4.1.2. Synthesis of the Benzothiopyranes and Derivatives
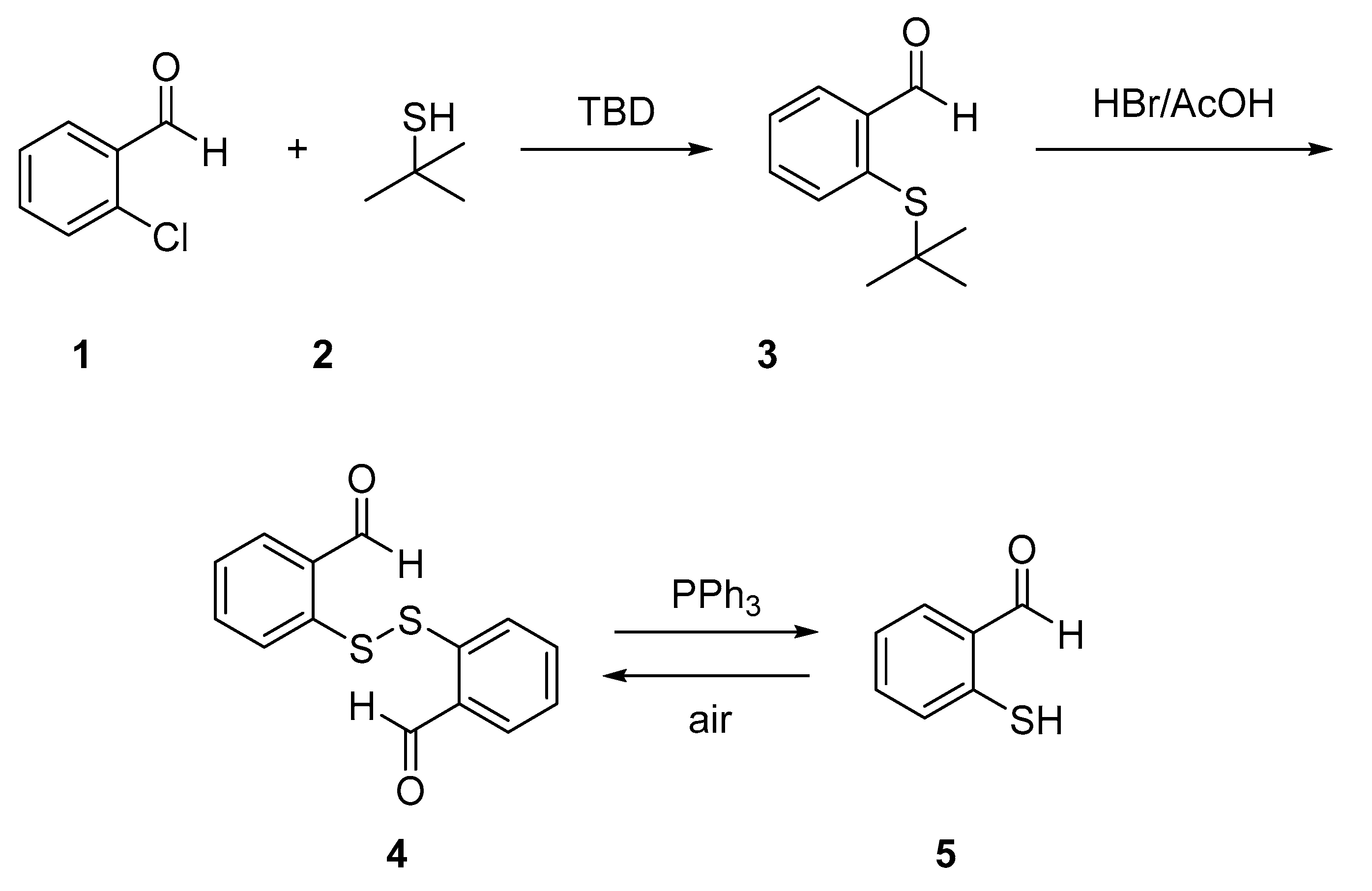
2-(tert-Butylthio)benzaldehyde (3)
In a screw-capped flask equipped with a stir bar, DMSO (2.0 mL, 0.028 mol, 4 equivalent) and potassium hydroxide (KOH; 470 mg, 8.4 mmol,1.1 equivalent) were mixed under nitrogen and stirred at room temperature. After 30 min, the KOH was crushed with a spatula, and 2-methyl-2-propanethiol (610 µL, 0.0106 mol, 1.5 eq) was added, and the mixture was stirred for 20 min. 2-Chlorobenzaldehyde (810 μL, 7.2 mmol, 1.0 equivalent) was added, and the reaction was heated to 110 °C for 90 min. After that, the reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (2 × 30 mL). The combined organic layers were washed with water (30 mL) and brine (30 mL), dried (anhydrous Na2SO4), and concentrated on a rotary evaporator the residue was purified by chromatography using a mixture of 10% ethyl acetate in hexanes to afford 2-(tert-butylthio)benzaldehyde 15 (980 mg, 70% yield) along with some unreacted 2-chlorobenzaldehyde. 1H-NMR (300 MHz, CDCl3) 10.78 (s, 1H), 7.98 (d, J = 4.3 Hz, 1H), 7.62–7.50 (m, 1H), 7.45–7.30 (m, 1H), 1.30 (s, 9H). GC-MS: m/z (%) = 194 (27) [M+], 161 (8), 138 (100), 77 (5).
2,2′-Disulfanediyldibenzaldehyde (4)
A 25-mL round-bottom flask containing 2-(tert-butylthio)benzaldehyde (3, 194 mg, 1.0 mmol, 1 equivalent) was placed in an ice bath. Acetic acid (343 μL, 6 mmol, 6.0 equivalents), 48% aqueous HBr (343 μL, 3 mmol, 3.0 equivalents) and DMSO (73 μL, 1.0 mmol, 1.0 equivalent) were then added to the resulting cooled mixture. The reaction mixture was then allowed to warm to room temperature and then was stirred overnight. The reaction mixture, containing a solid precipitate, was diluted with cold water (2 mL), filtered, dried and purified by column chromatography using 20% ethyl acetate in hexanes as eluent to afford 2,2-disulfanediyldibenzaldehyde (4). Yield 73%. White solid. m.p.:.: 149–151 °C. 1H-NMR (300 MHz, CDCl3) δ 10.19 (s, 2H), 7.85 (dd, J = 7.5, 1.4 Hz, 2H), 7.75 (d, J = 7.8 Hz, 2H), 7.47 (td, J = 7.7, 1.6 Hz, 2H), 7.37 (dd, J = 10.6, 4.2 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 192.1, 140.2, 135.0, 134.5, 134.0, 126.8, 126.5. IR ν: 1664, 1582, 1555. GC-MS: m/z (%) = 274 (14) [M+], 137 (100), 109 (54). HRMS-ESI (m/z): calcd. for C14H10O2S2 [M + Na]+ 297.0014, found: 297.0025.
