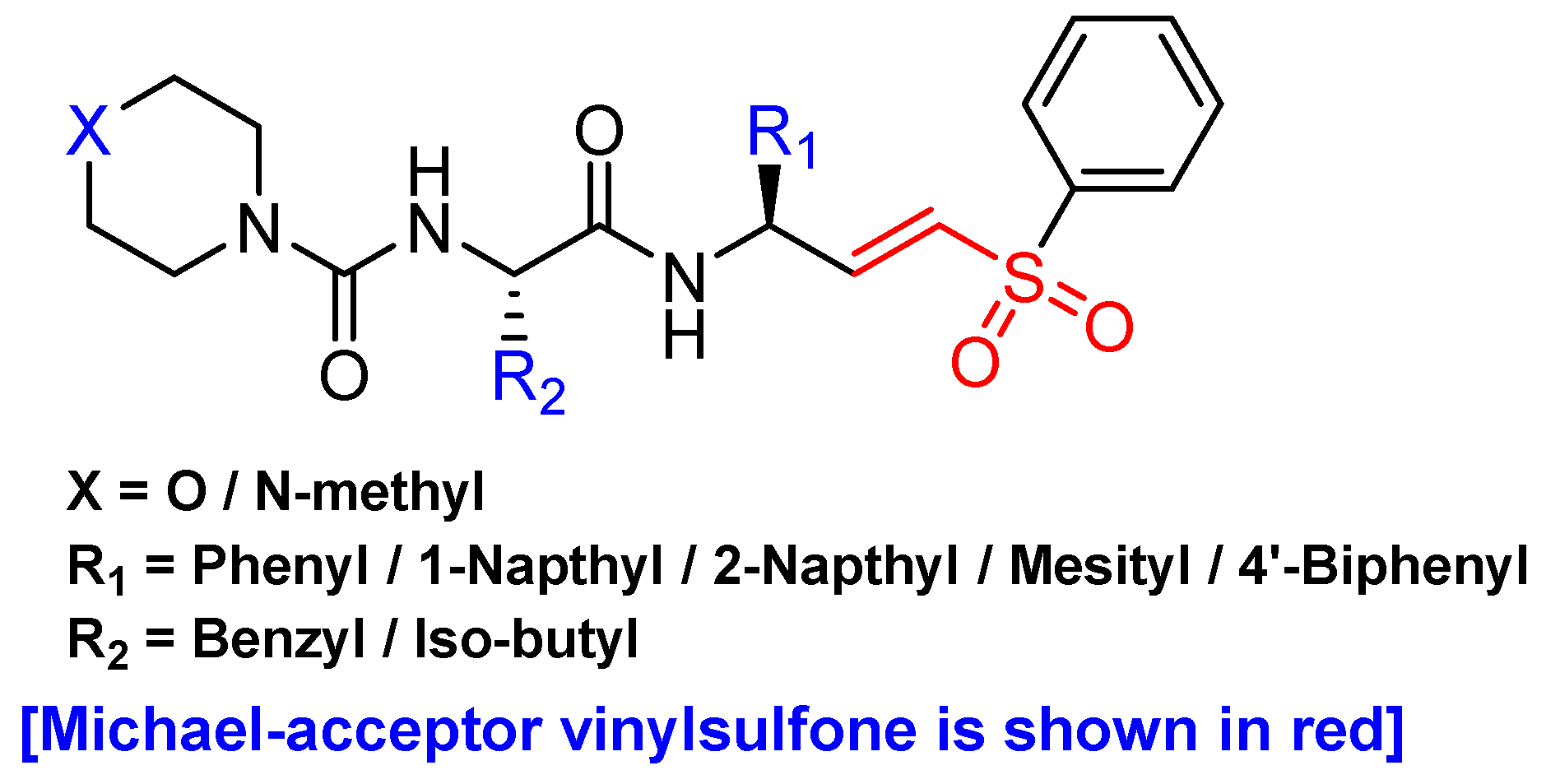
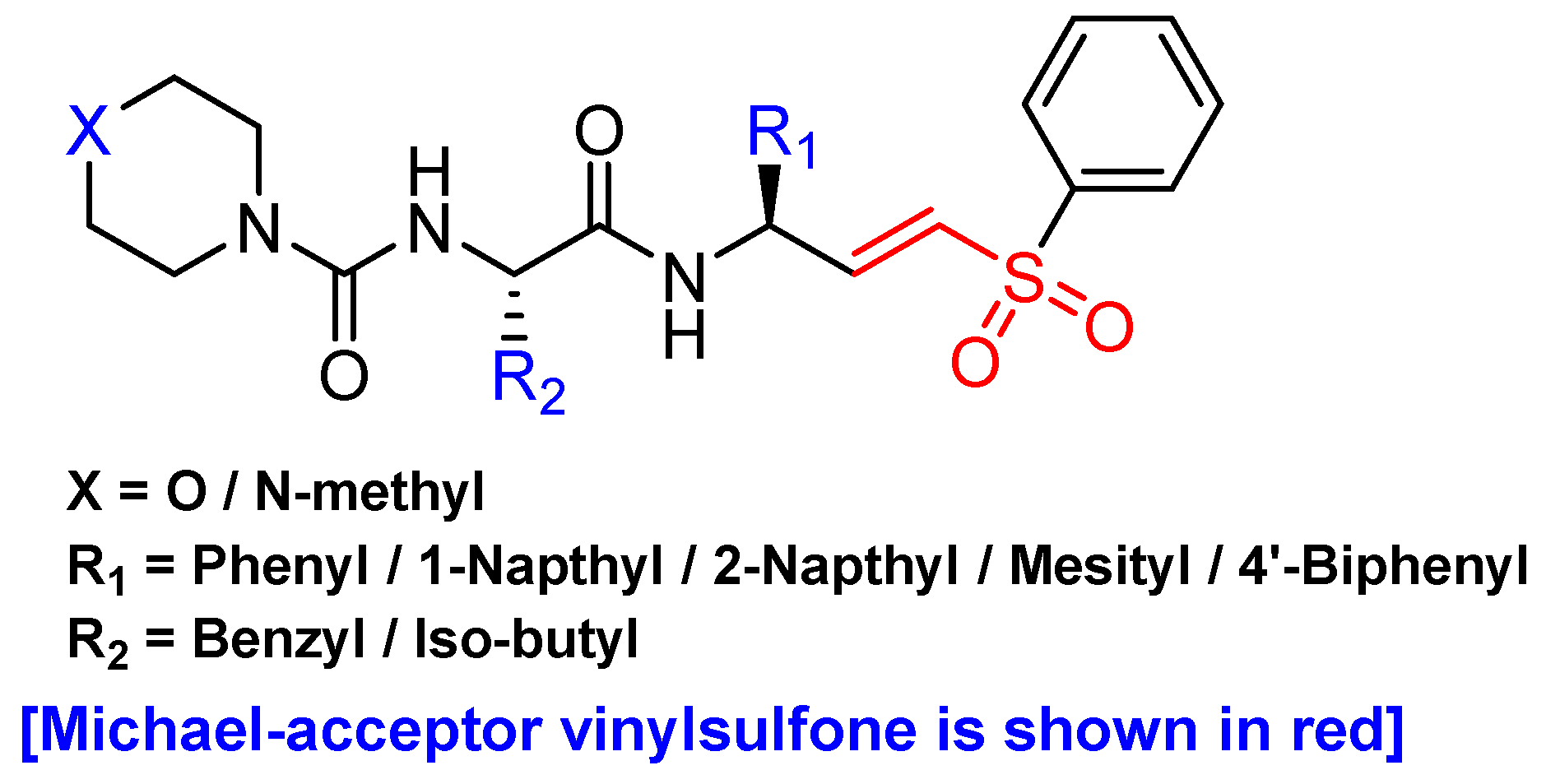
3.6. Peptidyl Aryl Vinylsulfones
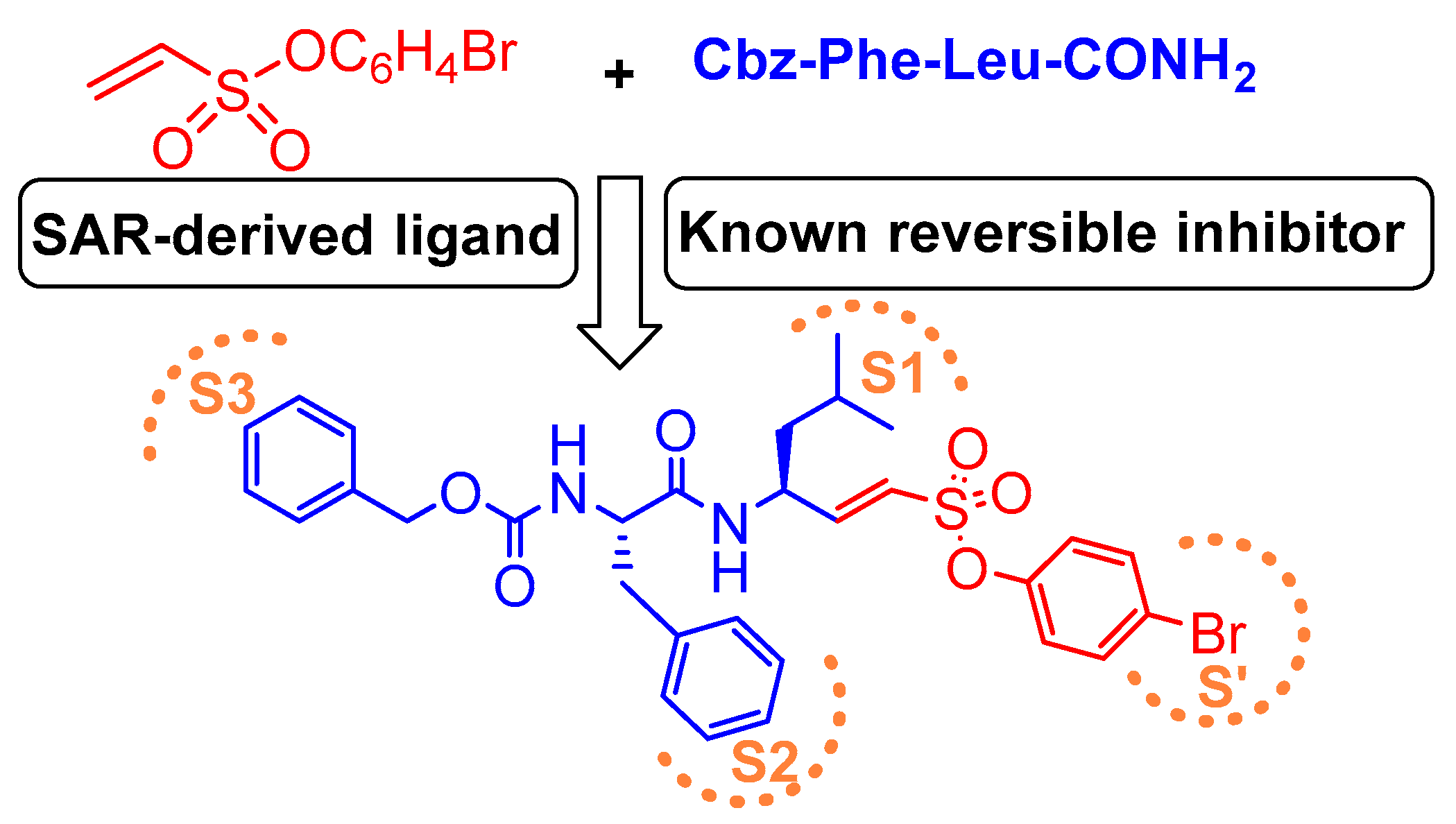
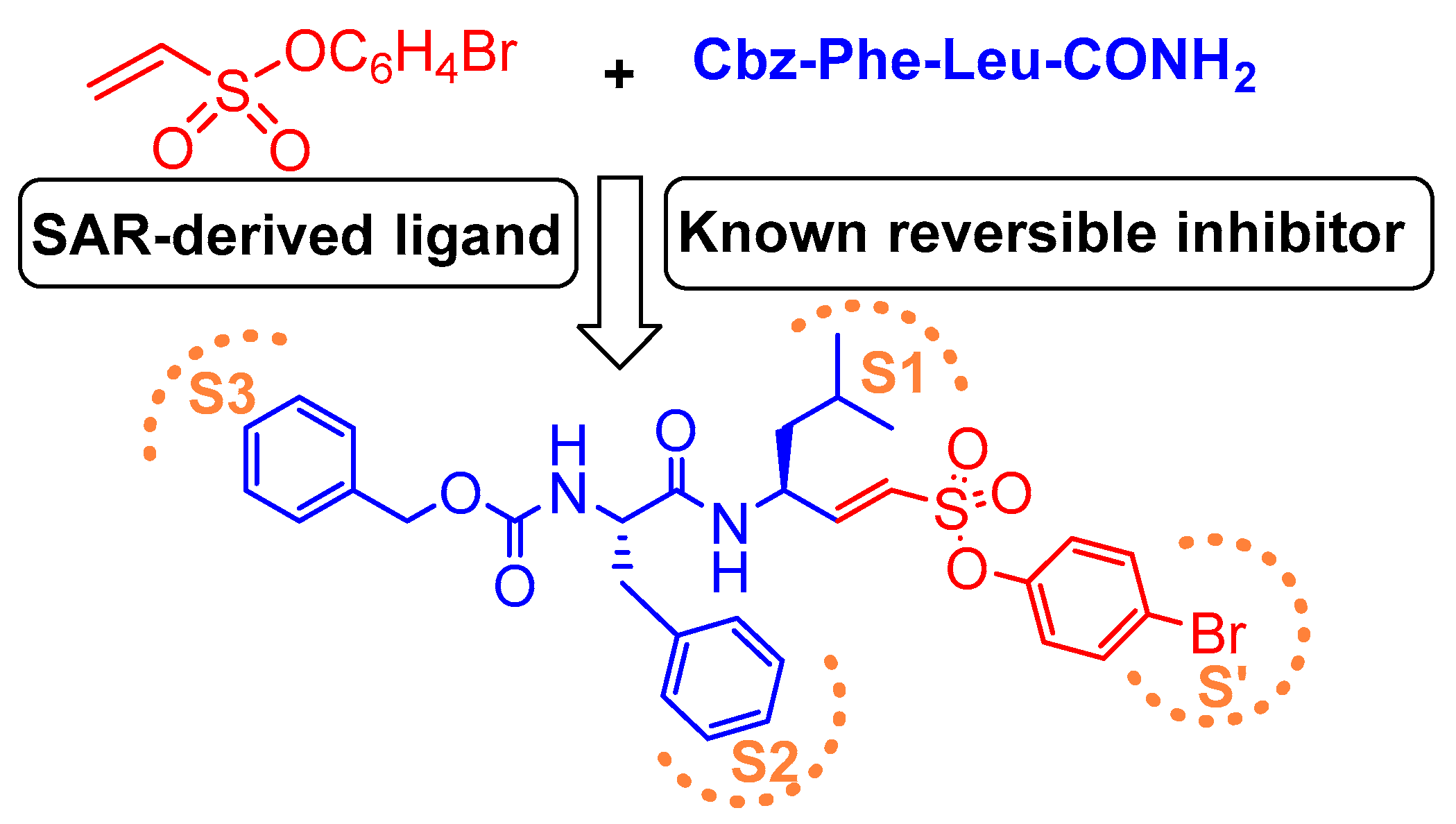
3.7. Peptidyl Aryl Vinylsulfonate
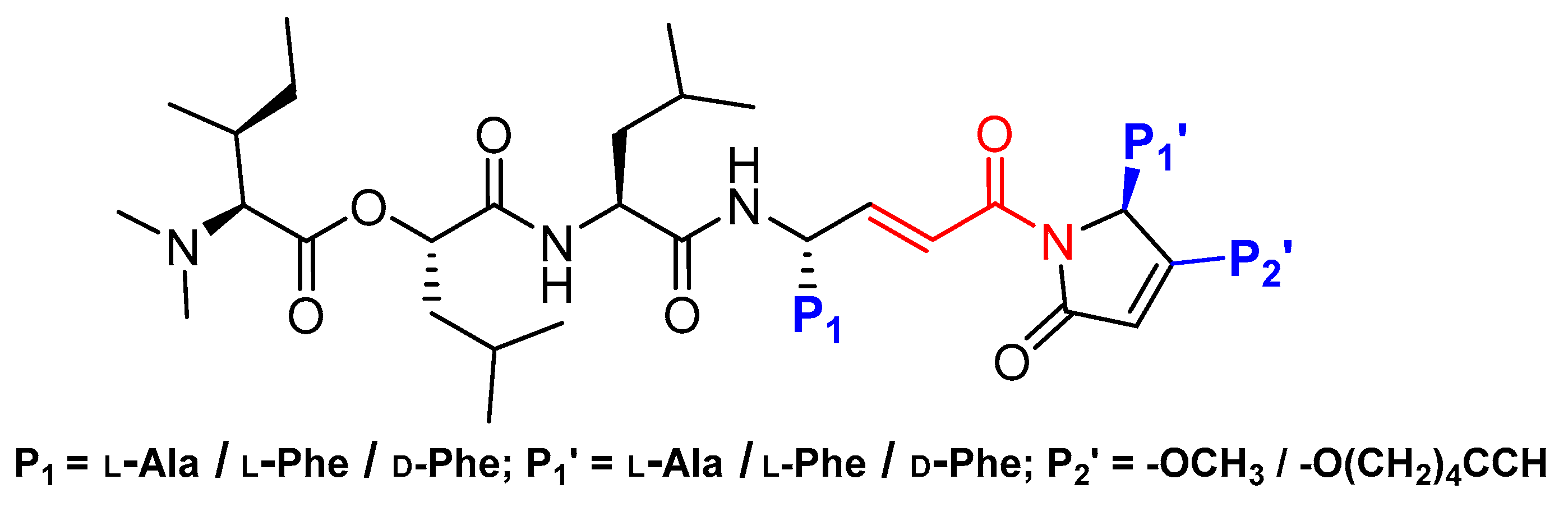
3.8. Gallinamide A Analogs
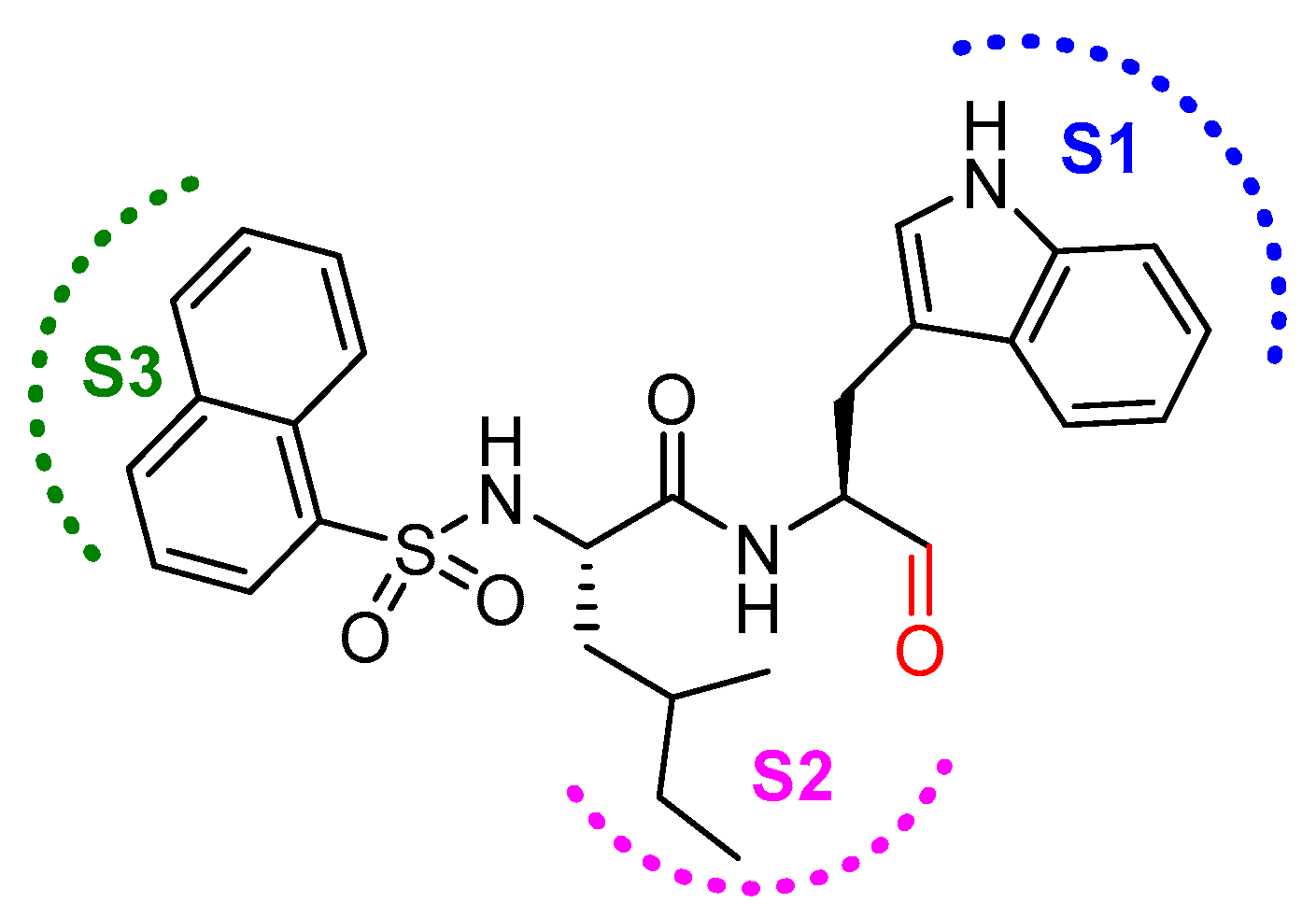
3.9. Peptidyl Aldehydes
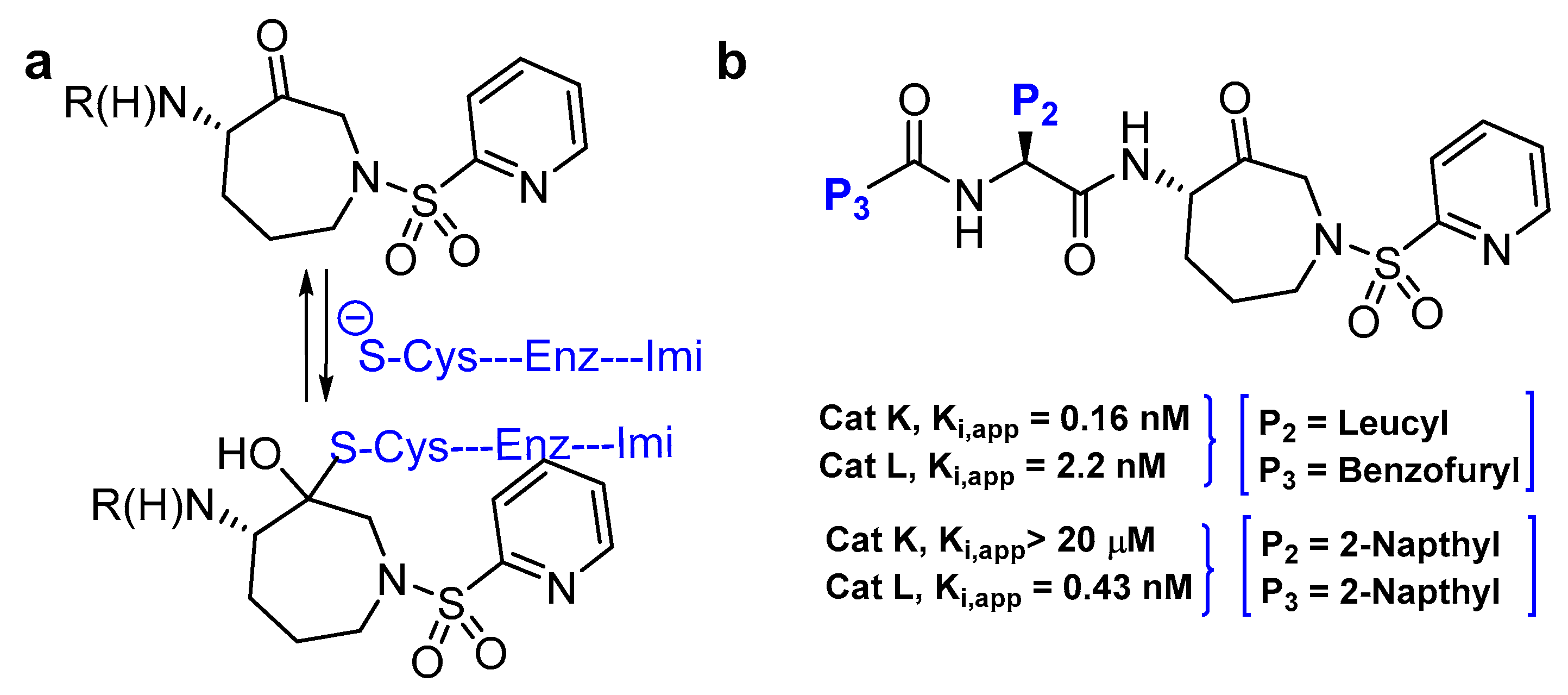
3.10. Azepanone-based Inhibitors
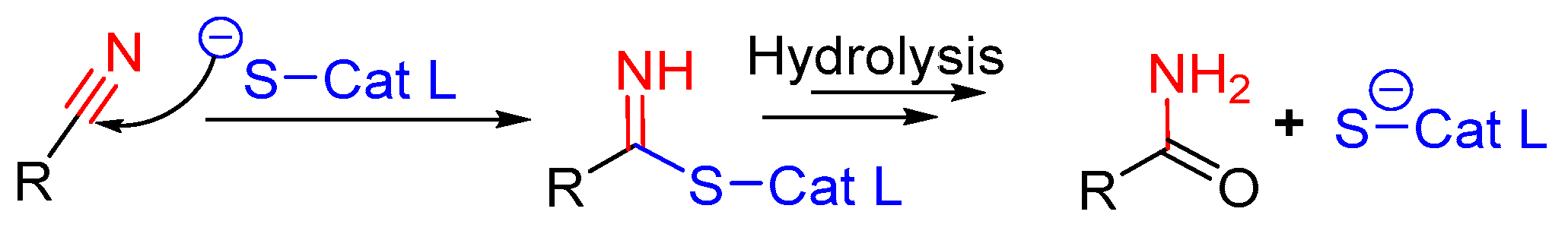
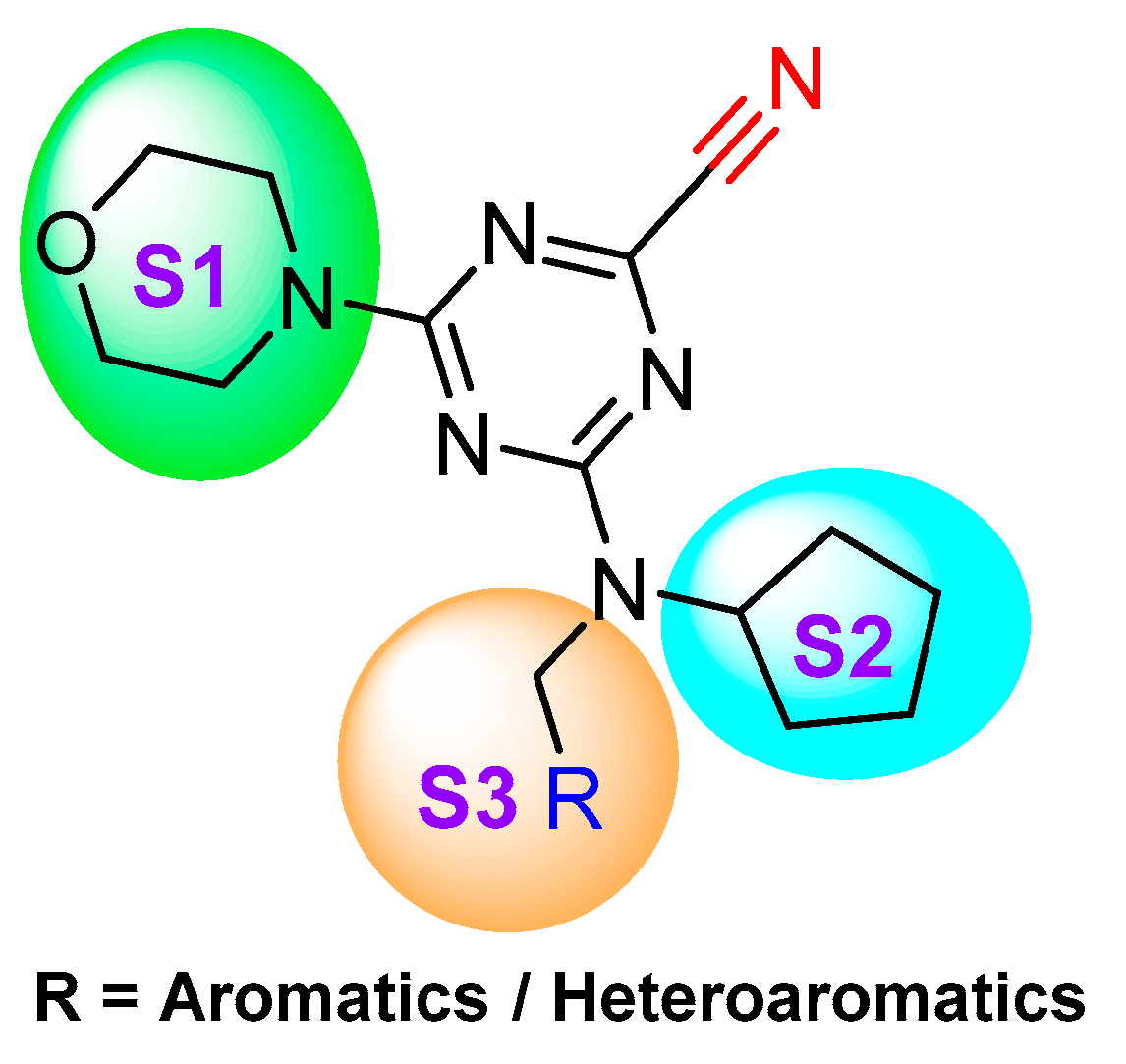
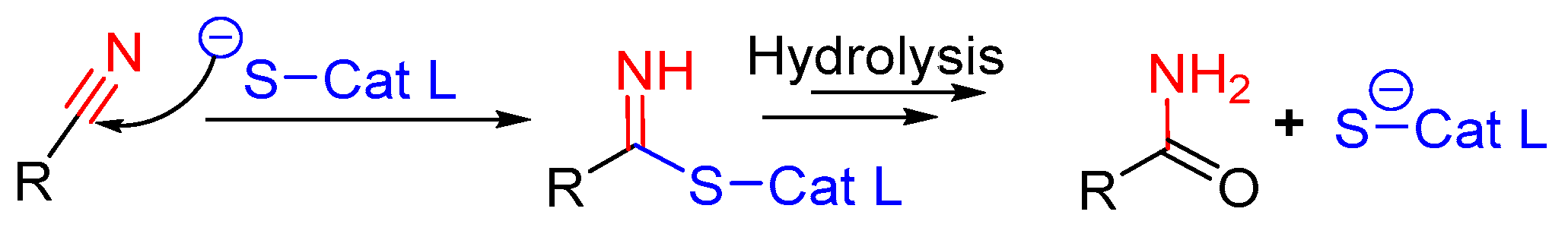
3.11. Nitrile-Containing Inhibitors
3.12. Thiosemicarbazone
3.13. Propeptide Mimics
3.14. Thiocarbazate, Oxocarbazate and Azapeptides
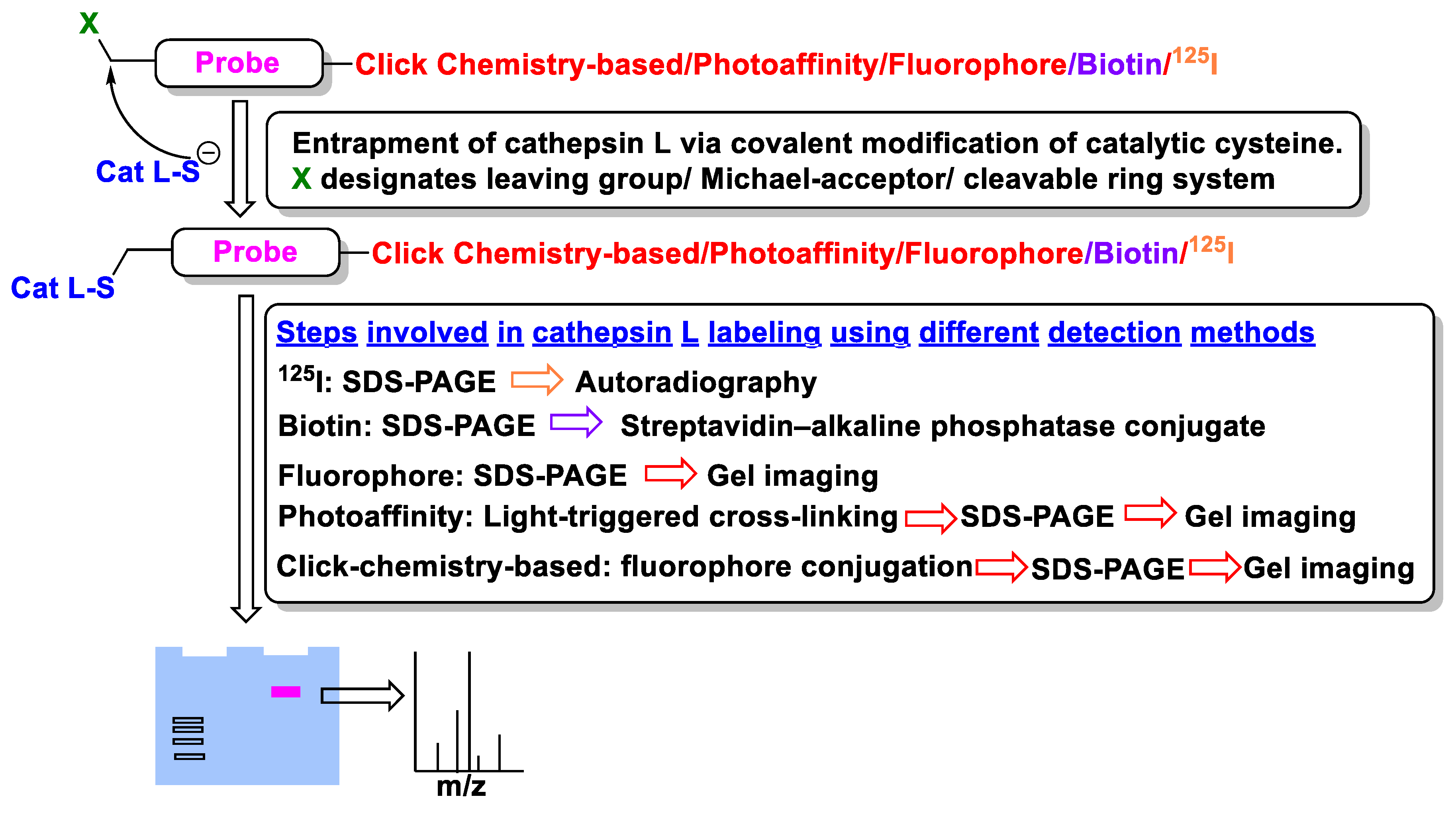
4. Molecular Probes
4.1. Radio-Labelled
4.2. Affinity-Based
4.3. Photoaffinity-Based
4.4. Fluorescence-Based
4.4.1. Two-Photon Based
4.4.2. One-Photon Based
4.5. Clickable and Tagless
4.6. Mass Cytometry-Compatible Activity-Based Probes
5. Final Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MA | Methacrylate |
| Phe | Phenylalanine |
| Gly | Glycine |
| Z/Cbz | Carboxybenzyl |
| His | Histidine |
| NBz | Nitrobenzyl |
| iAm | isoamyl |
| SAR | Structure Activity Relationship |
| AOMK | Acyloxymethanes/Acyloxymethyl Ketones |
| Leu | Leucine |
| Met | Methionine |
| Azi | Aziridine |
| FRET | Fluorescence Resonance Energy Transfer |
| catABP | Clickable and Tagless Activity-based Probe |
References
- de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Schroder, B.A.; Wrocklage, C.; Hasilik, A.; Saftig, P. The proteome of lysosomes. Proteomics 2010, 10, 4053–4076. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Deveraux, Q.; Turk, B.; Sali, A. Comprehensive search for cysteine cathepsins in the human genome. Biol. Chem. 2004, 385, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [Green Version]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Vizovisek, M.; Fonovic, M.; Turk, B. Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Boil. J. Int. Soc. Matrix Biol. 2019, 75, 141–159. [Google Scholar] [CrossRef]
- Kramer, L.; Turk, D.; Turk, B. The Future of Cysteine Cathepsins in Disease Management. Trends Pharmacol. Sci. 2017, 38, 873–898. [Google Scholar] [CrossRef]
- Siklos, M.; BenAissa, M.; Thatcher, G.R. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Loser, R.; Pietzsch, J. Cysteine cathepsins: Their role in tumor progression and recent trends in the development of imaging probes. Front. Chem. 2015, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Jakos, T.; Pislar, A.; Jewett, A.; Kos, J. Cysteine Cathepsins in Tumor-Associated Immune Cells. Front. Immunol. 2019, 10, 2037. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Roth, W.; Wong, P.; Nelson, A.; Farr, A.; Deussing, J.; Villadangos, J.A.; Ploegh, H.; Peters, C.; Rudensky, A.Y. Cathepsin L: Critical role in Ii degradation and CD4 T cell selection in the thymus. Science 1998, 280, 450–453. [Google Scholar] [CrossRef]
- Manchanda, M.; Das, P.; Gahlot, G.P.S.; Singh, R.; Roeb, E.; Roderfeld, M.; Datta Gupta, S.; Saraya, A.; Pandey, R.M.; Chauhan, S.S. Cathepsin L and B as Potential Markers for Liver Fibrosis: Insights From Patients and Experimental Models. Clin. Transl. Gastroenterol. 2017, 8, e99. [Google Scholar] [CrossRef]
- Maehr, R.; Mintern, J.D.; Herman, A.E.; Lennon-Dumenil, A.M.; Mathis, D.; Benoist, C.; Ploegh, H.L. Cathepsin L is essential for onset of autoimmune diabetes in NOD mice. J. Clin. Investig. 2005, 115, 2934–2943. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Vaag, A.; Carlsson, E.; Hansson, M.; Ahren, B.; Groop, L. Impaired cathepsin L gene expression in skeletal muscle is associated with type 2 diabetes. Diabetes 2003, 52, 2411–2418. [Google Scholar] [CrossRef] [Green Version]
- Kitamoto, S.; Sukhova, G.K.; Sun, J.; Yang, M.; Libby, P.; Love, V.; Duramad, P.; Sun, C.; Zhang, Y.; Yang, X.; et al. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation 2007, 115, 2065–2075. [Google Scholar] [CrossRef]
- Sun, M.; Chen, M.; Liu, Y.; Fukuoka, M.; Zhou, K.; Li, G.; Dawood, F.; Gramolini, A.; Liu, P.P. Cathepsin-L contributes to cardiac repair and remodelling post-infarction. Cardiovasc. Res. 2011, 89, 374–383. [Google Scholar] [CrossRef] [Green Version]
- He, W.; McCarroll, C.S.; Nather, K.; Elliott, E.B.A.; Nicklin, S.A.; Loughrey, C.M. The Cathepsin-L Inhibitor Caa0225 Protects against Myocardial Ischaemia-Reperfusion Injury. Heart 2015, 101, A1. [Google Scholar] [CrossRef]
- Potts, W.; Bowyer, J.; Jones, H.; Tucker, D.; Freemont, A.J.; Millest, A.; Martin, C.; Vernon, W.; Neerunjun, D.; Slynn, G.; et al. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int. J. Exp. Pathol. 2004, 85, 85–96. [Google Scholar] [CrossRef]
- Furuyama, N.; Fujisawa, Y. Distinct roles of cathepsin K and cathepsin L in osteoclastic bone resorption. Endocr. Res. 2000, 26, 189–204. [Google Scholar] [CrossRef]
- Yasuma, T.; Oi, S.; Choh, N.; Nomura, T.; Furuyama, N.; Nishimura, A.; Fujisawa, Y.; Sohda, T. Synthesis of Peptide Aldehyde Derivatives as Selective Inhibitors of Human Cathepsin L and Their Inhibitory Effect on Bone Resorption. J. Med. Chem. 1998, 41, 4301–4308. [Google Scholar] [CrossRef] [PubMed]
- Onishi, K.; Li, Y.; Ishii, K.; Hisaeda, H.; Tang, L.; Duan, X.; Dainichi, T.; Maekawa, Y.; Katunuma, N.; Himeno, K. Cathepsin L is crucial for a Th1-type immune response during Leishmania major infection. Microbes Infect. 2004, 6, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Sever, S.; Altintas, M.M.; Nankoe, S.R.; Moller, C.C.; Ko, D.; Wei, C.; Henderson, J.; del Re, E.C.; Hsing, L.; Erickson, A.; et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J. Clin. Investig. 2007, 117, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Oh, J.; Shirato, I.; Asanuma, K.; Hug, A.; Mundel, T.M.; Honey, K.; Ishidoh, K.; Kominami, E.; Kreidberg, J.A.; et al. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J. Biol. Chem. 2004, 279, 34827–34832. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 2015, 155, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Lankelma, J.M.; Voorend, D.M.; Barwari, T.; Koetsveld, J.; Van der Spek, A.H.; De Porto, A.P.; Van Rooijen, G.; Van Noorden, C.J. Cathepsin L, target in cancer treatment? Life Sci. 2010, 86, 225–233. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Allan, E.R.; Yates, R.M. Redundancy between Cysteine Cathepsins in Murine Experimental Autoimmune Encephalomyelitis. PLoS ONE 2015, 10, e0128945. [Google Scholar] [CrossRef]
