
Towards the Preparation of Stable Cyclic Amino(ylide)Carbenes


Abstract
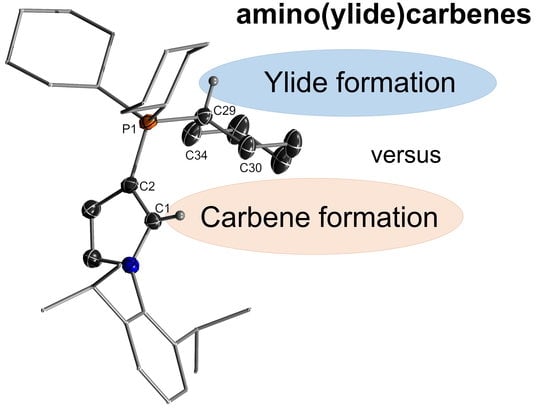
:
1. Introduction
2. Results
2.1. Attempted Synthesis of CAYCs with Trialklyl Onium Groups
2.2. Computational Studies
3. Discussion and Conclusions
4. Materials and Methods
4.1. Crystal Structure Determination
4.2. Computational Studies
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef]
- Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
- Naumann, S.; Dove, A.P. N-Heterocyclic carbenes for metal-freepolymerization catalysis: An update. Polym. Int. 2016, 65, 16–27. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-Heterocyclic Carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- Marion, N.; Díez-González, S.; Nolan, S.P. N-Heterocyclic Carbenes as Organocatalysts. Angew. Chem. Int. Ed. 2007, 46, 2988–3000. [Google Scholar] [CrossRef]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018, 118, 9678–9842. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Soleilhavoup, M.; Bertrand, G. Carbene-stabilized main group radicals and radical ions. Chem. Sci. 2013, 4, 3020–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, R.; Jazzar, R.; Bertrand, G. A crystalline monosubstituted carbene. Nat. Chem. 2018, 10, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J.; Goerlich, J.R.; Marshall, W.J. A Stable Thiazol-2-ylidene and Its Dimer. Liebigs Ann. -Recl. 1997, 1997, 365–374. [Google Scholar] [CrossRef]
- Martin, D.; Baceiredo, A.; Gornitzka, H.; Schoeller, W.W.; Bertrand, G. A stable P-heterocyclic carbene. Angew. Chem. Int. Ed. Engl. 2005, 44, 1700–1703. [Google Scholar] [CrossRef]
- Alcarazo, M.; Suárez, R.M.; Goddard, R.; Fürstner, A.A. A New Class of Singlet Carbene Ligands. Chem. Eur. J. 2010, 16, 9746–9749. [Google Scholar] [CrossRef]
- Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Stable cyclic (alkyl)(amino)carbenes as rigid or flexible, bulky, electron-rich ligands for transition-metal catalysts: A quaternary carbon atom makes the difference. Angew. Chem. Int. Ed. Engl. 2005, 44, 5705–5709. [Google Scholar] [CrossRef] [Green Version]
- Lavallo, V.; Mafhouz, J.; Canac, Y.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Synthesis, reactivity, and ligand properties of a stable alkyl carbene. J. Am. Chem. Soc. 2004, 126, 8670–8671. [Google Scholar] [CrossRef]
- Frey, G.D.; Lavallo, V.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science 2007, 316, 439–441. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, B.; Silvi, B.; Dewhurst, R.D.; Phukan, A.K. Theoretical Strategies Toward Stabilization of SingletRemote N-Heterocyclic Carbenes. J. Comput. Chem. 2016, 37, 1484–1490. [Google Scholar] [CrossRef]
- Andrada, D.M.; Holzmann, N.; Hamadi, T.; Frenking, G. Direct estimate of the internal π-donation to the carbene centre within N-heterocyclic carbenes and related molecules. Beilstein J. Org. Chem. 2015, 11, 2727–2736. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, B.; Phukan, A.K. Moving toward Ylide-Stabilized Carbenes. Chem. Eur. J. 2015, 21, 11603–11609. [Google Scholar] [CrossRef] [PubMed]
- Fekete, Á.; Nyulászi, L. Phosphorus stabilized carbenes: Theoretical predictions. J. Organomet. Chem. 2002, 643–644, 278–284. [Google Scholar] [CrossRef]
- Bharadwaz, P.; Chetia, P.; Phukan, A.K. Electronicand Ligand Properties of Skeletally Substituted Cyclic(Alkyl)(Amino)Carbenes(CAACs) and Their Reactivity towardsSmall Molecule Activation: A Theoretical Study. Chem. Eur. J. 2017, 23, 9926–9936. [Google Scholar] [CrossRef]