2-Mercaptobenzaldehyde (5)
In a 25 mL round bottom flask, 2,2-disulfanediyldibenzaldehyde (4, 177 mg, 0.65 mmol, 1 equivalent), was dissolved in a mixture of DMF (5.4 mL), MeOH (5.4 mL) and of water (3.0 mL; all solvents were deoxygenated before use) then tripheylphosphine (253 mg, 0.975 mmol, 1.5 equivalent) was added, and the mixture stirred a room temperature for 30 min. After that the reaction mixture was diluted with water (20 mL) and extracted with deoxygenated diethyl ether (2 × 30 mL). The combined organic layers were washed with deoxygenated water (30 mL) and brine (30 mL), dried (anhydrous Na2SO4), and concentrated on a rotary evaporator the residue was purified by chromatography using a mixture of 10% ethyl acetate in hexanes to afford 2-mercaptobenzaldehyde (5), in 73% yield. During the whole process, the presence of air in the mixture was avoided to prevent oxidation back to the disulfide 4. 1H-NMR (300 MHz, CDCl3) δ 10.02 (s, 1H), 7.73 (dd, J = 8.5, 7.7 Hz, 1H), 7.38 – 7.20 (m, 3H), 5.48 (s, 1H).
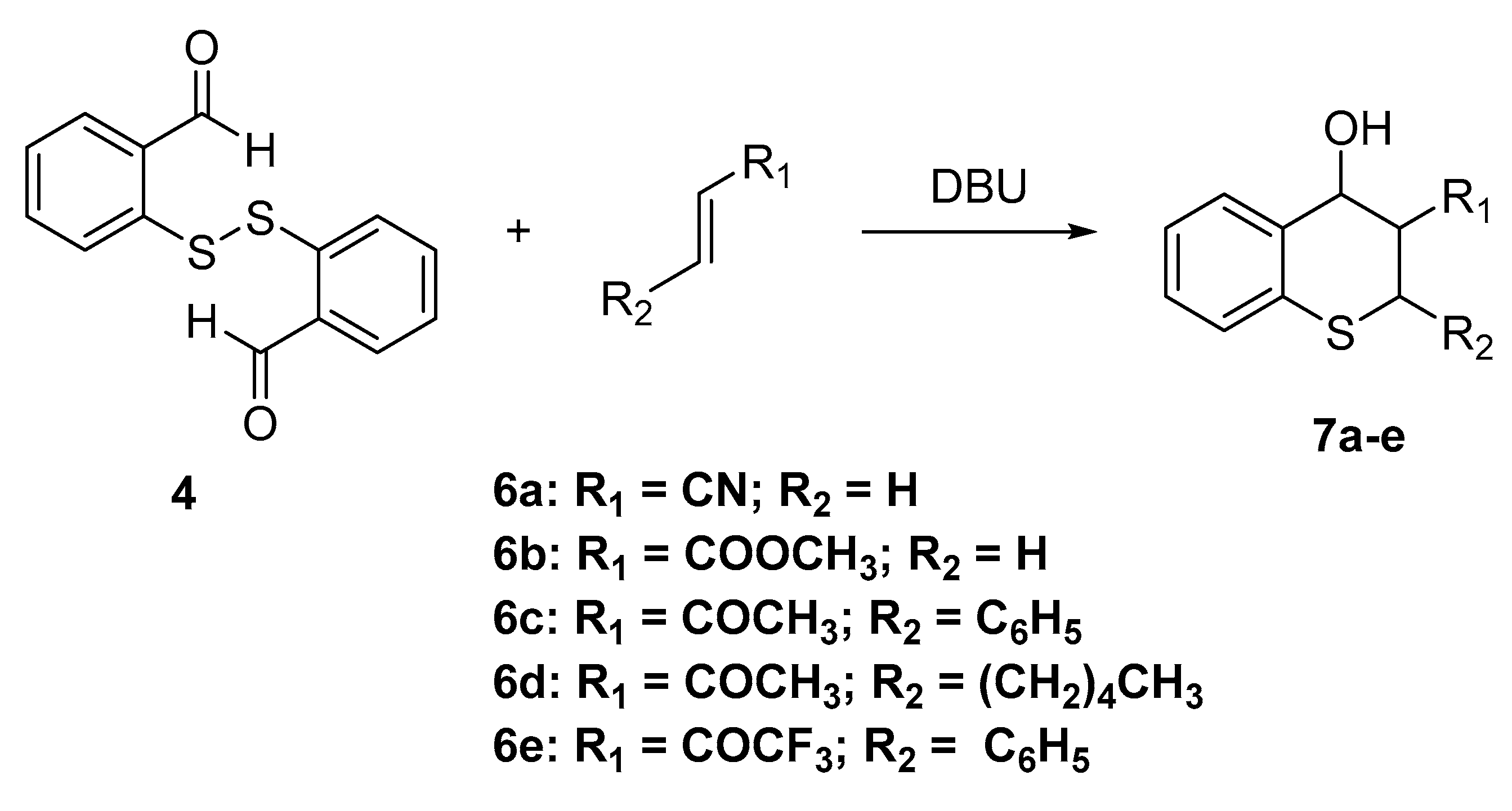
2H-Thiochromene-3-carbonitrile (8a)
In a round-bottom flask were placed 2,2-disulfanediyl-dibenzaldehyde (4, 137 mg, 0.5 mmol, 1 equivalent), acrylonitrile 58 mg (0.55 mmol, 2.2 equivalents) and DBU 84 mg (0.55 mmol, 1.1 equivalents) under nitrogen, and the mixture was heated with stirring at 80 °C for 24 h, After cooling to room temperature, the crude reaction mixture was then purified by column chromatography over silica gel using hexane/EtOAc (5:1) as eluent to afford 7a as a white solid (77 mg, 45%). Alternatively, 2H-thiochromene-3-carbonitrile (7a) was prepared by a different methodology, as follows:
Step 1. In a screw-capped flask, 2,2-disulfanediyl-dibenzaldehyde (4, 70 mg, 0.25 mmol, 1 equivalent); triphenylphosphine 131 mg (0.5 mmol, 2 equivalents) were dissolved in THF (5 mL), the mixture was stirred at room temperature under an argon atmosphere. After 20 min an excess of acrylonitrile (80 mg, 1.5 mmol, 6 equivalents) was added and after another 10 min of reaction a TLC plate showed the formation of 2-mercaptobenzaldehyde (5) and the mixture of stereoisomers of 4-hydroxy-thiochromane-3-carbonitrile. The reaction mixture is stirred overnight, and after that time, the only products are the 4-hydroxythiochromane-3-carbonitriles.