- Cermak, S.; Kosicek, M.; Mladenovic-Djordjevic, A.; Smiljanic, K.; Kanazir, S.; Hecimovic, S. Loss of Cathepsin B and L Leads to Lysosomal Dysfunction, NPC-Like Cholesterol Sequestration and Accumulation of the Key Alzheimer’s Proteins. PLoS ONE 2016, 11, e0167428. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Lee, J.; Seo, H.Y.; Lim, J.S.; Kim, E.K. Cathepsin inhibition-induced lysosomal dysfunction enhances pancreatic beta-cell apoptosis in high glucose. PLoS ONE 2015, 10, e0116972. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Lang, L.; Reitman, M.; Tang, J.; Roberts, R.M.; Kornfeld, S. Lysosomal enzyme phosphorylation. Recognition of a protein-dependent determinant allows specific phosphorylation of oligosaccharides present on lysosomal enzymes. J. Biol. Chem. 1984, 259, 14663–14671. [Google Scholar]
- Braun, M.; Waheed, A.; von Figura, K. Lysosomal acid phosphatase is transported to lysosomes via the cell surface. EMBO J. 1989, 8, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kondo, C.; Katunuma, N. An Active 32-kDa Cathepsin L Is Secreted Directly from HT 1080 Fibrosarcoma Cells and Not via Lysosomal Exocytosis. PLoS ONE 2015, 10, e0145067. [Google Scholar] [CrossRef] [PubMed]
- Jerala, R.; Zerovnik, E.; Kidric, J.; Turk, V. pH-induced conformational transitions of the propeptide of human cathepsin L. A role for a molten globule state in zymogen activation. J. Biol. Chem. 1998, 273, 11498–11504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Kawabata, T.; Furuno, K.; Kato, K. Evidence that aspartic proteinase is involved in the proteolytic processing event of procathepsin L in lysosomes. Arch. Biochem. Biophys. 1989, 271, 400–406. [Google Scholar] [CrossRef]
- Abudula, A.; Rommerskirch, W.; Weber, E.; Gunther, D.; Wiederanders, B. Splice variants of human cathepsin L mRNA show different expression rates. Biol. Chem. 2001, 382, 1583–1591. [Google Scholar] [CrossRef]
- Jean, D.; Guillaume, N.; Frade, R. Characterization of human cathepsin L promoter and identification of binding sites for NF-Y, Sp1 and Sp3 that are essential for its activity. Biochem. J. 2002, 361, 173–184. [Google Scholar] [CrossRef]
- Goulet, B.; Nepveu, A. Complete and limited proteolysis in cell cycle progression. Cell Cycle 2004, 3, 986–989. [Google Scholar] [CrossRef]
- Goulet, B.; Sansregret, L.; Leduy, L.; Bogyo, M.; Weber, E.; Chauhan, S.S.; Nepveu, A. Increased expression and activity of nuclear cathepsin L in cancer cells suggests a novel mechanism of cell transformation. Mol. Cancer Res. MCR 2007, 5, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Goulet, B.; Baruch, A.; Moon, N.S.; Poirier, M.; Sansregret, L.L.; Erickson, A.; Bogyo, M.; Nepveu, A. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol. Cell 2004, 14, 207–219. [Google Scholar] [CrossRef]
- Coulombe, R.; Grochulski, P.; Sivaraman, J.; Menard, R.; Mort, J.S.; Cygler, M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996, 15, 5492–5503. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, R.; Li, Y.; Takebe, S.; Menard, R.; Mason, P.; Mort, J.S.; Cygler, M. Crystallization and preliminary X-ray diffraction studies of human procathepsin L. Proteins 1996, 25, 398–400. [Google Scholar] [CrossRef]
- Abrahamson, M.; Alvarez-Fernandez, M.; Nathanson, C.M. Cystatins. Biochem. Soc. Symp. 2003, 70, 179–199. [Google Scholar]
- Kotsyfakis, M.; Sa-Nunes, A.; Francischetti, I.M.; Mather, T.N.; Andersen, J.F.; Ribeiro, J.M. Antiinflammatory and immunosuppressive activity of sialostatin L, a salivary cystatin from the tick Ixodes scapularis. J. Biol. Chem. 2006, 281, 26298–26307. [Google Scholar] [CrossRef] [Green Version]
- Horka, H.; Staudt, V.; Klein, M.; Taube, C.; Reuter, S.; Dehzad, N.; Andersen, J.F.; Kopecky, J.; Schild, H.; Kotsyfakis, M.; et al. The tick salivary protein sialostatin L inhibits the Th9-derived production of the asthma-promoting cytokine IL-9 and is effective in the prevention of experimental asthma. J. Immunol. 2012, 188, 2669–2676. [Google Scholar] [CrossRef]
- Carmona, E.; Dufour, E.; Plouffe, C.; Takebe, S.; Mason, P.; Mort, J.S.; Menard, R. Potency and selectivity of the cathepsin L propeptide as an inhibitor of cysteine proteases. Biochemistry 1996, 35, 8149–8157. [Google Scholar] [CrossRef]
- Green, G.D.; Kembhavi, A.A.; Davies, M.E.; Barrett, A.J. Cystatin-like cysteine proteinase inhibitors from human liver. Biochem. J. 1984, 218, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Bromme, D.; Rinne, R.; Kirschke, H. Tight-binding inhibition of cathepsin S by cystatins. Biomed. Biochim. Acta 1991, 50, 631–635. [Google Scholar]
- Barrett, A.J.; Davies, M.E.; Grubb, A. The place of human gamma-trace (cystatin C) amongst the cysteine proteinase inhibitors. Biochem. Biophys. Res. Commun. 1984, 120, 631–636. [Google Scholar] [CrossRef]
- Balbin, M.; Hall, A.; Grubb, A.; Mason, R.W.; Lopez-Otin, C.; Abrahamson, M. Structural and functional characterization of two allelic variants of human cystatin D sharing a characteristic inhibition spectrum against mammalian cysteine proteinases. J. Biol. Chem. 1994, 269, 23156–23162. [Google Scholar] [PubMed]
- Bevec, T.; Stoka, V.; Pungercic, G.; Dolenc, I.; Turk, V. Major histocompatibility complex class II-associated p41 invariant chain fragment is a strong inhibitor of lysosomal cathepsin L. J. Exp. Med. 1996, 183, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.; Parkes, C.; Abrahamson, M.; Grubb, A.; Barrett, A.J. Human low-Mr kininogen contains three copies of a cystatin sequence that are divergent in structure and in inhibitory activity for cysteine proteinases. Biochem. J. 1986, 234, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullere-Esterl, W.; Iwanaga, S.; Nakanishi, S. Kininogens Revisited. Trends Biochem. Sci. 1986, 11, 336–339. [Google Scholar] [CrossRef]
