
GPR6 Structural Insights: Homology Model Construction and Docking Studies

 , ,
, ,
Abstract
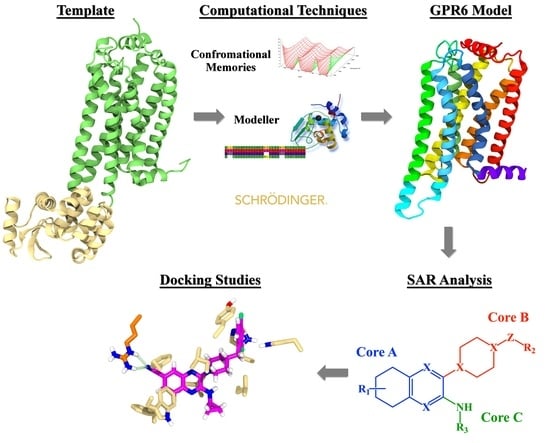
1. Introduction
2. Results and Discussion
2.1. GPR6 Inactive-State Model Development
2.1.1. TMH1
2.1.2. TMH6
2.1.3. TMH7
2.1.4. N-Terminus
2.1.5. EC2
2.1.6. Ionic Lock and Toggle Switch
2.1.7. Inactive State Model Minimization
2.1.8. Molecular Dynamics
2.2. Docking Study and SAR
2.2.1. Structure-Activity Relationship (SAR) Analysis for Pyrazine Analogs
- (1)
- C6 Cycloalkyl or C6 heterocycloalkyl as piperidinyl-, piperazinyl-, or morpholino were explored as linkers.
- (2)
- Methylene, oxygen, carbonyl, and flouromethylene were tolerated at the Z position.
- (3)
- Aromaticity is required at R2, with a phenyl ring preferred over heterocyclic rings or bicyclic rings. Electron-withdrawing substituents such as fluoro-, chloro- or electron-donating group substituents as methoxy on the aromatic ring are required for higher potency.
2.2.2. Conformational Analysis of Selected GPR6 Inverse Agonists
2.2.3. Docking Studies
2.3. Proposed Mutagenesis for Future Perspectives
3. Materials and Methods
3.1. Amino Acid Numbering System
3.2. Receptor Model Development
3.3. Conformational Memories Technique for Calculating TMH Conformations
3.4. Modeling Loops and N- and C-Termini Conformations
3.5. Receptor Minimization
3.6. Molecular Dynamics (MD)
3.7. Conformer Analysis of Pyrazine Analogs
3.8. Electrostatic Potential Map Calculation
3.9. Docking of Ligands
3.10. Calculation of Ligand/Receptor Interaction Energy
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GPCR | G-Protein Coupled Receptor |
| cAMP | cyclic AMP |
| CHO | Chinese hamster ovary |
| S1P1 | Sphingosine-1-phosphate receptor 1 |
| CM | Conformational Memories |
| TMH | Transmembrane helix |
| EC | Extracellular |
| IC | Intracellular |
| CB1 | Cannabinoid receptor type 1 |
| MD | Molecular dynamics |
| RMSD | root-mean-square deviation |
| SAR | Structure-Activity Relationship |
| MBER | Assisted Model Building with Energy Refinement |
References
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef]
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef]
- Song, Z.H.; Young, W.S.; Brownstein, M.J.; Bonner, T.I. Molecular cloning of a novel candidate G protein-coupled receptor from rat brain. FEBS Lett. 1994, 351, 375–379. [Google Scholar] [CrossRef]
- Eggerickx, D.; Denef, J.F.; Labbe, O.; Hayashi, Y.; Refetoff, S.; Vassart, G.; Parmentier, M.; Libert, F. Molecular cloning of an orphan G-protein-coupled receptor that constitutively activates adenylate cyclase. Biochem. J. 1995, 309, 837–843. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Fredriksson, R.; Schiöth, H.B. The Repertoire of G-Protein–Coupled Receptors in Fully Sequenced Genomes. Mol. Pharmacol. 2005, 67, 1414–1425. [Google Scholar] [CrossRef]
- Uhlenbrock, K.; Gassenhuber, H.; Kostenis, E. Sphingosine 1-phosphate is a ligand of the human gpr3, gpr6 and gpr12 family of constitutively active G protein-coupled receptors. Cell Signal. 2002, 14, 941–953. [Google Scholar] [CrossRef]
- Ignatov, A.; Lintzel, J.; Kreienkamp, H.J.; Schaller, H.C. Sphingosine-1-phosphate is a high-affinity ligand for the G protein-coupled receptor GPR6 from mouse and induces intracellular Ca2+ release by activating the sphingosine-kinase pathway. Biochem. Biophys. Res. Commun. 2003, 311, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Battey, J.; Benson, H.E.; Benya, R.V.; Bonner, T.I.; Davenport, A.P.; Eguchi, S.; Harmar, A.; Holliday, N.; Jensen, R.T.; et al. Class A Orphans (version 2019.5) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2019. [Google Scholar] [CrossRef]
- Shrader, S.H.; Song, Z.-H. Discovery of endogenous inverse agonists for G protein-coupled receptor 6. Biochem. Biophys. Res. Commun. 2019, 522, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Laun, A.S.; Song, Z.H. GPR3 and GPR6, novel molecular targets for cannabidiol. Biochem. Biophys. Res. Commun. 2017, 490, 17–21. [Google Scholar] [CrossRef]