Step 2. The mixture of 4-hydroxythiochromane-3-carbonitriles is heated overnight at reflux in dichloroethane with 20% mol of Amberlyst 15 after that, the solvent was evaporated on a rotary evaporator and diluted in the minimum amount of CH2Cl2 and purified by column chromatography using a mixture of 20% ethyl acetate in hexanes as eluent to afford 2H-thiochromene-3-carbonitrile 8a in 68% yield (calculated from the dimer). Mp. 96–98 °C. 1H-NMR (400 MHz, CDCl3) δ 7.30 − 7.19 (m, J = 7.5 Hz, 2H), 7.20 – 7.10 (m, 3H), 3.57 (s, 2H). 13C-NMR (101 MHz, CDCl3) δ 142.4, 132.9, 131.1, 130.3, 130.3, 127.7, 126.5, 118.5, 103.9, 26.0. IR ν: 2207, 1615, 1435, 753. HRMS-ESI (m/z): calcd. for C10H7NS [M + H]+ 174.0372, found: 174.0380.
Methyl 2H-thiochromene-3-carboxylate (8b)
In a screw-capped flask, 2,2-disulfanediyldibenzaldehyde (4, 137 mg, 0.5 mmol, 1 equivalent), methylacrylate (130 mg, 1.5 mmol, 3.0 equivalents) and DBU (230 mg, 1.5 mmol, 3.0 equivalents) were mixed under an argon atmosphere and then heated to 80 °C for 24 h, After cooling to room temperature the reaction mixture was dissolved with CH2Cl2 and poured directly onto a chromatography column that was eluted using 10% ethyl acetate in hexanes to afford methyl 2H-thiochromene-3-carboxylate (8b) in 48% yield as a yellowish solid with mp. 34–35 °C. 1H-NMR (400 MHz, CDCl3) δ 7.55 (s, 1H), 7.30 − 7.16 (m, 3H), 7.13 (d, J = 7.4 Hz, 3H), 3.84 (s, 3H), 3.73 (s, 2H). 13C-NMR (100 MHz, CDCl3) δ 166.4, 137.3, 134.0, 131.3, 130.6, 130.2, 127.1, 125.8, 123.0, 52.2, 24.0. IR ν: 2952, 1703, 1434, 1235, 751. HRMS-ESI (m/z): calcd. for C11H10O2S [M + H]+ 207.0474, found: 207.0185.
1-(2-Phenyl-2H-thiochromen-3-yl)ethan-1-one (8c)
In a 10 mL round bottom flask equipped with a reflux condenser were added (E)-4-phenylbut-3-en-2-one (146 mg, 1.0 mmol) and 2-(tert-butylthio)benzaldehyde (3, 250 mg, 1.3 mmol) and to this mixture was added concentrated hydrochloric acid (12M, 2.0 mL) and the mixture was placed in a preheated oil bath at 110 °C and held for two hours. Thereafter water (10 mL) was added and the mixture was extracted with dichloromethane (3 × 15 mL), the combined organic layers were washed with saturated NaHCO3 solution. After drying over Na2SO4, the solvent was evaporated, and the residue was purified by column chromatography to yield 106 mg (40%) of 1-(2-phenyl-2H-thio-chromen-3-yl)ethan-1-one 27, as a yellowish solid. Mp: 115–116 °C. 1H-NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.25 (dd, J = 3.9, 2.8 Hz, 1H), 7.21 – 7.13 (m, 5H), 5.36 (d, J = 1.6 Hz, 1H), 2.49 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 196.5, 142.0, 137.4, 134.0, 132.8, 131.1, 130.7, 130.4, 128.5, 127.6, 127.6, 126.5, 125.7, 38.8, 25.6. IR ν: 3031, 1656, 1622, 1196, 900. HRMS-ESI (m/z): calcd. for C17H14OS [M + H]+ 267.0838, found: 267.0345.
1-(2-Pentyl-2H-thiochromen-3-yl)ethan-1-one (8d)
In a 10 mL round bottom flask were added 2,2-disulfanediyldibenzaldehyde (4, 140 mg, 0.5 mmol), 2-Nonanone (145 mg, 1.0 mmol) and triphenylphosphine (262 mg, 1.0 mmol) dissolved in THF (5 mL) and the mixture was stirred under an argon atmosphere at room temperature overnight. Column chromatography of the crude residue after solvent removal gave 195 mg (70%) of a mixture of stereoisomers of 1-(4-hydroxy-2-pentylthiochroman-3-yl)ethan-1-one. Later 140 mg of the isomeric alcohols was dissolved in THF (5 mL containing Amberlyst 15 (20 % mol) in a 10 mL round bottom flask equipped with a reflux condenser and the mixture was heated to reflux overnight during 20 h. After cooling to room temperature the reaction mixture was dissolved with CH2Cl2 and the crude was purified by column chromatography over silica gel using hexane/EtOAc (9:1) as eluent to afford 40 mg (29%) of 1-(2-pentyl-2H-thiochromen-3-yl)ethan-1-one (8d) as a yellowish oil. 1H-NMR (400 MHz, CDCl3) δ 7.45 (s, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.34 − 7.24 (m, 2H), 7.20 (td, J = 7.3, 1.4 Hz, 1H), 4.11 (dd, J = 8.0, 5.6 Hz, 1H), 2.52 (s, 3H), 1.47 (dt, J = 10.8, 3.7 Hz, 2H), 1.35 − 1.15 (m, 6H), 0.88 (t, J = 6.8 Hz, 3H. 13C-NMR (75 MHz, CDCl3) δ 196.8, 136.4, 133.3, 130.8, 130.6, 128.1, 125.5, 36.2, 34.4, 31.2, 25.7, 25.5, 22.5, 14.1. Note: the signals at 130.6 and 133.3 each correspond to two overlapped peaks. IR ν: 2927, 1635, 1407, 1388, 756. HRMS-ESI (m/z): calcd. for C16H20OS [M + H]+ 261.1308, found: 261.1320.