- Ni, J.; Fernandez, M.A.; Danielsson, L.; Chillakuru, R.A.; Zhang, J.; Grubb, A.; Su, J.; Gentz, R.; Abrahamson, M. Cystatin F is a glycosylated human low molecular weight cysteine proteinase inhibitor. J. Biol. Chem. 1998, 273, 24797–24804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrault, P.M.; Samsonov, S.A.; Weber, G.; Coquet, L.; Nazmi, K.; Bolscher, J.G.; Lalmanach, A.C.; Jouenne, T.; Bromme, D.; Pisabarro, M.T.; et al. Antimicrobial Peptide LL-37 Is Both a Substrate of Cathepsins S and K and a Selective Inhibitor of Cathepsin L. Biochemistry 2015, 54, 2785–2798. [Google Scholar] [CrossRef]
- Ueno, T.; Takahashi, K. A cathepsin L-specific inhibitor preferentially inhibits degradation of autophagosomal LC3 and GABARAP in HeLa and Huh-7 cells. Autophagy 2009, 5, 878–879. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Sever, S. Podocyte biology and pathogenesis of kidney disease. Annu. Rev. Med. 2013, 64, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Sever, S.; Chang, J.; Gu, C. Dynamin rings: Not just for fission. Traffic 2013, 14, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Cocchiaro, P.; De Pasquale, V.; Della Morte, R.; Tafuri, S.; Avallone, L.; Pizard, A.; Moles, A.; Pavone, L.M. The Multifaceted Role of the Lysosomal Protease Cathepsins in Kidney Disease. Front. Cell Dev. Biol. 2017, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, N.I.; Sharma, M.; Vassmer, D.; Chapman, B.; Paul, B.; Zhou, J.; Brantley, J.G.; Wallace, D.P.; Maser, R.L.; Vanden Heuvel, G.B. Acceleration of polycystic kidney disease progression in cpk mice carrying a deletion in the homeodomain protein Cux1. Am. J. Physiol. Ren. Physiol. 2008, 295, F1725–F1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcalay, N.I.; Vanden Heuvel, G.B. Regulation of cell proliferation and differentiation in the kidney. Front. Biosci. 2009, 14, 4978–4991. [Google Scholar] [CrossRef] [PubMed]
- Stypmann, J.; Glaser, K.; Roth, W.; Tobin, D.J.; Petermann, I.; Matthias, R.; Monnig, G.; Haverkamp, W.; Breithardt, G.; Schmahl, W.; et al. Dilated cardiomyopathy in mice deficient for the lysosomal cysteine peptidase cathepsin L. Proc. Natl. Acad. Sci. USA 2002, 99, 6234–6239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, I.; Mayer, C.; Stypmann, J.; Biniossek, M.L.; Tobin, D.J.; Engelen, M.A.; Dandekar, T.; Grune, T.; Schild, L.; Peters, C.; et al. Lysosomal, cytoskeletal, and metabolic alterations in cardiomyopathy of cathepsin L knockout mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1266–1268. [Google Scholar] [CrossRef] [PubMed]
- Spira, D.; Stypmann, J.; Tobin, D.J.; Petermann, I.; Mayer, C.; Hagemann, S.; Vasiljeva, O.; Gunther, T.; Schule, R.; Peters, C.; et al. Cell type-specific functions of the lysosomal protease cathepsin L in the heart. J. Biol. Chem. 2007, 282, 37045–37052. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Kumar, M.; Manchanda, M.; Singh, R.; Thakur, B.; Rani, N.; Arava, S.; Narang, R.; Arya, D.S.; Chauhan, S.S. Clinical significance of cathepsin L and cathepsin B in dilated cardiomyopathy. Mol. Cell. Biochem. 2017, 428, 139–147. [Google Scholar] [CrossRef]
- Tang, Q.; Cai, J.; Shen, D.; Bian, Z.; Yan, L.; Wang, Y.X.; Lan, J.; Zhuang, G.Q.; Ma, W.Z.; Wang, W. Lysosomal cysteine peptidase cathepsin L protects against cardiac hypertrophy through blocking AKT/GSK3beta signaling. J. Mol. Med. 2009, 87, 249–260. [Google Scholar] [CrossRef]
- Dalet-Fumeron, V.; Guinec, N.; Pagano, M. In vitro activation of pro-cathepsin B by three serine proteinases: Leucocyte elastase, cathepsin G, and the urokinase-type plasminogen activator. FEBS Lett. 1993, 332, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Felbor, U.; Dreier, L.; Bryant, R.A.; Ploegh, H.L.; Olsen, B.R.; Mothes, W. Secreted cathepsin L generates endostatin from collagen XVIII. EMBO J. 2000, 19, 1187–1194. [Google Scholar] [CrossRef]
- Everts, V.; Korper, W.; Hoeben, K.A.; Jansen, I.D.; Bromme, D.; Cleutjens, K.B.; Heeneman, S.; Peters, C.; Reinheckel, T.; Saftig, P.; et al. Osteoclastic bone degradation and the role of different cysteine proteinases and matrix metalloproteinases: Differences between calvaria and long bone. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2006, 21, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, E.; Dimmeler, S. Homing of progenitor cells to ischemic tissues. Antioxid. Redox Signal. 2011, 15, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, E.; Koyanagi, M.; Dimmeler, S. Enhancing the outcome of cell therapy for cardiac repair: Progress from bench to bedside and back. Circulation 2010, 121, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, E.; Urbich, C.; Dimmeler, S. Homing and engraftment of progenitor cells: A prerequisite for cell therapy. J. Mol. Cell. Cardiol. 2008, 45, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Dernbach, E.; Rossig, L.; Zeiher, A.M.; Dimmeler, S. High glucose reduces cathepsin L activity and impairs invasion of circulating progenitor cells. J. Mol. Cell. Cardiol. 2008, 45, 429–436. [Google Scholar] [CrossRef]
- Wartmann, T.; Mayerle, J.; Kahne, T.; Sahin-Toth, M.; Ruthenburger, M.; Matthias, R.; Kruse, A.; Reinheckel, T.; Peters, C.; Weiss, F.U.; et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology 2010, 138, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, X.; Fei, X.; Xia, L.; Qin, Z.; Liang, Z. Parkinson’s disease involves autophagy and abnormal distribution of cathepsin L. Neurosci. Lett. 2011, 489, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Koike, M.; Shibata, M.; Sasaki, M. Autophagic neuron death. Methods Enzymol. 2009, 453, 33–51. [Google Scholar]
- Ohkouchi, S.; Shibata, M.; Sasaki, M.; Koike, M.; Safig, P.; Peters, C.; Nagata, S.; Uchiyama, Y. Biogenesis and proteolytic processing of lysosomal DNase II. PLoS ONE 2013, 8, e59148. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Horie, S.; Nakamura, O.; Maruyama, K.; Takase, H.; Usui, Y.; Takeuchi, M.; Ishidoh, K.; Koike, M.; Uchiyama, Y.; et al. Acquisition of T regulatory function in cathepsin L-inhibited T cells by eye-derived CTLA-2alpha during inflammatory conditions. J. Immunol 2009, 183, 5013–5022. [Google Scholar] [CrossRef] [Green Version]
- Yasothornsrikul, S.; Greenbaum, D.; Medzihradszky, K.F.; Toneff, T.; Bundey, R.; Miller, R.; Schilling, B.; Petermann, I.; Dehnert, J.; Logvinova, A.; et al. Cathepsin L in secretory vesicles functions as a prohormone-processing enzyme for production of the enkephalin peptide neurotransmitter. Proc. Natl. Acad. Sci. USA 2003, 100, 9590–9595. [Google Scholar] [CrossRef] [Green Version]