- Laun, A.S.; Shrader, S.H.; Song, Z.-H. Novel inverse agonists for the orphan G protein-coupled receptor 6. Heliyon 2018, 4, 933. [Google Scholar] [CrossRef] [PubMed]
- Breivogel, C.S.; McPartland, J.M.; Parekh, B. Investigation of non-CB1, non-CB2 WIN55212-2-sensitive G-protein-coupled receptors in the brains of mammals, birds, and amphibians. J. Recept. Signal Transduct. 2018, 38, 316–326. [Google Scholar] [CrossRef]
- Morales, P.; Isawi, I.; Reggio, P.H. Towards a better understanding of the cannabinoid-related orphan receptors GPR3, GPR6, and GPR12. Drug Metab. Rev. 2018, 50, 74–93. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive activity among orphan class-A G protein coupled receptors. PLoS ONE 2015, 10, e0138463. [Google Scholar] [CrossRef]
- Oeckl, P.; Hengerer, B.; Ferger, B. G-protein coupled receptor 6 deficiency alters striatal dopamine and cAMP concentrations and reduces dyskinesia in a mouse model of Parkinson’s disease. Exp. Neurol. 2014, 257, 1–9. [Google Scholar] [CrossRef]
- Oeckl, P.; Ferger, B. Increased susceptibility of G-protein coupled receptor 6 deficient mice to MPTP neurotoxicity. Neuroscience 2016, 337, 218–223. [Google Scholar] [CrossRef]
- Hitchcock, S.; Monenschein, H.; Reichard, H.; Sun, H.; Kikuchi, S.; Macklin, T.; Hopkins, M. Quinoxaline Derivatives as GPR6 Modulators. WO2014028479A1, 20 February 2014. [Google Scholar]
- Hitchcock, S.; Hopkins, M.; Kikuchi, S.; Monenschein, H.; Reichard, H.; Sun, H.; Macklin, T. Pyridopyrazines Modulators of GPR6. WO2015123505A1, 20 August 2015. [Google Scholar]
- Brown, J.; Hitchcock, S.; Hopkins, M.; Kikuchi, S.; Monenschein, H.; Reichard, H.; Schleicher, K.; Sun, H.; Macklin, T. Tetrahydropyridopyrazines as Modulators of GPR6. WO2015095728A1, 25 June 2015. [Google Scholar]
- Adams, M.E.; Brown, J.W.; Hitchcock, S.; Kikuchi, S.; Lam, B.; Monenschein, H.; Reichard, H.; Sun, H. Pyrazines as Modulators of GPR6. WO2015123533A1, 20 August 2015. [Google Scholar]
- Green, J.; Hopkines, M.; Jones, B.; Kiryanov, A.; Kuehler, J.; Monenschein, H.; Murphy, S.; Nixey, T.; Sun, H. Piperidinyl-and Piperazinyl-Substituted Heteroaromatic Carboxamides as Modulators of GPR6. WO2018183145A1, 4 October 2018. [Google Scholar]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Le Gras, S.; Keime, C.; Anthony, A.; Lotz, C.; De Longprez, L.; Brouillet, E.; Cassel, J.-C.; Boutillier, A.-L.; Merienne, K. Altered enhancer transcription underlies Huntington’s disease striatal transcriptional signature. Sci. Rep. 2017, 7, 42875. [Google Scholar] [CrossRef]
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-induced LRP1B and GPR6 Proteins Expressed Early in Alzheimer Disease Mouse Models, Are Essential for the C1q-mediated Protection against Amyloid-β Neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef]
- Lobo, M.K.; Cui, Y.; Ostlund, S.B.; Balleine, B.W.; Yang, X.W. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nat. Neurosci. 2007, 10, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Beeley, N.R.A.; Behan, D.P.; Chalmers, D.T.; Menzaghi, F.; Strah-Pleynet, S. Small Molecule Modulators of G Protein-Coupled Receptor Six. WO2001062765A2, 30 August 2001. [Google Scholar]
- Hanson, M.A.; Roth, C.B.; Jo, E.; Griffith, M.T.; Scott, F.L.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.M.; Schuerer, S.C.; et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012, 335, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, F.; Weinstein, H. Conformational Memories and the Exploration of Biologically Relevant Peptide Conformations: An Illustration for the Gonadotropin-Releasing Hormone. J. Am. Chem. Soc. 1996, 118, 5580–5589. [Google Scholar] [CrossRef]
- Guarnieri, F.; Wilson, S.R. Conformational memories and a simulated annealing program that learns: Application to LTB4. J. Comput. Chem. 1995, 16, 648–653. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Deupi, X.; Olivella, M.; Haaksma, E.E.J.; Pardo, L. Serine and threonine residues bend α-helices in the χ1= g- conformation. Biophys. J. 2000, 79, 2754–2760. [Google Scholar] [CrossRef]