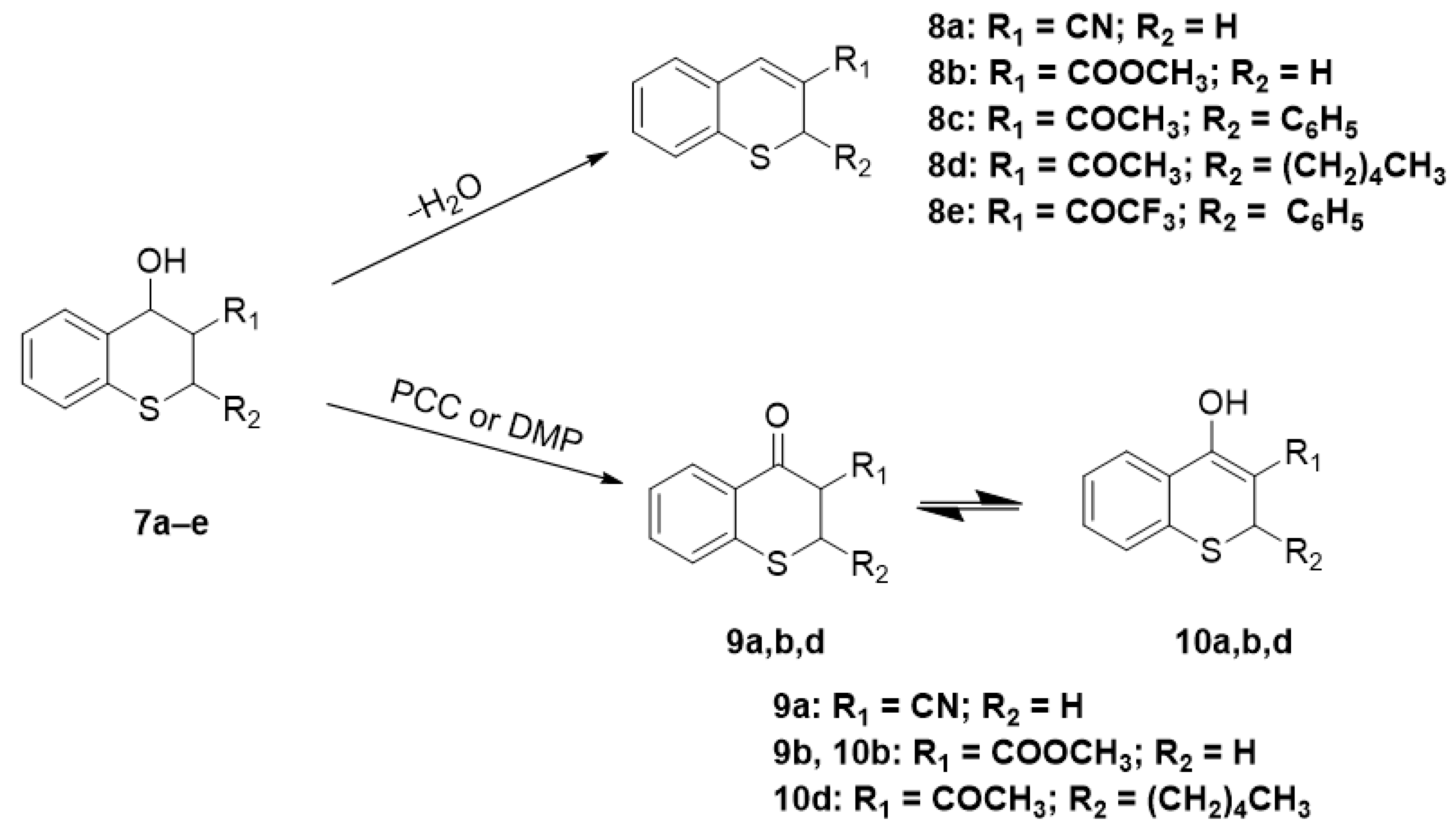
4-Oxo-thiochromane-3-carbonitrile (9a)
According with Step 1 of the procedure for the synthesis of compound 8a we prepared a mixture of stereoisomers of 4-hydroxythiochromane-3-carbonitrile, then this mixture (191 mg, 1.0 mmol) was dissolved in dichloromethane (5.0 mL) and mixed with the Dess-Martin periodinane reagent (430 mg, 1 mmol). After 1 h of stirring at room temperature the crude mixture was purified by column chromatography over silica gel using hexane/EtOAc (4:1) as eluent to afford the desired 4-oxothiochromane-3-carbonitrile (9a, 60 mg, 30%) as a white solid with mp: 79–81°C. 1H-NMR (300 MHz, CDCl3) δ 1H NMR (300 MHz, CDCl3) δ 8.17 (dd, J = 8.0, 1.4 Hz, 1H), 7.61–7.44 (m, 1H), 7.42–7.21 (m, 2H), 4.18 (dd, J = 11.6, 3.7 Hz, 1H), 3.67 (dd, J = 13.5, 11.7 Hz, 1H), 3.57–3.40 (m, 1H). 13C-NMR (75 MHz, CDCl3) δ 184.6, 141.0, 134.7, 130.5, 128.9, 127.7, 126.0, 115.1, 41.8, 29.4. IR ν: 2918, 2256, 1678, 1582, 1430. HRMS-ESI (m/z): calcd. for C10H7NOS [M + Na]+ 212.0141, found: 212.0153.