- Schurigt, U.; Eilenstein, R.; Gajda, M.; Leipner, C.; Sevenich, L.; Reinheckel, T.; Peters, C.; Wiederanders, B.; Brauer, R. Decreased arthritis severity in cathepsin L-deficient mice is attributed to an impaired T helper cell compartment. Inflamm. Res. 2012, 61, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Hsing, L.C.; Kirk, E.A.; McMillen, T.S.; Hsiao, S.H.; Caldwell, M.; Houston, B.; Rudensky, A.Y.; LeBoeuf, R.C. Roles for cathepsins S, L, and B in insulitis and diabetes in the NOD mouse. J. Autoimmun. 2010, 34, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, B.J.; Lindholt, J.S.; Wang, J.; Cheng, X.; Shi, G.P. Plasma levels of cathepsins L, K, and V and risks of abdominal aortic aneurysms: A randomized population-based study. Atherosclerosis 2013, 230, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Rabaglino, M.B.; Wood, C.E.; Siemann, D.W. Cathepsin L in tumor angiogenesis and its therapeutic intervention by the small molecule inhibitor KGP94. Clin. Exp. Metastasis 2016, 33, 461–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Hanahan, D. Multiple roles for cysteine cathepsins in cancer. Cell Cycle 2004, 3, 1516–1619. [Google Scholar] [CrossRef]
- Gottesman, M.M. Transformation-dependent secretion of a low molecular weight protein by murine fibroblasts. Proc. Natl. Acad. Sci. USA 1978, 75, 2767–2771. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Turk, D.; Guncar, G. Lysosomal cysteine proteases (cathepsins): Promising drug targets. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.; Leonetti, F.; Greenbaum, D.C.; Lecaille, F.; Bogyo, M.; Bromme, D.; Ellman, J.A.; Craik, C.S. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 2006, 281, 12824–12832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishima, A.; Imai, Y.; Nomura, T.; Fujisawa, Y.; Yamamoto, Y.; Sugawara, T. The crystal structure of human cathepsin L complexed with E-64. FEBS Lett. 1997, 407, 47–50. [Google Scholar] [CrossRef]
- Chowdhury, S.F.; Sivaraman, J.; Wang, J.; Devanathan, G.; Lachance, P.; Qi, H.; Menard, R.; Lefebvre, J.; Konishi, Y.; Cygler, M.; et al. Design of noncovalent inhibitors of human cathepsin L. From the 96-residue proregion to optimized tripeptides. J. Med. Chem. 2002, 45, 5321–5329. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, R.T.; Sivaraman, J. Structural basis for reversible and irreversible inhibition of human cathepsin L by their respective dipeptidyl glyoxal and diazomethylketone inhibitors. J. Struct. Biol. 2011, 173, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Hardegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Halogen bonding at the active sites of human cathepsin L and MEK1 kinase: Efficient interactions in different environments. ChemMedChem 2011, 6, 2048–2054. [Google Scholar] [CrossRef]
- Giroud, M.; Dietzel, U.; Anselm, L.; Banner, D.; Kuglstatter, A.; Benz, J.; Blanc, J.B.; Gaufreteau, D.; Liu, H.; Lin, X.; et al. Repurposing a Library of Human Cathepsin L Ligands: Identification of Macrocyclic Lactams as Potent Rhodesain and Trypanosoma brucei Inhibitors. J. Med. Chem. 2018, 61, 3350–3369. [Google Scholar] [CrossRef]
- Barrett, A.J.; Kembhavi, A.A.; Brown, M.A.; Kirschke, H.; Knight, C.G.; Tamai, M.; Hanada, K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 1982, 201, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Goursalin, B.J.; Lachance, P.; Bonneau, P.R.; Storer, A.C.; Kirschke, H.; Broemme, D. E-64 Analogs as Inhibitors of Cathepsin L and Cathepsin S: Importance of the S2-P2 Interactions for Potency and Selectivity. Bioorg. Chem. 1994, 22, 227–241. [Google Scholar] [CrossRef]
- Katunuma, N.; Murata, E.; Kakegawa, H.; Matsui, A.; Tsuzuki, H.; Tsuge, H.; Turk, D.; Turk, V.; Fukushima, M.; Tada, Y.; et al. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999, 458, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Tsuge, H.; Nishimura, T.; Tada, Y.; Asao, T.; Turk, D.; Turk, V.; Katunuma, N. Inhibition mechanism of cathepsin L-specific inhibitors based on the crystal structure of papain-CLIK148 complex. Biochem. Biophys. Res. Commun. 1999, 266, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Crawford, C.; Mason, R.W.; Wikstrom, P.; Shaw, E. The design of peptidyldiazomethane inhibitors to distinguish between the cysteine proteinases calpain II, cathepsin L and cathepsin B. Biochem. J. 1988, 253, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.; Demuth, H.U.; Barth, A. N,O-Diacylhydroxylamines as enzyme-activated inhibitors for serine proteases. Die Pharm. 1983, 38, 249. [Google Scholar]
- Demuth, H.U.; Baumgrass, R.; Schaper, C.; Fischer, G.; Barth, A. Dipeptidylpeptidase IV--inactivation with N-peptidyl-O-aroyl hydroxylamines. J. Enzym. Inhib. 1988, 2, 129–142. [Google Scholar] [CrossRef]
- Demuth, H.U.; Schönlein, C.; Barth, A. Potent and selective inactivation of proteinases with N-peptidyl-O-acylhydroxylamines. Biochim. Biophys. Acta 1989, 996, 19–22. [Google Scholar] [CrossRef]
- Demuth, H.U.; Neumann, U.; Barth, A. Reactions between dipeptidyl peptidase IV and diacyl hydroxylamines: Mechanistic investigations. J. Enzym. Inhib. 1989, 2, 239–248. [Google Scholar] [CrossRef]
- Brömme, D.; Schierhorn, A.; Kirschke, H.; Wiederanders, B.; Barth, A.; Fittkau, S.; Demuth, H.U. Potent and selective inactivation of cysteine proteinases with N-peptidyl-O-acyl hydroxylamines. Biochem. J. 1989, 263, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Broemme, D.; Neumann, U.; Kirschke, H.; Demuth, H.-U. Novel N-peptidyl-O-acyl hydroxamates: Selective inhibitors of cysteine proteinases. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1993, 1202, 271. [Google Scholar] [CrossRef]
- Brömme, D.; Kirschke, H. N-peptidyl-O-carbamoyl amino acid hydroxamates: Irreversible inhibitors for the study of the S2′ specificity of cysteine proteinases. FEBS Lett. 1993, 322, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Gour-Salin, B.J.; Lachance, P.; Magny, M.C.; Plouffe, C.; Menard, R.; Storer, A.C. E64 [trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane] analogues as inhibitors of cysteine proteinases: Investigation of S2 subsite interactions. Biochem. J. 1994, 299, 389–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krantz, A. Peptidyl (acyloxy)methanes as quiescent affinity labels for cysteine proteinases. Methods Enzymol. 1994, 244, 656. [Google Scholar] [PubMed]