- Nakafuji, S.-Y.; Kobayashi, J.; Kawashima, T. Generation and coordinating properties of a carbene bearing a phosphorus ylide: An intensely electron-donating ligand. Angew. Chem. Int. Ed. Engl. 2008, 47, 1141–1144. [Google Scholar] [CrossRef]
- Fürstner, A.; Alcarazo, M.; Radkowski, K.; Lehmann, C.W. Carbenes stabilized by ylides: Pushing the limits. Angew. Chem. Int. Ed. Engl. 2008, 47, 8302–8306. [Google Scholar] [CrossRef] [Green Version]
- Asay, M.; Donnadieu, B.; Baceiredo, A.; Soleilhavoup, M.; Bertrand, G. Cyclic (amino)bis(ylide)carbene as an anionic bidentate ligand for transition-metal complexes. Inorg. Chem. 2008, 47, 3949–3951. [Google Scholar] [CrossRef] [Green Version]
- Asay, M.; Inoue, S.; Driess, M. Aromatic ylide-stabilized carbocyclic silylene. Angew. Chem. Int. Ed. Engl. 2011, 50, 9589–9592. [Google Scholar] [CrossRef]
- Alvarado-Beltran, I.; Baceiredo, A.; Saffon-Merceron, N.; Branchadell, V.; Kato, T. Cyclic Amino(Ylide) Silylene: A Stable Heterocyclic Silylene with Strongly Electron-Donating Character. Angew. Chem. Int. Ed. Engl. 2016, 55, 16141–16144. [Google Scholar] [CrossRef]
- Mohapatra, C.; Scharf, L.; Scherpf, T.; Mallick, B.; Feichtner, K.-S.; Schwarz, C.; Gessner, V.H. Isolation of a Diylide-Stabilized Stannylene and Germylene: Enhanced Donor Strength through Coplanar Lone Pair Alignment. Angew. Chem. Int. Ed. Engl. 2019. [Google Scholar] [CrossRef]
- Hartke, K.; Teuber, D.; Gerber, H.-D. Indole- and pyrrole-sulfonium ylides. Tetrahedron 1988, 44, 3261–3270. [Google Scholar] [CrossRef]
- Kobayashi, J.; Nakafuji, S.-Y.; Yatabe, A.; Kawashima, T. A novel ylide-stabilized carbene; formation and electron donating ability of an amino(sulfur-ylide)carbene. Chem. Commun. 2008, 6233–6235. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, A.G.; Schulz, T.; Torborg, C.; Spannenberg, A.; Neumann, H.; Beller, M. Palladium-catalyzed hydroxylation of aryl halides under ambient conditions. Angew. Chem. Int. Ed. Engl. 2009, 48, 7595–7599. [Google Scholar] [CrossRef]
- Meng, G.; Kakalis, L.; Nolan, S.P.; Szostak, M. A simple 1H NMR method for determining the σ-donor properties of N-heterocyclic carbenes. Tetrahedron Lett. 2019, 60, 378–381. [Google Scholar] [CrossRef]
- Li, J.J. Name Reactions, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 146–147. [Google Scholar]
- Li, A.-H.; Dai, L.-X.; Aggarwal, V.K. Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement. Chem. Rev. 1997, 97, 2341–2372. [Google Scholar] [CrossRef]
- Alvarez, A.; Guzman, A.; Ruiz, A.; Velarde, E.; Muchowski, J.M. Synthesis of 3-arylpyrroles and 3-pyrrolylacetylenes by palladium-catalyzed coupling reactions. J. Org. Chem. 1992, 57, 1653–1656. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced rigid-bond restraints. Acta Cryst. Sect. A: Fundam. Cryst. Cryst. 2012, 68, 448–451. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A Fundam. Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A: Fundam. Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Pennington, W.T. DIAMOND – Visual Crystal Structure Information System. J. Appl. Cryst. 1999, 32, 1028–1029. [Google Scholar] [CrossRef] [Green Version]
- The GIMP Team. Available online: https://www.gimp.org/news/2019/04/07/gimp-2-10-10-released/ (accessed on 25 January 2020).