Methyl 4-oxo-thiochromane-3-carboxylate (9b, 10b)
In a screw-capped flask, 2,2-disulfanediyl-dibenzaldehyde (4, 70 mg, 0.25 mmol, 1 equivalent) and triphenylphosphine (131 mg (0.5 mmol, 2 equivalents), were dissolved in THF (5 mL), and the mixture was stirred at room temperature under an argon atmosphere. After 20 min an excess of acrylonitrile (80 mg, 1.5 mmol, 6 equivalents) was added and after another 10 min of reaction a TLC plate showed the formation of 2-mercaptobenzaldehyde (5) and a mixture of 3-cyano-4-hydroxy-2H-thiochroman isomers. The reaction mixture was stirred overnight, and after that, the only products are the mixture of stereoisomers of 3-cyano-4-hydroxy-2H-thiochroman. A portion of the above mixture (112 mg) was dissolved in dichloromethane (5.0 mL) and mixed with the Dess-Martin periodinane reagent (430 mg, 1 mmol). After 1 h of stirring at room temperature, the crude mixture was purified by column chromatography to afford 40 mg (36%) of the desired methyl 4-oxo-thiochromane-3-carboxylate as a white solid with mp: 85–86 °C. Methyl 4-oxo-thiochromane-3-carboxylate (9b) exists in solution in equilibria with its tautomeric methyl 4-hydroxy-2H-thiochromene-3-carboxylate enol form 10b. The 1H-NMR spectrum in CDCl3 revealed the that the ratio of tautomers 9b and 10b was 1:5. Proton H5 appears as a doublet at 8.17 ppm, J = 7.9 Hz, the proton H5 of the enol form has a chemical shift of 7.89 d, J = 7.7 Hz the integration areas are 0.2 and 1.0 respectively, which indicates that the enol form corresponds to 83% of the mixture. The same ratio can be calculated with the protons of the methoxy group of the ester. 1H-NMR (300 MHz, CDCl3) δ 12.71a (s), 8.17b (d, J = 7.9 Hz), 7.89a (d, J = 7.7 Hz), 7.44b (t, J = 7.6 Hz), 7.37–7.19 a,b (m), 3.89 a (s), 3.85 b (s), 3.76 a (s,), 3.38 b (dd, J = 13.5, 3.6 Hz) (a is the enol form, b is the keto form); 13C-NMR (75 MHz, CDCl3) δ 171.6, 165.9, 137.2, 131.0, 129.8, 129.2, 127.3, 126.8, 125.6, 125.3, 93.7, 77.5, 77.1, 76.7, 54.0, 52.7, 52.1, 23.3; IR ν: 3012, 2951, 1718, 1681, 1645, 1608, 1583, 1553, 1437. HRMS-ESI (m/z): calcd. for C11H10O3S [M + Na]+ 245.0243, found: 245.0254.
1-(4-Hydroxy-2-pentyl-2H-thiochromen-3-yl)ethan-1-one (10d)
In a 10 mL round bottom flask were added 2,2-disulfanediyldibenzaldehyde 17, (140 mg, 0.5 mmol), 2-nonanone, (145 mg, 1.0 mmol) and triphenylphosphine (262 mg, 1.0 mmol), and the mixture was stirred under an argon atmosphere at room temperature overnight. Column chromatography of the crude residue after solvent removal gave 195 mg (70%) of a mixture of stereoisomers of 1-(4-hydroxy-2-pentylthiochroman-3-yl)ethan-1-one (10d). Next, 90 mg of this mixture were dissolved in dichloromethane (5.0 mL) and mixed with the Dess-Martin