- Torkar, A.; Lenarcic, B.; Lah, T.; Dive, V.; Devel, L. Identification of new peptide amides as selective cathepsin L inhibitors: The first step towards selective irreversible inhibitors? Bioorg. Med. Chem. Lett. 2013, 23, 2968–2973. [Google Scholar] [CrossRef] [PubMed]
- Martichonok, V.; Plouffe, C.; Storer, A.C.; Menard, R.; Jones, J.B. Aziridine Analogs of [[trans-(Epoxysuccinyl)-L-leucyl]amino]-4-guanidinobutane (E-64) as Inhibitors of Cysteine Proteases. J. Med. Chem. 1995, 38, 3078. [Google Scholar] [CrossRef] [PubMed]
- Schirmeister, T. New Peptidic Cysteine Protease Inhibitors Derived from the Electrophilic α-Amino Acid Aziridine-2,3-dicarboxylic Acid. J. Med. Chem. 1999, 42, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Mendieta, L.; Pico, A.; Tarrago, T.; Teixido, M.; Castillo, M.; Rafecas, L.; Moyano, A.; Giralt, E. Novel peptidyl aryl vinyl sulfones as highly potent and selective inhibitors of cathepsins L and B. ChemMedChem 2010, 5, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Dana, D.; De, S.; Rathod, P.; Davalos, A.R.; Novoa, D.A.; Paroly, S.; Torres, V.M.; Afzal, N.; Lankalapalli, R.S.; Rotenberg, S.A.; et al. Development of a highly potent, selective, and cell-active inhibitor of cysteine cathepsin L-A hybrid design approach. Chem. Commun. 2014, 50, 10875–10878. [Google Scholar] [CrossRef]
- Boudreau, P.D.; Miller, B.W.; McCall, L.-I.; Almaliti, J.; Reher, R.; Hirata, K.; Le, T.; Siqueira-Neto, J.L.; Hook, V.; Gerwick, W.H. Design of Gallinamide A Analogs as Potent Inhibitors of the Cysteine Proteases Human Cathepsin L and Trypanosoma cruzi Cruzain. J. Med. Chem. 2019, 62, 9026–9044. [Google Scholar] [CrossRef]
- Lynas, J.F.; Hawthorne, S.J.; Walker, B. Development of peptidyl α-keto-β-aldehydes as new inhibitors of cathepsin L—Comparisons of potency and selectivity profiles with cathepsin B. Bioorg. Med. Chem. Lett. 2000, 10, 1771–1773. [Google Scholar] [CrossRef]
- Marquis, R.W.; James, I.; Zeng, J.; Trout, R.E.; Thompson, S.; Rahman, A.; Yamashita, D.S.; Xie, R.; Ru, Y.; Gress, C.J.; et al. Azepanone-based inhibitors of human cathepsin L. J. Med. Chem. 2005, 48, 6870–6878. [Google Scholar] [CrossRef]
- Hardegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Systematic investigation of halogen bonding in protein-ligand interactions. Angew. Chem. Int. Ed. Engl. 2011, 50, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Giroud, M.; Harder, M.; Kuhn, B.; Haap, W.; Trapp, N.; Schweizer, W.B.; Schirmeister, T.; Diederich, F. Fluorine Scan of Inhibitors of the Cysteine Protease Human Cathepsin L: Dipolar and Quadrupolar Effects in the pi-Stacking of Fluorinated Phenyl Rings on Peptide Amide Bonds. ChemMedChem 2017, 12, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Tichý, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.; Giroud, M.; Schirmeister, T.; Abel, R.; et al. Prospective Evaluation of Free Energy Calculations for the Prioritization of Cathepsin L Inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Parker, E.N.; Odutola, S.O.; Wang, Y.; Strecker, T.E.; Mukherjee, R.; Shi, Z.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Synthesis and biological evaluation of a water-soluble phosphate prodrug salt and structural analogues of KGP94, a lead inhibitor of cathepsin L. Bioorg. Med. Chem. Lett. 2017, 27, 1304–1310. [Google Scholar] [CrossRef]
- Parker, E.N.; Song, J.; Kishore Kumar, G.D.; Odutola, S.O.; Chavarria, G.E.; Charlton-Sevcik, A.K.; Strecker, T.E.; Barnes, A.L.; Sudhan, D.R.; Wittenborn, T.R.; et al. Synthesis and biochemical evaluation of benzoylbenzophenone thiosemicarbazone analogues as potent and selective inhibitors of cathepsin L. Bioorg. Med. Chem. 2015, 23, 6974–6992. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.D.K.; Chavarria, G.E.; Charlton-Sevcik, A.K.; Yoo, G.K.; Song, J.; Strecker, T.E.; Siim, B.G.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Functionalized benzophenone, thiophene, pyridine, and fluorene thiosemicarbazone derivatives as inhibitors of cathepsin L. Bioorg. Med. Chem. Lett. 2010, 20, 6610–6615. [Google Scholar] [CrossRef]
- Kishore Kumar, G.D.; Chavarria, G.E.; Charlton-Sevcik, A.K.; Arispe, W.M.; Macdonough, M.T.; Strecker, T.E.; Chen, S.E.; Siim, B.G.; Chaplin, D.J.; Trawick, M.L.; et al. Design, synthesis, and biological evaluation of potent thiosemicarbazone based cathepsin L inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1415–1419. [Google Scholar] [CrossRef]
- Shenoy, R.T.; Chowdhury, S.F.; Kumar, S.; Joseph, L.; Purisima, E.O.; Sivaraman, J. A Combined Crystallographic and Molecular Dynamics Study of Cathepsin L Retrobinding Inhibitors. J. Med. Chem. 2009, 52, 6335–6346. [Google Scholar] [CrossRef]
- Myers, M.C.; Shah, P.P.; Diamond, S.L.; Huryn, D.M.; Smith, A.B., 3rd. Identification and synthesis of a unique thiocarbazate cathepsin L inhibitor. Bioorg. Med. Chem. Lett. 2008, 18, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.C.; Shah, P.P.; Beavers, M.P.; Napper, A.D.; Diamond, S.L.; Smith, A.B., 3rd; Huryn, D.M. Design, synthesis, and evaluation of inhibitors of cathepsin L: Exploiting a unique thiocarbazate chemotype. Bioorganic Med. Chem. Chem. Lett. 2008, 18, 3646–3651. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.P.; Myers, M.C.; Beavers, M.P.; Purvis, J.E.; Jing, H.; Grieser, H.J.; Sharlow, E.R.; Napper, A.D.; Huryn, D.M.; Cooperman, B.S.; et al. Kinetic characterization and molecular docking of a novel, potent, and selective slow-binding inhibitor of human cathepsin L. Mol. Pharmacol. 2008, 74, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.P.; Wang, T.; Kaletsky, R.L.; Myers, M.C.; Purvis, J.E.; Jing, H.; Huryn, D.M.; Greenbaum, D.C.; Smith, A.B., 3rd; Bates, P.; et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 2010, 78, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Vicik, R.; Helten, H.; Schirmeister, T.; Engels, B. Rational design of aziridine-containing cysteine protease inhibitors with improved potency: Studies on inhibition mechanism. ChemMedChem 2006, 1, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Bromme, D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [Google Scholar] [CrossRef] [PubMed]