- Deupi, X.; Olivella, M.; Govaerts, C.; Ballesteros, J.A.; Campillo, M.; Pardo, L. Ser and Thr residues modulate the conformation of pro-kinked transmembrane alpha-helices. Biophys. J. 2004, 86, 105–115. [Google Scholar] [CrossRef]
- Deupi, X.; Dolker, N.; Luz Lopez-Rodriguez, M.; Campillo, M.; Ballesteros, J.; Pardo, L. Structural Models of Class A G Protein-Coupled Receptors as a Tool for Drug Design: Insights on Transmembrane Bundle Plasticity. Curr. Top. Med. Chem. 2007, 7, 991–998. [Google Scholar] [CrossRef]
- Curran, A.R.; Engelman, D.M. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 2003, 13, 412–417. [Google Scholar] [CrossRef]
- Hall, S.E.; Roberts, K.; Vaidehi, N. Position of helical kinks in membrane protein crystal structures and the accuracy of computational prediction. J. Mol. Graph. Model. 2009, 27, 944–950. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Analysis and refinement of criteria for predicting the structure and relative orientations of transmembranal helical domains. Biophys. J. 1992, 62, 107–109. [Google Scholar] [CrossRef][Green Version]
- Sansom, M.S.; Weinstein, H. Hinges, swivels and switches: The role of prolines in signalling via transmembrane α-helices. Trends Pharmacol. Sci. 2000, 21, 445–451. [Google Scholar] [CrossRef]
- Cordes, F.S.; Bright, J.N.; Sansom, M.S.P. Proline-induced distortions of transmembrane helices. J. Mol. Biol. 2002, 323, 951–960. [Google Scholar] [CrossRef]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef]
- Visiers, I.; Braunheim, B.B.; Weinstein, H. Prokink: A protocol for numerical evaluation of helix distortions by proline. Protein Eng. 2000, 13, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef]
- Wheatley, M. Glycosylation of G-protein-coupled receptors for hormones central to normal reproductive functioning: Its occurrence and role. Hum. Reprod. Update 1999, 5, 356–364. [Google Scholar] [CrossRef]
- Hurst, D.P.; Grossfield, A.; Lynch, D.L.; Feller, S.; Romo, T.D.; Gawrisch, K.; Pitman, M.C.; Reggio, P.H. A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-coupled Receptor. J. Biol. Chem. 2010, 285, 17954–17964. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. Transduction of Receptor Signals by ß-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Ranjan, R.; Dwivedi, H.; Baidya, M.; Kumar, M.; Shukla, A.K. Novel Structural Insights into GPCR–β-Arrestin Interaction and Signaling. Trends Cell Biol. 2017, 27, 851–862. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Jensen, A.D.; Liapakis, G.; Rasmussen, S.G.F.; Shi, L.; Gether, U.; Javitch, J.A. Activation of the β 2 -Adrenergic Receptor Involves Disruption of an Ionic Lock between the Cytoplasmic Ends of Transmembrane Segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. [Google Scholar] [CrossRef]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef]
- Rahmeh, R.; Damian, M.; Cottet, M.; Orcel, H.; Mendre, C.; Durroux, T.; Sharma, K.S.; Durand, G.; Pucci, B.; Trinquet, E.; et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 6733–6738. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Methods for the Development of In Silico GPCR Models. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 593, pp. 405–448. [Google Scholar]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.S.; Ha, S.; Ball, R.G.; Tsou, N.N.; Castonguay, L.A.; Doss, G.A.; Fong, T.M.; Shen, C.P.; Jing, C.X.; Goulet, M.T.; et al. Conformational analysis and receptor docking of N-[(1S,2S)-3-(4-chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2-{[5-(trifluoromethyl) pyridin-2-yl]oxy}propanamide (taranabant, MK-0364), a novel, acyclic cannabinoid-1 receptor inverse agonist. J. Med. Chem. 2008, 51, 2108–2114. [Google Scholar] [CrossRef]