periodinane reagent (192 mg, 0.45 mmol) and water (1.0 mL). After stirring for 1 h at room temperature the crude mixture was purified by column chromatography to afford 55 mg (60%) of the desired product as a yellowish oil which exists in solution only as its enol form 1-(4-hydroxy-2-pentyl-2H-thiochromen-3-yl)ethan-1-one. 1H-NMR (400 MHz, CDCl3) δ 7.97 (ddd, J = 7.9, 1.4, 0.5 Hz, 1H), 7.36–7.25 (m, 2H), 7.22 (ddd, J = 7.9, 7.0, 1.6 Hz, 1H), 3.67 (dd, J = 9.6, 5.0 Hz, 1H), 2.31 (s, J = 1.8 Hz, 3H), 1.77–1.65 (m, 1H), 1.63–1.43 (m, 2H), 1.36–1.09 (m, 5H), 0.85 (t, J = 7.0 Hz, 3H). 13C-NMR (101 MHz, MeOH) δ 195.4, 174.0, 135.8, 132.0, 129.5, 128.2, 127.7, 125.5, 108.8, 39.6, 36.4, 31.1, 26.6, 24.4, 22.5, 14.0. IR ν: 2928, 2856, 1634, 1593, 1545, 1380. HRMS-ESI (m/z): calcd. for C16H20O2S [M + Na]+ 299.1076, found: 299.1088.
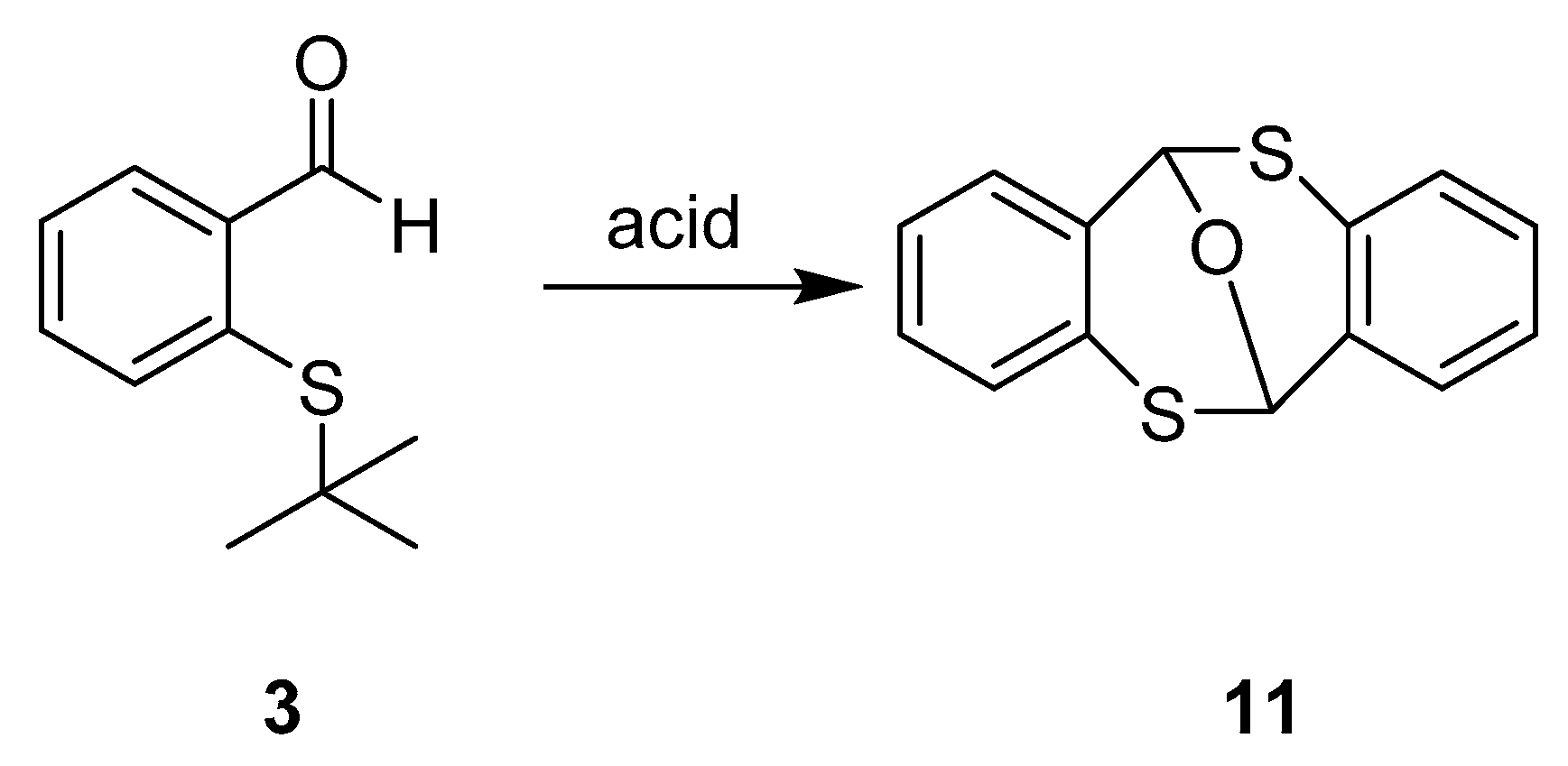
6H,12H-6,12-Epoxydibenzo[b,f][1,5]dithiocine (11)
In a 10 mL round bottom flask equipped with a reflux condenser containing concentrated hydrochloric acid (12M, 3.0 mL) was added 2-(tert-butylthio)benzaldehyde (3, 582 mg, 3.0 mmol). This mixture was placed in a preheated oil bath at 110 °C and held for 2 h, after which water (10 mL) was added and the mixture was extracted with dichloromethane (3 × 15 mL) and the combined organic layers were washed with saturated NaHCO3 solution. After drying over Na2S04, the solvent was evaporated, and the residue was purified by column chromatography over silica gel using hexane/EtOAc (4:1) as eluent to yield 282 mg, (72%) of 6H,12H-6,12-epoxydibenzo[b,f][1,5]dithiocine with m.p.: 160–161 °C. 1H-NMR (300 MHz, CDCl3) δ 7.36–7.31 (m, 1H), 7.17 –7.05 (m, 3H), 6.40 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 132.7, 129.0, 128.3, 127.9, 125.2, 74.7. IR ν: 3051, 1433, 1262, 1083, 956, 726. GC-MS: m/z (%) = 258 (40) [M+], 153 (100), 121 (15), 77 (28).
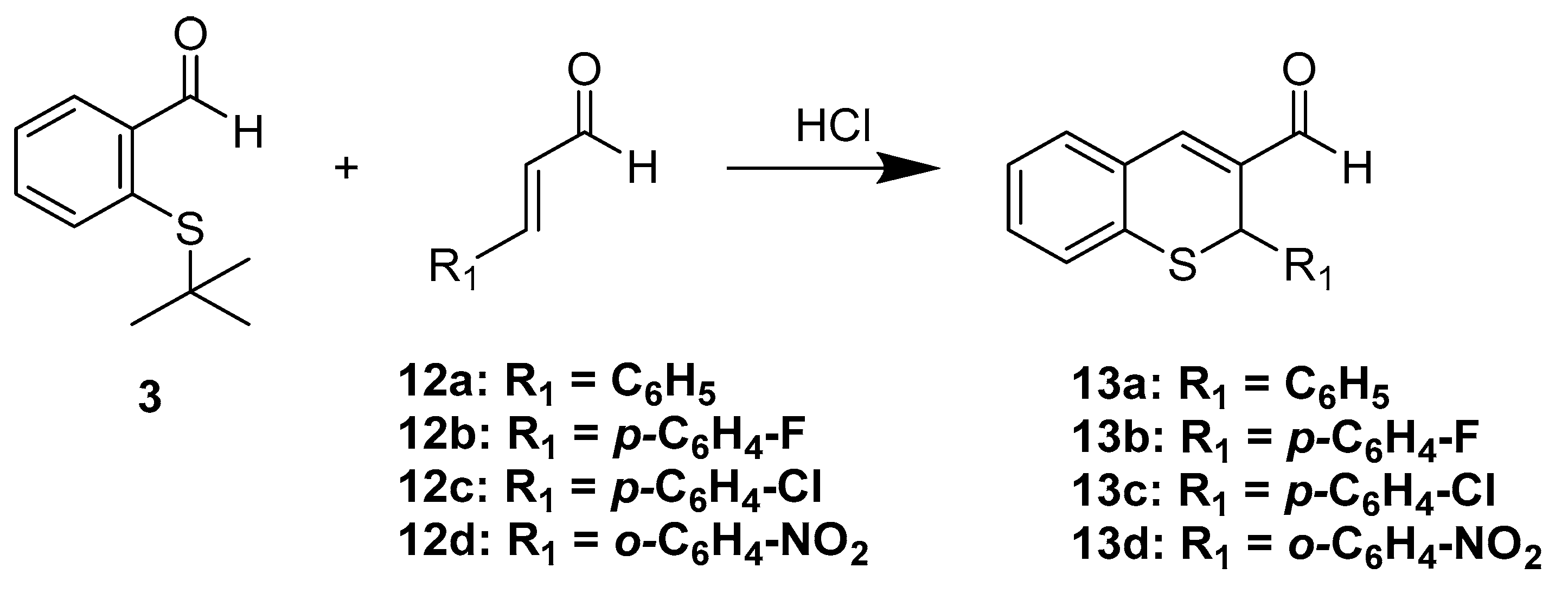
4.1.3. General Procedure for the Synthesis of 2-aryl-2H-thiochromene-3-carbaldehydes 13a–d
In a 10 mL round bottom flask equipped with a reflux condenser were added the corresponding cinnamaldehyde (1.0 mmol) and 2-(tert-butylthio)benzaldehyde (3, 1.3 mmol) and to this mixture was added concentrated hydrochloric acid (12M, 2.0 mL). Then, the mixture was placed in a preheated oil bath at 110°C and held for two hours, after which water (10 mL) was added and the mixture was extracted with dichloromethane (3×15 mL), and the combined organic layers were washed with saturated NaHCO3 solution. After drying over Na2SO4, the solvent was evaporated, and the residue was purified by column chromatography over silica gel using hexane/EtOAc (4:1) as eluent to yield the corresponding 2-aryl-2H-thiochromene-3-carbaldehyde 13a–d.