- Bromme, D.; Klaus, J.L.; Okamoto, K.; Rasnick, D.; Palmer, J.T. Peptidyl vinyl sulphones: A new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem. J. 1996, 315, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Reddick, J.J.; Cheng, J.M.; Roush, W.R. Relative rates of Michael reactions of 2 ‘-(phenethyl)thiol with vinyl sulfones, vinyl sulfonate esters, and vinyl sulfonamides relevant to vinyl sulfonyl cysteine protease inhibitors. Org. Lett. 2003, 5, 1967–1970. [Google Scholar] [CrossRef] [PubMed]
- Roush, W.R.; Gwaltney, S.L.; Cheng, J.M.; Scheidt, K.A.; McKerrow, J.H.; Hansell, E. Vinyl sulfonate esters and vinyl sulfonamides: Potent, irreversible inhibitors of cysteine proteases. J. Am. Chem. Soc. 1998, 120, 10994–10995. [Google Scholar] [CrossRef]
- Miller, B.; Friedman, A.J.; Choi, H.; Hogan, J.; McCammon, J.A.; Hook, V.; Gerwick, W.H. The marine cyanobacterial metabolite gallinamide A is a potent and selective inhibitor of human cathepsin L. J. Nat. Prod. 2014, 77, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.-T.; Sigeizumi, S.; Yamaguchi, K.; Sugimoto, K.; Kobori, T.; Tsuji, T.; Kondo, K. Peptidyl aldehyde derivatives as potent and selective inhibitors of cathepsin L. Bioorg. Med. Chem. Lett. 1995, 5, 1501. [Google Scholar] [CrossRef]
- Woo, J.T.; Yamaguchi, K.; Hayama, T.; Kobori, T.; Sigeizumi, S.; Sugimoto, K.; Kondo, K.; Tsuji, T.; Ohba, Y.; Tagami, K.; et al. Suppressive effect of N-(benzyloxycarbonyl)-L-phenylalanyl-L-tyrosinal on bone resorption in vitro and in vivo. Eur. J. Pharmacol. 1996, 300, 131–135. [Google Scholar] [CrossRef]
- Marquis, R.W.; Ru, Y.; LoCastro, S.M.; Zeng, J.; Yamashita, D.S.; Oh, H.J.; Erhard, K.F.; Davis, L.D.; Tomaszek, T.A.; Tew, D.; et al. Azepanone-based inhibitors of human and rat cathepsin K. J. Med. Chem. 2001, 44, 1380–1395. [Google Scholar] [CrossRef] [PubMed]
- Kerns, J.K.; Nie, H.; Bondinell, W.; Widdowson, K.L.; Yamashita, D.S.; Rahman, A.; Podolin, P.L.; Carpenter, D.C.; Jin, Q.; Riflade, B.; et al. Azepanone-based inhibitors of human cathepsin S: Optimization of selectivity via the P2 substituent. Bioorg. Med. Chem. Lett. 2011, 21, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong le, T.; Falgueyret, J.P.; Kimmel, D.B.; Lamontagne, S.; Leger, S.; LeRiche, T.; et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.A.; McCrea, J.B.; Witter, R.; Zajic, S.; Stoch, S.A. Clinical and translational pharmacology of the cathepsin K inhibitor odanacatib studied for osteoporosis. Br. J. Clin. Pharmacol. 2019, 85, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
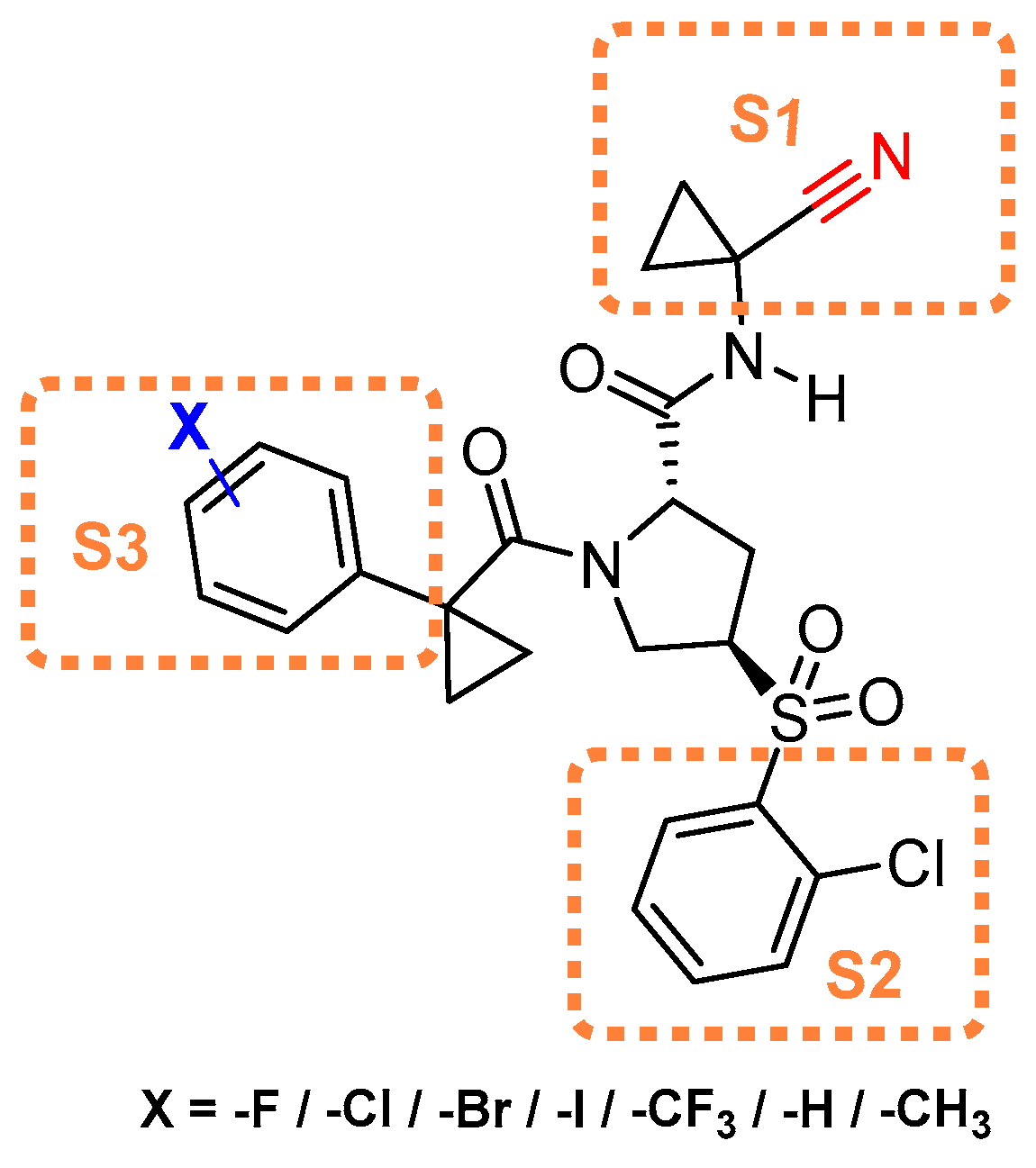
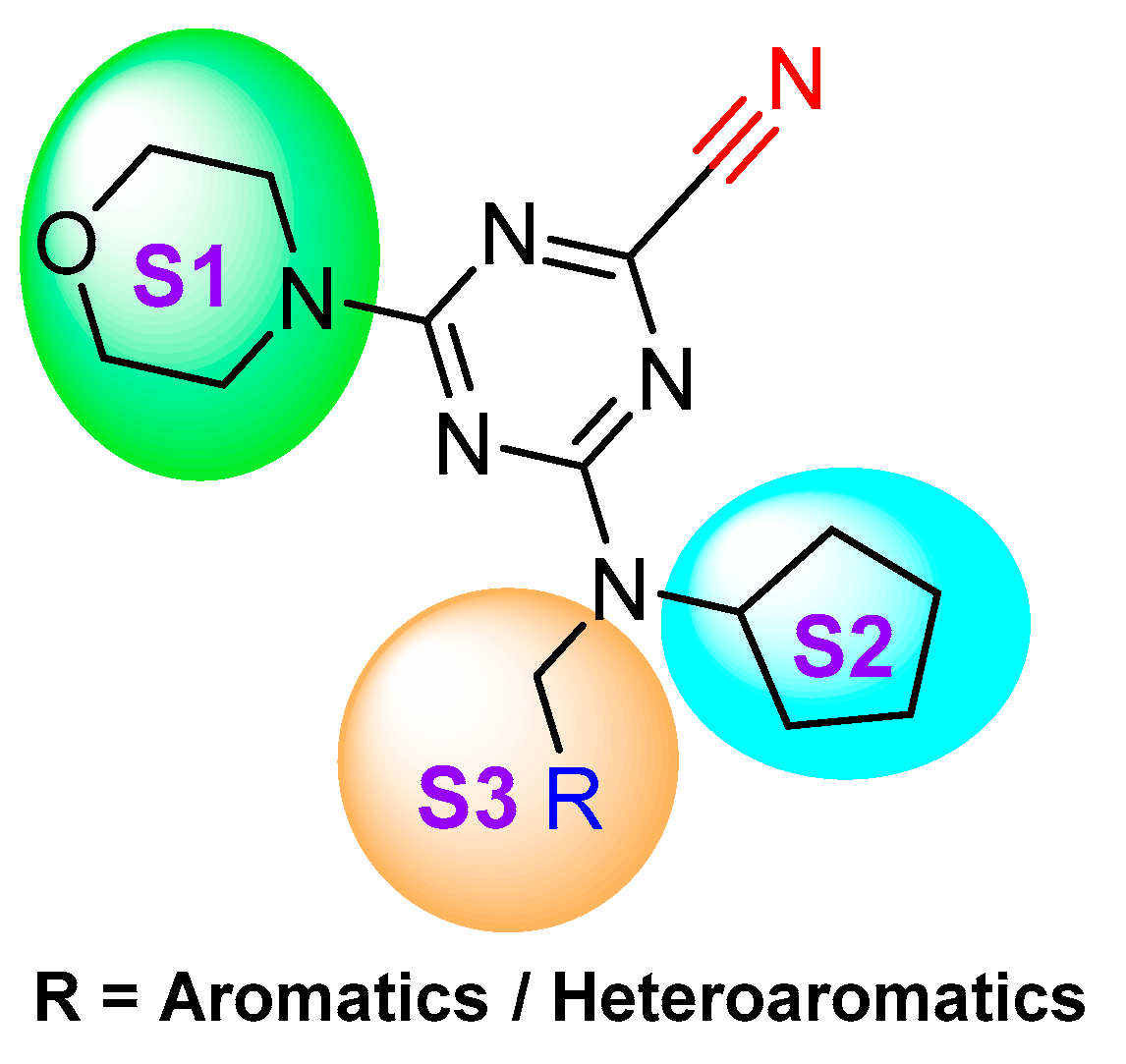
- Giroud, M.; Ivkovic, J.; Martignoni, M.; Fleuti, M.; Trapp, N.; Haap, W.; Kuglstatter, A.; Benz, J.; Kuhn, B.; Schirmeister, T.; et al. Inhibition of the Cysteine Protease Human Cathepsin L by Triazine Nitriles: Amide⋅⋅⋅Heteroarene π-Stacking Interactions and Chalcogen Bonding in the S3 Pocket. ChemMedChem 2017, 12, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Falgueyret, J.P.; Oballa, R.M.; Okamoto, O.; Wesolowski, G.; Aubin, Y.; Rydzewski, R.M.; Prasit, P.; Riendeau, D.; Rodan, S.B.; Percival, M.D. Novel, nonpeptidic cyanamides as potent and reversible inhibitors of human cathepsins K and L. J. Med. Chem. 2001, 44, 94–104. [Google Scholar] [CrossRef] [PubMed]
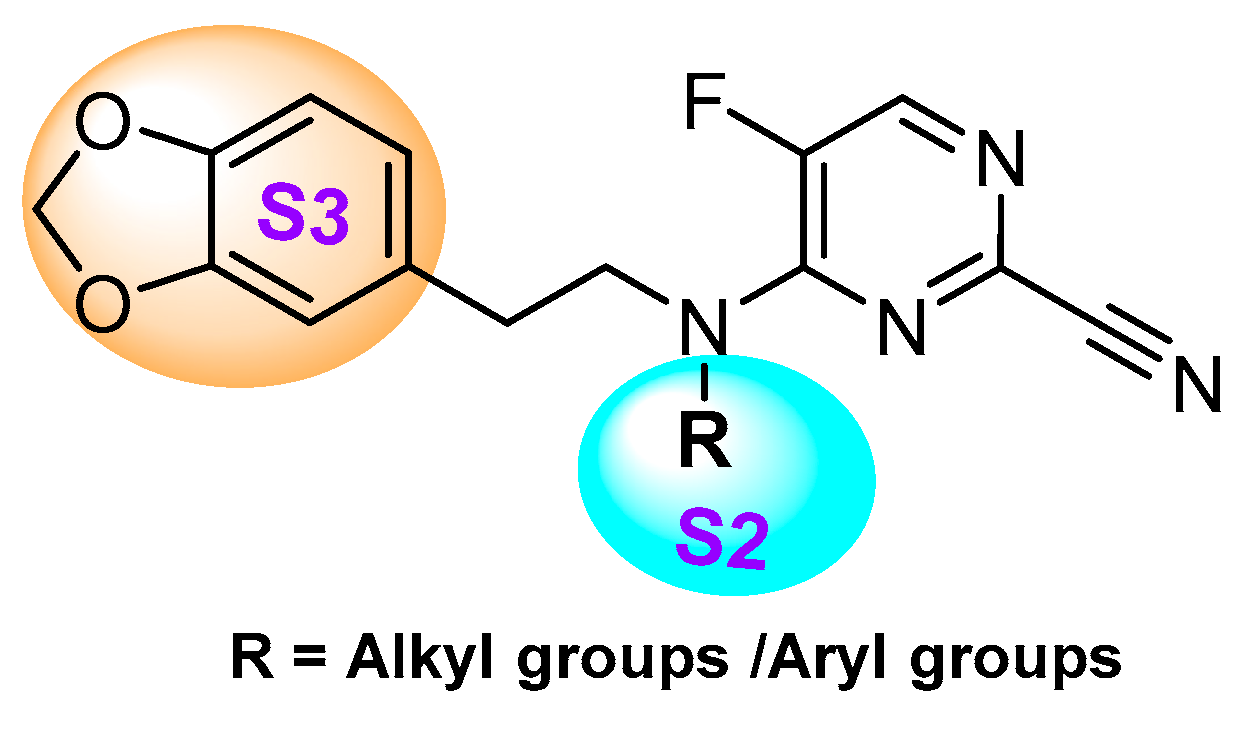
- Asaad, N.; Bethel, P.A.; Coulson, M.D.; Dawson, J.E.; Ford, S.J.; Gerhardt, S.; Grist, M.; Hamlin, G.A.; James, M.J.; Jones, E.V.; et al. Dipeptidyl nitrile inhibitors of Cathepsin L. Bioorg. Med. Chem. Lett. 2009, 19, 4280–4283. [Google Scholar] [CrossRef]
- Mallari, J.P.; Shelat, A.; Kosinski, A.; Caffrey, C.R.; Connelly, M.; Zhu, F.; McKerrow, J.H.; Guy, R.K. Discovery of trypanocidal thiosemicarbazone inhibitors of rhodesain and TbcatB. Bioorg. Med. Chem. Lett. 2008, 18, 2883–2885. [Google Scholar] [CrossRef] [Green Version]
- Magrath, J.; Abeles, R.H. Cysteine protease inhibition by azapeptide esters. J. Med. Chem. 1992, 35, 4279. [Google Scholar] [CrossRef]
- Beavers, M.P.; Myers, M.C.; Shah, P.P.; Purvis, J.E.; Diamond, S.L.; Cooperman, B.S.; Huryn, D.M.; Smith, A.B., 3rd. Molecular docking of cathepsin L inhibitors in the binding site of papain. J. Chem. Inf. Mode. 2008, 48, 1464–1472. [Google Scholar] [CrossRef]
- Jean, D.; Rousselet, N.; Frade, R. Cathepsin L expression is up-regulated by hypoxia in human melanoma cells: Role of its 5′-untranslated region. Biochem. J. 2008, 413, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro-IL-1beta Synthesis and NLRP3-Mediated IL-1beta Activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogyo, M. Finding enzymes that are actively involved in cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 2379–2380. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Zhou, B.; Liang, F.; Wang, W.Q.; Huang, Z.; Zhang, Z.Y. Activity-based probes for protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 2004, 101, 7943–7948. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Zhou, B.; Liang, F.; Yang, H.; Wang, W.Q.; Zhang, Z.Y. Global analysis of protein tyrosine phosphatase activity with ultra-sensitive fluorescent probes. J. Proteome Res. 2006, 5, 1898–1905. [Google Scholar] [CrossRef]
- Yim, J.J.; Tholen, M.; Klaassen, A.; Sorger, J.; Bogyo, M. Optimization of a Protease Activated Probe for Optical Surgical Navigation. Mol. Pharm. 2018, 15, 750–758. [Google Scholar] [CrossRef]
- Chan, C.H.; Liesenfeld, L.F.; Ferreiro-Neira, I.; Cusack, J.C., Jr. Preclinical Evaluation of Cathepsin-Based Fluorescent Imaging System for Cytoreductive Surgery. Ann. Surg. Oncol. 2017, 24, 931–938. [Google Scholar] [CrossRef]
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38. [Google Scholar] [CrossRef]
- Edgington, L.E.; Verdoes, M.; Bogyo, M. Functional imaging of proteases: Recent advances in the design and application of substrate-based and activity-based probes. Curr. Opin. Chem. Biol. 2011, 15, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Sanman, L.E.; Bogyo, M. Activity-based profiling of proteases. Annu. Rev. Biochem. 2014, 83, 249–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, K.; Carroll, R.; Steiner, D.F. Identification of a 31,500 molecular weight islet cell protease as cathepsin B. Proc. Natl. Acad. Sci. USA 1983, 80, 3245–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, K.; Hutton, J.C.; Steiner, D.F. Cathepsin B-related proteases in the insulin secretory granule. J. Biol. Chem. 1984, 259, 6041. [Google Scholar] [PubMed]