- Kotsikorou, E.; Navas, F.; Roche, M.J.; Gilliam, A.F.; Thomas, B.F.; Seltzman, H.H.; Kumar, P.; Song, Z.H.; Hurst, D.P.; Lynch, D.L.; et al. The importance of hydrogen bonding and aromatic stacking to the affinity and efficacy of cannabinoid receptor CB2 antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3, 3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carbo. J. Med. Chem. 2013, 56, 6593–6612. [Google Scholar] [CrossRef][Green Version]
- Lingerfelt, M.A.; Zhao, P.; Sharir, H.P.; Hurst, D.P.; Reggio, P.H.; Abood, M.E. Identification of Crucial Amino Acid Residues Involved in Agonist Signaling at the GPR55 Receptor. Biochemistry 2017, 56, 473–486. [Google Scholar] [CrossRef]
- McAllister, S.D.; Hurst, D.P.; Barnett-Norris, J.; Lynch, D.; Reggio, P.H.; Abood, M.E. Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: The importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J. Biol. Chem. 2004, 279, 48024–48037. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Academic Press: Cambridge, MA, USA, 1995; pp. 366–428. [Google Scholar]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoubi, R.P.; Hurst, D.H.; Reggio, P. Structural Insights from Recent CB1 X-Ray Crystal Structures. In Recent Advances in Cannabinoid Research; IntechOpen: London, UK, 2018. [Google Scholar]
- Reggio, P.H. Computational methods in drug design: Modeling G protein-coupled receptor monomers, dimers, and oligomers. AAPS J. 2006, 8, E322–E336. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Buck, M.; Bouguet-Bonnet, S.; Pastor, R.W.; MacKerell, A.D. Importance of the CMAP Correction to the CHARMM22 Protein Force Field: Dynamics of Hen Lysozyme. Biophys. J. 2006, 90, L36–L38. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5.6.1–5.6.30. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Gutiérrez-De-Terán, H.; Massink, A.; Rodríguez, D.; Liu, W.; Han, G.W.; Joseph, J.S.; Katritch, I.; Heitman, L.H.; Xia, L.; Ijzerman, A.P.; et al. The role of a sodium ion binding site in the allosteric modulation of the A2A adenosine G protein-coupled receptor. Structure 2013, 21, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Fenalti, G.; Abola, E.E.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Lerouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Caliman, A.D.; McCammon, J.A. Allosteric effects of sodium ion binding on activation of the M3 muscarinic G-protein-coupled receptor. Biophys. J. 2015, 108, 1796–1806. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Marcu, J.; Shore, D.M.; Kapur, A.; Trznadel, M.; Makriyannis, A.; Reggio, P.H.; Abood, M.E. Novel insights into CB1 cannabinoid receptor signaling: A key interaction identified between the extracellular-3 loop and transmembrane helix 2. J. Pharmacol. Exp. Ther. 2013, 345, 189–197. [Google Scholar] [CrossRef]
Sample Availability: Not available. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Helix | Hinge Residue | Bend Angle (deg) | Wobble Angle (deg) | Face Shift (deg) |
|---|---|---|---|---|
| TMH1 | T1.44 | 13.0 | 172.4 | 3.0 |
| TMH6 | T6.43 | 9.9 | 113.0 | 5.8 |
| TMH7 | P7.41 | 12.7 | −20.1 | 31.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isawi, I.H.; Morales, P.; Sotudeh, N.; Hurst, D.P.; Lynch, D.L.; Reggio, P.H. GPR6 Structural Insights: Homology Model Construction and Docking Studies. Molecules 2020, 25, 725. https://doi.org/10.3390/molecules25030725
Isawi IH, Morales P, Sotudeh N, Hurst DP, Lynch DL, Reggio PH. GPR6 Structural Insights: Homology Model Construction and Docking Studies. Molecules. 2020; 25(3):725. https://doi.org/10.3390/molecules25030725
Chicago/Turabian StyleIsawi, Israa H., Paula Morales, Noori Sotudeh, Dow P. Hurst, Diane L. Lynch, and Patricia H. Reggio. 2020. "GPR6 Structural Insights: Homology Model Construction and Docking Studies" Molecules 25, no. 3: 725. https://doi.org/10.3390/molecules25030725
APA StyleIsawi, I. H., Morales, P., Sotudeh, N., Hurst, D. P., Lynch, D. L., & Reggio, P. H. (2020). GPR6 Structural Insights: Homology Model Construction and Docking Studies. Molecules, 25(3), 725. https://doi.org/10.3390/molecules25030725