2-Phenyl-2H-thiochromene-3-carbaldehyde (13a)
Starting from cinnamaldehyde, according to the general procedure described above, 2-phenyl-2H-thiochromene-3-carbaldehyde (13a, 175 mg, 70%) was obtained as a yellowish solid with mp: 195–196 °C. 1H-NMR (200 MHz, CDCl3) δ 9.70 (s, 1H), 7.55 (s, 1H), 7.43 (m, 1H), 7.35–7.19 (m, 5H), 7.10 (s, 1H), 4.80 (s, 1H). IR ν: 3040, 2820, 1660, 1622, 1139. HRMS-ESI (m/z): calcd. for C16H12OS [M + Na]+ 275.0501, found: 275.0510.
2-(4-Fluorophenyl)-2H-thiochromene-3-carbaldehyde (13b)
Starting from 4-fluorocinnamaldehyde (150 mg, 1.0 mmol) according to the general procedure described above 2-(4-fluorophenyl)-2H-thiochromene-3-carbaldehyde (13b, 160 mg, 59% yield) was obtained as a yellow solid with m.p.: 96–98 °C. 1H-NMR (400 MHz, CDCl3) δ 9.66 (s, 1H), 7.47 (s, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.35–7.25 (m, 2H), 7.24–7.12 (m, 3H), 6.86 (dd, J = 12.6, 4.8 Hz, 2H), 5.19 (s, 1H). 13C-NMR (101 MHz, CDCl3) δ 191.3, 162.5 (d, J = 246.5 Hz), 145.4, 137.7, 134.9, 134.0, 132.1, 131.3, 129.9, 128.4 (d, J = 8.2 Hz), 127.9, 126.2, 115.7 (d, J = 21.6 Hz), 37.8. IR ν: 3050, 2826, 1665, 1625, 1503, 1139, 845. HRMS-ESI (m/z): calcd. for C16H11FOS [M + Na]+ 293.0407, found: 293.0416.
2-(4-Chlorophenyl)-2H-thiochromene-3-carbaldehyde (13c)
Starting from 4-chlorocinnamaldehyde (174 mg, 1.0 mmol) according to the general procedure described above 2-(4-chlorophenyl)-2H-thiochromene-3-carbaldehyde (13c, 250 mg, 87% yield) was obtained as a yellow solid with m.p.: 119–120 °C. 1H-NMR (400 MHz, CDCl3) δ 9.67 (s, 1H), 7.48 (s, 1H), 7.42 (d, J = 7.4 Hz, 1H), 7.34–7.25 (m, 2H), 7.21 (td, J = 7.3, 1.1 Hz, 1H), 7.17–7.11 (m, 4H), 5.17 (s, 1H). 13C-NMR (101 MHz, CDCl3) δ 191.1, 145.3, 140.3, 140.1, 134.5, 133.6, 132.0, 131.1, 129.7, 128.8, 127.9, 127.7, 126.1, 37.7. IR ν: 2845, 1667, 1627, 1580. 1138, 761. HRMS-ESI (m/z): calcd. for C16H11ClOS [M + Na]+ 309.0111, found: 309.0120.
2-(2-Nitrophenyl)-2H-thiochromene-3-carbaldehyde (13d)
Starting from 2-nitrocinnamaldehyde (1.0 mmol) according to the general procedure described above 2-(4-chlorophenyl)-2H-thiochromene-3-carbaldehyde (13d, 410 mg, 69% yield) was obtained as a yellow solid with m.p.: 130–132 °C. 1H-NMR (400 MHz, CDCl3) δ 9.66 (d, J = 1.7 Hz, 1H), 8.01 (d, J = 7.1 Hz, 1H), 7.67 (s, 1H), 7.45 (d, J = 7.4 Hz, 1H), 7.35 (p, J = 7.5 Hz, 2H), 7.29 (d, J = 7.5 Hz, 1H), 7.26–7.15 (m, 2H), 7.10 (d, J = 7.1 Hz, 1H), 6.01 (s, 1H). 13C-NMR (101 MHz, CDCl3) δ 190.7, 147.1, 146.4, 135.8, 133.9, 133.5, 133.2, 132.3, 131.2, 129.2, 128.5, 128.4, 127.8, 126.2, 125.9, 33.4. IR ν: 2992, 1661, 1623, 1519, 1142, 740. GC-MS: m/z (%) = 297 (4) [M+], 280 (43), 251 (60), 235 (44), 221 (100.). HRMS-ESI (m/z): calcd. for C16H11NO3S [M + Na]+ 320.0352, found: 320.0362.
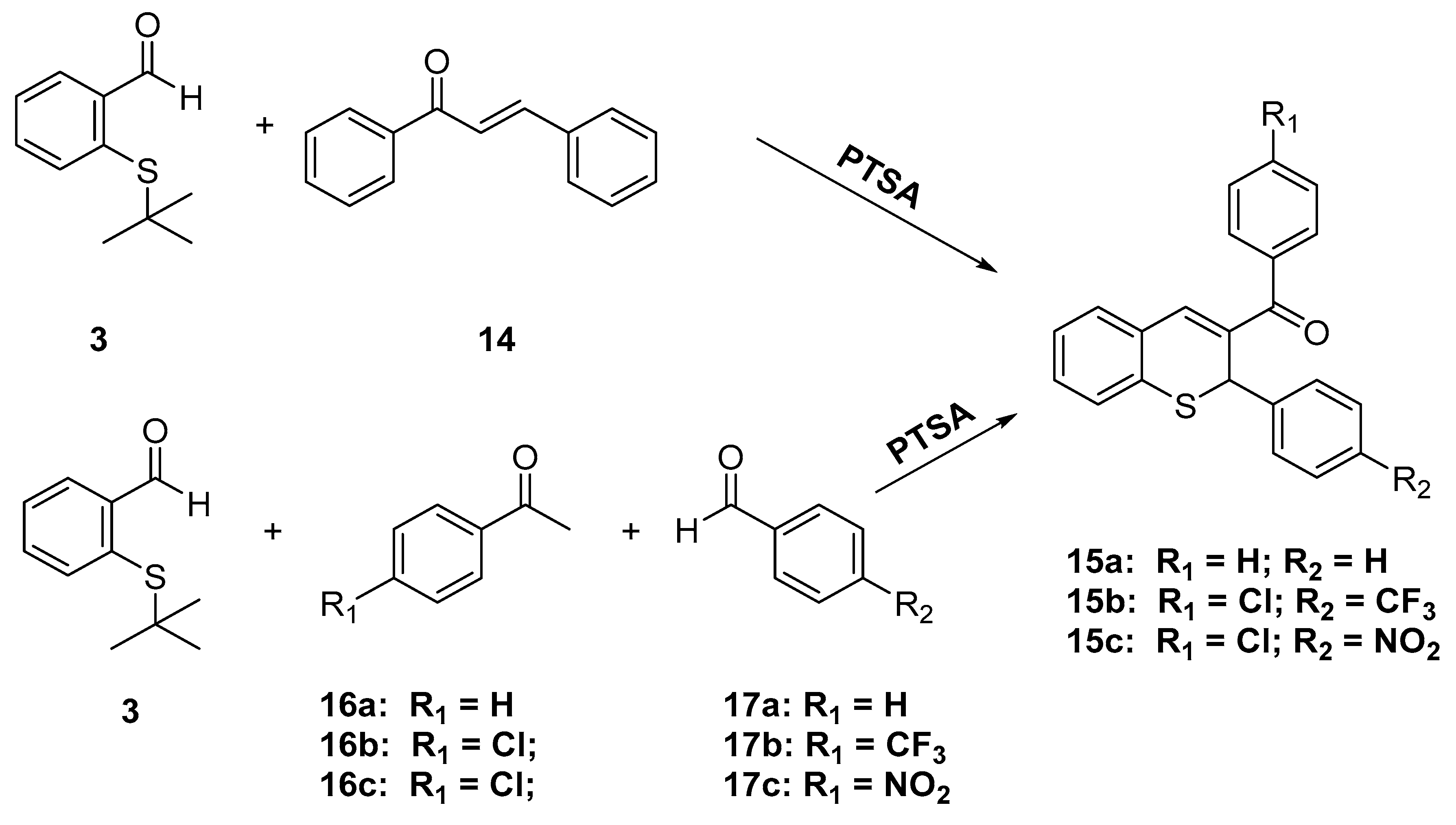
Phenyl(2-phenyl-2H-thiochromen-3-yl)methanone (15a)