- Mason, R.W.; Wilcox, D.; Wikstrom, P.; Shaw, E.N. The identification of active forms of cysteine proteinases in Kirsten-virus-transformed mouse fibroblasts by use of a specific radiolabeled inhibitor. Biochem. J. 1989, 257, 125. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.W.; Bartholomew, L.T.; Hardwick, B.S. The use of benzyloxycarbonyl [125I]iodotyrosylalanyldiazomethane as a probe for active cysteine proteinases in human tissues. Biochem. J. 1989, 263, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, D.; Mason, R.W. Inhibition of cysteine proteinases in lysosomes and whole cells. Biochem. J. 1992, 285, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Xing, R.; Addington, A.K.; Mason, R.W. Quantification of cathepsins B and L in cells. Biochem. J. 1998, 332, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Gelhaus, C.; Vicik, R.; Hilgenfeld, R.; Schmidt, C.L.; Leippe, M.; Schirmeister, T. Synthesis and antiplasmodial activity of a cysteine protease-inhibiting biotinylated aziridine-2,3-dicarboxylate. Biol. Chem. 2004, 385, 435–438. [Google Scholar] [CrossRef]
- Vicik, R.; Busemann, M.; Gelhaus, C.; Stiefl, N.; Scheiber, J.; Schmitz, W.; Schulz, F.; Mladenovic, M.; Engels, B.; Leippe, M.; et al. Aziridide-based inhibitors of cathepsin L: Synthesis, inhibition activity, and docking studies. ChemMedChem 2006, 1, 1126–1141. [Google Scholar] [CrossRef]
- Torkar, A.; Bregant, S.; Devel, L.; Novinec, M.; Lenarcic, B.; Lah, T.; Dive, V. A novel photoaffinity-based probe for selective detection of cathepsin L active form. Chembiochem Eur. J. Chem. Biol. 2012, 13, 2616–2621. [Google Scholar] [CrossRef]
- Na, Z.; Li, L.; Uttamchandani, M.; Yao, S.Q. Microarray-guided discovery of two-photon (2P) small molecule probes for live-cell imaging of cysteinyl cathepsin activities. Chem. Commun. 2012, 48, 7304–7306. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Rut, W.; Vizovisek, M.; Groborz, K.; Kasperkiewicz, P.; Finlay, D.; Vuori, K.; Turk, D.; Turk, B.; Salvesen, G.S.; et al. Selective imaging of cathepsin L in breast cancer by fluorescent activity-based probes. Chem. Sci. 2018, 9, 2113–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dana, D.; Garcia, J.; Bhuiyan, A.I.; Rathod, P.; Joo, L.; Novoa, D.A.; Paroly, S.; Fath, K.R.; Chang, E.J.; Pathak, S.K. Cell penetrable, clickable and tagless activity-based probe of human cathepsin L. Bioorg. Chem. 2019, 85, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Groborz, K.; Rut, W.; Pore, M.; Snipas, S.J.; Vizovisek, M.; Turk, B.; Kuhn, P.; Drag, M.; Salvesen, G.S. The Activome: Multiplexed probing of activity of proteolytic enzymes using mass cytometry-compatible activity-based probes (TOF-probes). bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Blum, G.; von Degenfeld, G.; Merchant, M.J.; Blau, H.M.; Bogyo, M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat. Chem. Biol. 2007, 3, 668–677. [Google Scholar] [CrossRef]
- Blum, G.; Mullins, S.R.; Keren, K.; Fonovic, M.; Jedeszko, C.; Rice, M.J.; Sloane, B.F.; Bogyo, M. Dynamic imaging of protease activity with fluorescently quenched activity-based probes. Nat. Chem. Biol. 2005, 1, 203–209. [Google Scholar] [CrossRef]
- Vogel, A.M.; Gerster, T. Expression of a zebrafish Cathepsin L gene in anterior mesendoderm and hatching gland. Dev. Genes Evol. 1997, 206, 477–479. [Google Scholar] [CrossRef]
- Crosier, P.S.; Bardsley, A.; Horsfield, J.A.; Krassowska, A.K.; Lavallie, E.R.; Collins-Racie, L.A.; Postlethwait, J.H.; Yan, Y.L.; McCoy, J.M.; Crosier, K.E. In situ hybridization screen in zebrafish for the selection of genes encoding secreted proteins. Dev. Dyn. 2001, 222, 637–644. [Google Scholar] [CrossRef]
- Gardiner, M.R.; Daggett, D.F.; Zon, L.I.; Perkins, A.C. Zebrafish KLF4 is essential for anterior mesendoderm/pre-polster differentiation and hatching. Dev. Dyn. 2005, 234, 992–996. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Altman, Y.; D’Angelo, M.; Salvesen, G.S.; Drag, M. Toolbox of Fluorescent Probes for Parallel Imaging Reveals Uneven Location of Serine Proteases in Neutrophils. J. Am. Chem. Soc. 2017, 139, 10115–10125. [Google Scholar] [CrossRef]
- Choi, K.Y.; Swierczewska, M.; Lee, S.; Chen, X. Protease-activated drug development. Theranostics 2012, 2, 156–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, M.A.; Moya, I.A.; Bhilocha, S.; McMillan, C.C.; Vigliarolo, B.G.; Zehbe, I.; Phenix, C.P. Prodrug-inspired probes selective to cathepsin B over other cysteine cathepsins. J. Med. Chem. 2014, 57, 6092–6104. [Google Scholar] [CrossRef] [PubMed]
- Weinstain, R.; Segal, E.; Satchi-Fainaro, R.; Shabat, D. Real-time monitoring of drug release. Chem. Commun. 2010, 46, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Ueki, N.; Lee, S.; Sampson, N.S.; Hayman, M.J. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat. Commun. 2013, 4, 2735. [Google Scholar] [CrossRef] [Green Version]
- Ljunggren, A.; Redzynia, I.; Alvarez-Fernandez, M.; Abrahamson, M.; Mort, J.S.; Krupa, J.C.; Jaskólski, M.; Bujacz, G. Crystal Structure of the Parasite Protease Inhibitor Chagasin in Complex with a Host Target Cysteine Protease. J. Mol. Boil. 2007, 371, 137–153. [Google Scholar] [CrossRef] [Green Version]
- Fairhead, M.; Johnson, K.A.; Kowatz, T.; McMahon, S.A.; Carter, L.G.; Oke, M.; Liu, H.; Naismith, J.H.; van der Walle, C.F. Crystal structure and silica condensing activities of silicatein alpha-cathepsin L chimeras. Chem. Commun. 2008, 15, 1765–1767. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.F.; Joseph, L.; Kumar, S.; Tulsidas, S.R.; Bhat, S.; Ziomek, E.; Ménard, R.; Sivaraman, J.; Purisima, E.O. Exploring Inhibitor Binding at the S′ Subsites of Cathepsin L. J. Med. Chem. 2008, 51, 1361–1368. [Google Scholar] [CrossRef]
- Adams-Cioaba, M.A.; Krupa, J.C.; Xu, C.; Mort, J.S.; Min, J. Structural basis for the recognition and cleavage of histone H3 by cathepsin L. Nat. Commun. 2011, 2, 197. [Google Scholar] [CrossRef] [Green Version]
- Renko, M.; Türk, D. Unreduced Cathepsin L in Complex with Stefin A. 2010. Available online: https://www.rcsb.org/structure/3KSE (accessed on 4 February 2020).