- Spek, A.L. Structure validation in chemical crystallography. Acta. Cryst. Sect. D: Biol. Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Producer. GaussView, Version 3.0; Gaussian, Inc.: Pittburgh, PA, USA, 2000; Available online: https://gaussview.software.informer.com/3.0/ (accessed on 25 January 2020).
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee, MO, USA, 2016; Available online: https://gaussian.com/gaussview6/ (accessed on 25 January 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: https://gaussian.com/glossary/g09/ (accessed on 25 January 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/gaussian16/ (accessed on 25 January 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: https://gaussian.com/gaussian16/ (accessed on 25 January 2020).
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef]
- Deglmann, P.; Furche, F. Efficient characterization of stationary points on potential energy surfaces. J. Am. Chem. Soc. 2002, 117, 9535–9538. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Parameters | 11 | 12 | |
|---|---|---|---|
| bond length (Å) | C1–C2 | 1.358(4) | 1.379(2) |
| C1-N1 | 1.358(4) | 1.364(1) | |
| C2–P1 | 1.765(3) | 1.790(1) | |
| P1–C29 | 1.830(3) | 1.679(1) | |
| bond angle (°) | P1–C29–C30 | 110.9(2) | 122.20(10) |
| P1–C29–C34 | 111.1(2) | 120.68(10) | |
| C30–C29–C34 | 110.0(2) | 113.02(11) | |
| ∑ | 332.0(6) | 355.90(31) | |
| Onium Group | II (kJ/mol) | TSI→II (kJ/mol) | TSI→III (kJ/mol) | III (kJ/mol) | TSIII→IV (kJ/mol) | IV (kJ/mol) | |
|---|---|---|---|---|---|---|---|
| α-deprotonation | β-deprotonation | ||||||
| SCy2 | −61.4 | 44.1 | 66.9 (concerted) | −239.9 | |||
| SOCy2 | −61.7 | 50.6 | 102.7 (concerted) | −203.5 | |||
| 100.1eq | 107.4eq | 119.1eq | |||||
| PCy3 | −59.9 | 57.5 | 134.4 (concerted) | −106.0 | |||
| 115.6eq | 98.9eq | 176.2eq | |||||
| 80.7ax | 81.2ax | 90.4ax | |||||
| SMe2 | −52.2 | 54.8 | – | ||||
| SOMe2 | −85.0 | 60.3 | – | ||||
| PMe3 | −53.9 | 56.5 | – | ||||
| StBu2 | – | 54.0 (concerted) | −236.3 | ||||
| SOtBu2 | – | 35.9 (concerted) | −248.0 | ||||
| PtBu3 | – | 71.1 | 40.9 | 58.5 | −185.5 | ||
| SAd2 | – | 123.7 | −8.3 | ||||
| SOAd2 | – | 102.4 | 7.4 | ||||
| PAd3 | – | 91.6 | 91.2 | 108.4 | 47.1 | ||
| S(CH2)2 | – | 38.0a | −48.6 a | −40.4 a | −275.0 | ||
| S(CH2)3 | −11.9 | 65.5 | 105.0 | 75.4 | 83.0 | −238.8 | |
| S(CH2)4 | −33.6 | 56.7 | 90.5 (concerted) | −180.0 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinert, H.; Schwarz, C.; Kroll, A.; Gessner, V.H. Towards the Preparation of Stable Cyclic Amino(ylide)Carbenes. Molecules 2020, 25, 796. https://doi.org/10.3390/molecules25040796
Steinert H, Schwarz C, Kroll A, Gessner VH. Towards the Preparation of Stable Cyclic Amino(ylide)Carbenes. Molecules. 2020; 25(4):796. https://doi.org/10.3390/molecules25040796
Chicago/Turabian StyleSteinert, Henning, Christopher Schwarz, Alexander Kroll, and Viktoria H. Gessner. 2020. "Towards the Preparation of Stable Cyclic Amino(ylide)Carbenes" Molecules 25, no. 4: 796. https://doi.org/10.3390/molecules25040796
APA StyleSteinert, H., Schwarz, C., Kroll, A., & Gessner, V. H. (2020). Towards the Preparation of Stable Cyclic Amino(ylide)Carbenes. Molecules, 25(4), 796. https://doi.org/10.3390/molecules25040796