- Ehmke, V.; Winkler, E.; Banner, D.W.; Haap, W.; Schweizer, W.B.; Rottmann, M.; Kaiser, M.; Freymond, C.; Schirmeister, T.; Diederich, F. Optimization of Triazine Nitriles as Rhodesain Inhibitors: Structure-Activity Relationships, Bioisosteric Imidazopyridine Nitriles, and X-ray Crystal Structure Analysis with Human Cathepsin L. ChemMedChem 2013, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Banner, D.; Benz, J.; Stihle, M.; Kuglstatter, A. CATHEPSIN L IN COMPLEX WITH (2S,4R)-4-(2-Chloro-4-methoxy-benzenesulfonyl)-1-[3-(5-chloro-pyridin-2-yl)-azetidine-3-carbonyl]-pyrrolidine-2-carboxylic acid (1-cyano-cyclopropyl)-amide. 2016. Available online: https://www.rcsb.org/structure/5F02 (accessed on 4 February 2020). [CrossRef]
- Sosnowski, P.; Turk, D. Caught in the act: The crystal structure of cleaved cathepsin L bound to the active site of Cathepsin L. FEBS Lett. 2016, 590, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
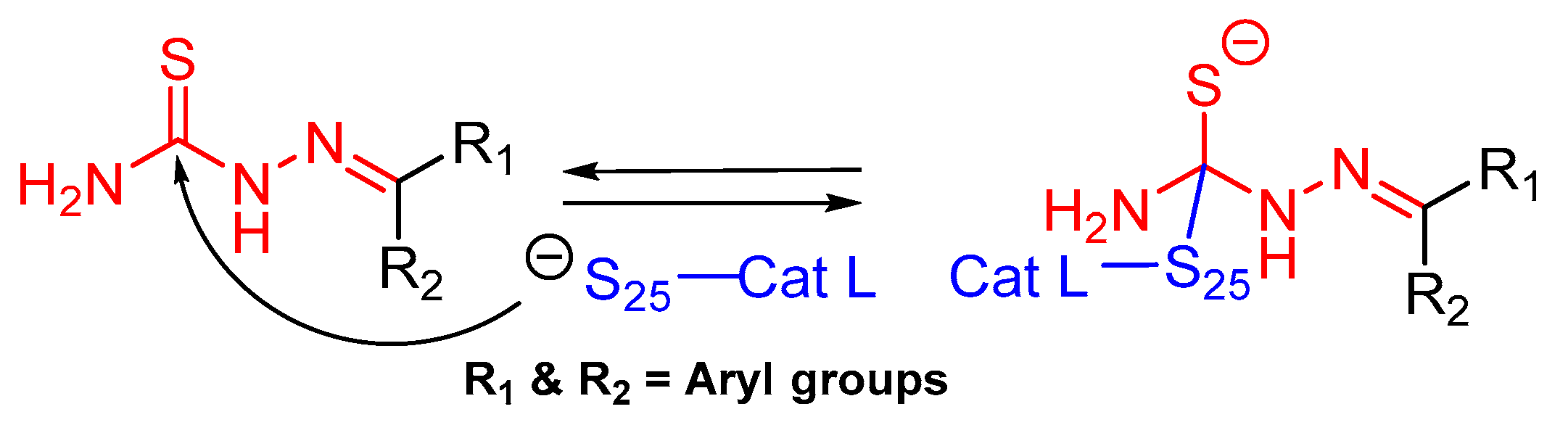{kind=link}
{kind=link}
{kind=link}
| Inhibitory Ligand | Inhibition Constant (Ki) |
|---|---|
| Recombinant Cathepsin L Propeptide | 0.088 nM [47] (pH = 5.5) |
| Cystatin A | 1.3 nM [48,49] |
| Cystatin B | 0.23 nM [48,49] |
| Cystatin C | <0.005 nM [50] |
| Cystatin D | 18 nM [51] |
| P41 of MHC Class II Molecule | 2 pM [52] |
| L-Kinenogen | 1.7 pM [53,54] |
| Cystatin F | 0.31 nM [55] |
| Sialostatin L | 95 pM [45] |
| Antimicrobial Peptide LL-37 | 150 nM [56] |
| # | Chemical Structure | Inhibitor Class | Inhibitor Efficacy (Cathepsin L) | Selectivity Factor # | Mechanism of Inhibition (Demonstrated Utilities) | References |
|---|---|---|---|---|---|---|
| 1 | 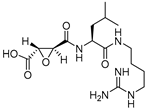 | Epoxy Succinyl | 2nd Order (M−1 s−1) 96250 | Cat B: 1.1 Cat H: 24 | Irreversible (In vitro) | Barrett et al., 1982 [99] |
| 2 | 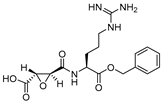 | Epoxy Succinyl | 2nd Order (103 M−1 s−1) 73 | Cat S: 89 | Irreversible (In vitro) | Gour-Salin et al., 1994 [111] |
| 3 | 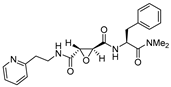 | Epoxy Succinyl | 10−6 M (100% inhibition) | Cat B: NI Cat S: 3 Cat K: NI | Irreversible (In vitro cellular) | Katunuma et al.,1999 [101] |
| 4 | 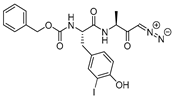 | Peptidyldiazomethane | 2nd Order (M−1 s−1) 1128000 | Cat B: 41 Calpain: NI | Irreversible (In vitro) | Crawford et al., 1988 [103] |
| 5 | 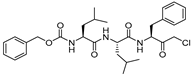 | Peptidylchloromethane (chloromethylketone) | 2nd Order (M−1 s−1) 21500000 | Cat B: 113 Calpain: <200 | Irreversible (In vitro) | Crawford et al., 1988 [103] |
| 6 | 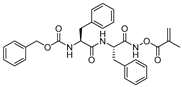 | N-peptidyl-O-acyl hydroxylamines | 2nd Order (M−1 s−1) 1222000 | Cat B: 436 Cat S: 58 Cat H: ND | Irreversible (In vitro) | Bromme et al., 1989 [108] |
| 7 |  | N-peptidyl-O-carbamoyl amino acid hydroxamates | 2nd Order (M−1 s−1) 931800 | Cat B: 59 Cat S: 10 Papain: 184 | Irreversible (In vitro) | Bromme et al., 1993 [110] |
| 8 | 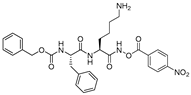 | N-peptidyl-O-acyl hydroxamates | 2nd Order (M−1 s−1) 3538000 | Cat S: 7 Cat B: 101 Cat H: 4655 SP a: II | Irreversible (In vitro) | Bromme et al., 1993 [109] |
| 9 |  | Peptidyl (acyloxy)methanes | 2nd Order (M−1 s−1) 10700000 | Cat S: 7 Cat B: 4 | Irreversible (In vitro) | Krantz, 1994 [112] |
| 10 | 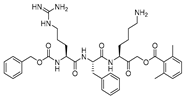 | Peptidyl acyloxymethyl ketone | 2nd Order (M−1 s−1) 875000 | Cat B: 11 | Irreversible (In vitro) | Torkar et al., 2013 [113] |
| 11 | 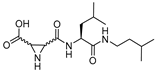 | Peptidyl aziridine | 2nd Order (M−1 s−1) (L) ~ 20000 (D) ~ 110000 | Cat B ~ 2(L) Cat B ~ 2(D) | Irreversible (In vitro) | Martichonok et al., 1995 [114] |
| 12 | 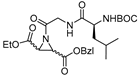 | N-acylated aziridines (type I) | 2nd Order (M−1 min−1) (S,S+R,R): 3227 | Cat B: 13 Papain: 22 | Irreversible (In vitro) | Schirmeister, 1999 [115] |
| 13 | 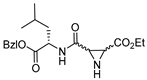 | N-unsubstituted aziridines (type II) | 2nd Order (M−1 min−1) (R,R): 16261 (S,S): 3130 | Cat B: 10(R,R) Cat B: 76(S,S) | Irreversible (In vitro) | Schirmeister, 1999 [115] |
| 14 |  | N-acylated bispeptidyl aziridines (type III) | 2nd Order (M−1 min−1) (R,R): 5896 (S,S): 1210 | Cat B: 3(R,R) Cat B: 3(S,S) | Irreversible (In vitro) | Schirmeister, 1999 [115] |
| 15 | 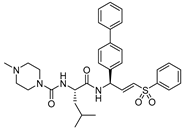 | Peptidyl aryl vinyl sulfones | IC50 (nM) 2.6 | Cat B: 404 | Irreversible (In vitro) | Mendieta et al., 2010 [116] |
| 16 |  | Peptidyl arylvinylsulfonate | 2nd Order (M−1 s−1) 4300000 | Cat K: 100 Cat B: 44000 Cat S: 13 Cat H: NI Cat D: NI Cat G: NI hPTP1B: NI Trypsin: NI | Irreversible (In vitro cellular) | Dana et al., 2014 [117] |
| 17 | 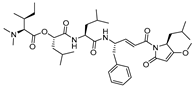 | Gallinamide A-analog | 2nd Order (M−1 s−1) 8730000 | ND | Irreversible (In vitro cellular) | Boudreau et al., 2019 [118] |
| 18 |  | Peptidyl aldehydes | IC50 (nM) 0.6 | Cat B: 357 | Covalent and reversible (In vitro) | Lynas et al., 2000 [119] |
| 19 | 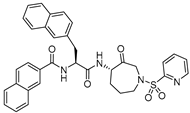 | Azepanone-based Inhibitors | Ki,app (nM) 0.43 | Cat K>20000 Cat S: 36 Cat B: 349 | Covalent and reversible (In vitro) | Marquis et al., 2005 [120] |
| 20 |  | Nitrile group containing inhibitor | IC50 (nM) 22 | NA | Covalent and reversible (In vitro) | Hardegger et al., 2011 [121] |
| 21 | 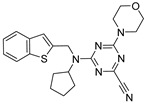 | Nitrile group containing inhibitor | Ki (nM) 4 | ND | Covalent and reversible (In vitro) | Giroud et al., 2017 [122] |
| 22 |  | Nitrile group containing inhibitor | Ki (nM) 12 | ND | Covalent and reversible (In vitro) | Kuhn et al., 2017 [123] |
| 23 | 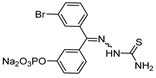 | Thiosemicarbazone | IC50 (nM) 189 (active inhibitor) b | Cat B: >50 | Covalent and reversible (In vitro cellular) | Parker et al., 2017 [124], Parker et al., 2015 [125] Kumar et al., 2010 [126] Kumar et al., 2010 [127] |
| 24 | 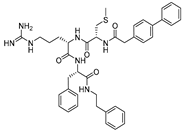 | Cat L propeptide mimic | IC50 (nM) 19 | Cat K: 310 Cat B: 210 | Reversible (In vitro) | Chowdhury et al., 2002 [95] Shenoy et al., 2009 [128] |
| 25 | 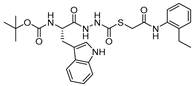 | Thiocarbazate | IC50 (nM) * 1 | Cat V: 11 Cat S: 14 Cat B: 50 Cat K: 137 | Reversible (In vitro) | Myers et al., 2008 [129,130,131] |
| 26 |  | Oxocarbazate | IC50 (nM) * 0.4 | Cat B: 714 | Reversible (In vitro cellular) | Myers et al., 2008 [130] Shah et al., 2010 [132] |
| 27 |  | Aza-peptide | IC50 (µM) 3.0 | ND | Reversible (In vitro) | Myers et al., 2008 [130] |
| # | Chemical Structure | Probe Class | Efficacy (Cathepsin L) | Selectivity Factor | Mechanism of Probe Action (Demonstrated Utilities) | References |
|---|---|---|---|---|---|---|
| P1 |  | Radio-labelled | 2nd order rate constant (M−1s−1) 240000 | Cat B: 23 | Covalent and irreversible (In vitro Cellular) | Mason et al., 1989 [165] Wilcox et al., 1992 [166] |
| P2 |  | Radio-labelled | 2nd order rate constant (M−1s−1) 60900 | Cat B: 2.4 Cat S: NI # | Covalent and irreversible (In vitro Cellular) | Xing et al., 1998 [167] |
| P3 | 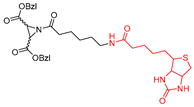 | Affinity-labeled | Inhibitor Constant(µM) (S,S) 1.4 | Cat B: 36 Papain: 4 | Covalent and irreversible (In vitro) | Gelhaus et al., 2004 [168] Vicik et al., 2006 [169] |
| P4 | 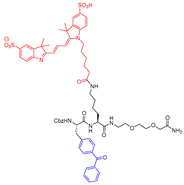 | Photoaffinity-based | Inhibitor Constant (µM) 3.6 (able to detect picomolar amount of protein) | Cat B: 9 Cat K: 3 Cat S: 0.3 Cat V: 0.15 [P4 did show remarkable selectivity in vivo and not in vitro] | Covalent and irreversible (In vitro) | Torkar et al., 2012 [170] |
| 5 |  | Two-photon FRET-based | N.D | N.D | Reversible (In vitro Cellular) | Na et al., 2012 [171] |
| 6 | 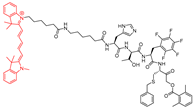 | Fluorescent | Cathepsin L specific (some degree of labelling was seen for cat V and B) | Cat V: 847 Cat B: 413 Cat S: 1431 Cat K: >1600 | Covalent and irreversible (In vitro Cellular) | Poreba et al., 2018 [172] |
| 7 | 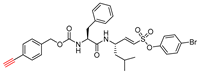 | Clickable and tagless | 2nd Order (M−1 s−1) 430000 | Cat B:9556 Cat K: 29 Cat H: NI Cat D: NI Cat G: NI hPTP1B: NI Trypsin: NI | Covalent and irreversible (In vitro Cellular) | Dana et al., 2019 [173] |
| 8 | 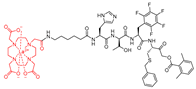 | TOF-based | 2nd Order (M−1 s−1) 229300 (mix-Gd) 232500 (159-Tb) 227000 (175-Lu) | Selectivity > 150-fold: cat B, V, and S N.I: cat K and Legumain | Covalent and irreversible (In vitro Cellular) | Poreba et al., 2019 [174] |
| PDB Entry | Method | Resolution (Å) | Reference |
|---|---|---|---|
| 1CJL | X-ray | 2.20 | [42] |
| 1CS8 | X-ray | 1.80 | [42] |
| 1ICF | X-ray | 2.00 | [42] |
| 1MHW | X-ray | 1.90 | [95] |
| 2NQD | X-ray | 1.75 | [185] |
| 2VHS | X-ray | 1.50 | [186] |
| 2XU1 | X-ray | 1.45 | [121] |
| 2XU3 | X-ray | 0.90 | [121] |
| 2XU4 | X-ray | 1.12 | [121] |
| 2XU5 | X-ray | 1.60 | [121] |
| 2YJ2 | X-ray | 1.15 | [97] |
| 2YJ8 | X-ray | 1.30 | [97] |
| 2YJ9 | X-ray | 1.35 | [97] |
| 2YJB | X-ray | 1.40 | [97] |
| 2YJC | X-ray | 1.14 | [97] |
| 3BC3 | X-ray | 2.20 | [187] |
| 3H89 | X-ray | 2.50 | [128] |
| 3H8B | X-ray | 1.80 | [128] |
| 3H8C | X-ray | 2.50 | [128] |
| 3HHA | X-ray | 1.27 | [147] |
| 3HWN | X-ray | 2.33 | [147] |
| 3IV2 | X-ray | 2.20 | [188] |
| 3K24 | X-ray | 2.50 | [188] |
| 3KSE | X-ray | 1.71 | [189] |
| 3OF8 | X-ray | 2.20 | [96] |
| 3OF9 | X-ray | 1.76 | [96] |
| 4AXL | X-ray | 1.92 | [190] |
| 4AXM | X-ray | 2.80 | [190] |
| 5F02 | X-ray | 1.43 | [191] |
| 5I4H | X-ray | 1.42 | [192] |
| 5MAE | X-ray | 1.00 | [145] |
| 5MAJ | X-ray | 1.00 | [145] |
| 5MQY | X-ray | 1.13 | [123] |
| 6EZP | X-ray | 1.37 | [98] |
| 6EZX | X-ray | 2.34 | [98] |
| 6F06 | X-ray | 2.02 | [98] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dana, D.; Pathak, S.K. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 2020, 25, 698. https://doi.org/10.3390/molecules25030698
Dana D, Pathak SK. A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules. 2020; 25(3):698. https://doi.org/10.3390/molecules25030698
Chicago/Turabian StyleDana, Dibyendu, and Sanjai K. Pathak. 2020. "A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L" Molecules 25, no. 3: 698. https://doi.org/10.3390/molecules25030698
APA StyleDana, D., & Pathak, S. K. (2020). A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules, 25(3), 698. https://doi.org/10.3390/molecules25030698