In a 10 mL round bottom flask equipped with a reflux condenser were added chalcone (1 mmol), 2-(tert-butylthio)benzaldehyde (3, 1.5 mmol) and p-toluenesulfonic acid monohydrate (60 mg) in toluene (2 mL). The reaction mixture was stirred at reflux for 2 h and progress of the reaction was monitored with TLC. Upon the consumption of the chalcone, the reaction mixture was directly subjected to column chromatography over silica gel using hexane/EtOAc (5:1) as eluent to give the desired compound as a yellow solid (45 mg, 46%) m.p.:. 151–153 °C. 1H-NMR (400 MHz, CDCl3) δ 7.71 (dd, J = 8.3, 1.3 Hz, 2H), 7.57 (ddd, J = 6.7, 2.7, 1.3 Hz, 1H), 7.51–7.44 (m, 2H), 7.41 (s, 1H), 7.33–7.18 (m, 8H), 7.14 (tdd, J = 7.7, 5.9, 1.6 Hz, 1H), 5.49 (s, 1H). 13C-NMR (101 MHz, CDCl3) δ 195.9, 142.1, 140.2, 138.3, 133.4, 132.9, 132.1, 131.3, 131.0, 130.6, 129.4, 128.9, 128.6, 127.9, 127.8, 126.8, 125.9, 40.4. IR ν: 3126, 2918, 1640, 1596, 1488, 1087, 756. HRMS-ESI (m/z): calcd. for C22H16OS [M + H]+ 329.0995, found: 329.1002.
4.1.4. General Procedure for the Synthesis of (4-chlorophenyl)(2-(aryl)-2H-thiochromen-3-yl)methanones 15b,c
In a 10 mL round bottom flask equipped with a reflux condenser were mixed 4’-chloro-acetophenone (155 mg, 1.0 mmol) and the appropriate substituted benzaldehyde (1.0 mmol) with Amberlyst-15 (250 mg) in toluene (5 mL). The reaction mixture was stirred at room temperature for 2 h, and the progress of the reaction was monitored with TLC until the formation of the corresponding chalcone was complete. Afterward 2-(tert-butylthio)benzaldehyde 3 (194 mg, 1.0 mmol) was added, and the mixture was allowed to react for another 2 h at 110 °C and then cooled to room temperature and subjected to column chromatography over silica gel using hexane/EtOAc (5:1) as eluent to afford the desired (4-chlorophenyl)(2-(aryl)-2H-thiochromen-3-yl)-methanones 15b,c.
(4-Chlorophenyl)(2-(4-(trifluoromethyl)phenyl)-2H-thiochromen-3-yl)methanone (15b)
Following the general procedure described above, starting from 4-(trifluoromethyl)-benzaldehyde (175 mg, 1 mmol) the desired 15b was obtained (200 mg, 46%) as a yellow solid m.p.: 105–106 °C. 1H-NMR (400 MHz, CDCl3) δ 7.66 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.5 Hz, 4H), 7.43 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.28 (t, J = 7.5 Hz, 3H), 7.18 (ddd, J = 7.2, 5.3, 3.7 Hz, 1H), 5.43 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 194.4, 145.6, 141.0, 138.7, 136.1, 132.33, 132.26, 131.7, 131.2, 130.8, 130.2, 129.9 (q, J = 32.7 Hz), 129.0, 127.9, 127.0, 126.3, 125.8 (q, J = 3.9 Hz), 123.8 (q, J = 270.4 Hz), 40.0. IR ν: 2952, 1612, 1585, 1323, 1114. HRMS-ESI (m/z): calcd. for C23H14ClF3OS [M + Na]+ 453.0298, found: 453.0304.
(4-Chlorophenyl)(2-(4-nitrophenyl)-2H-thiochromen-3-yl)methanone (15c)
Following the general procedure described above, starting from 4-nitrobenzaldehyde (152 mg, 1 mmol) the desired 15c (180 mg, 45%) was obtained as a yellow solid, m.p.: 93–94 °C. 1H-NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.7 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.52–7.38 (m, 5H), 7.34–7.25 (m, 3H), 7.22–7.16 (m, 1H), 5.42 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 13C-NMR (101 MHz, CDCl3) δ 194.3, 148.9, 147.4, 141.3, 138.9, 136.3, 135.9, 132.0, 131.9, 131.4, 130.8, 130.2, 129.1, 128.1, 127.6, 126.6, 124.2, 40.1. IR ν: 3026, 2849, 1656, 1607, 1585, 1515, 1338, 821. HRMS-ESI (m/z): calcd. for C22H14ClNO3S [M + Na]+ 408.0456, found: 408.0461.
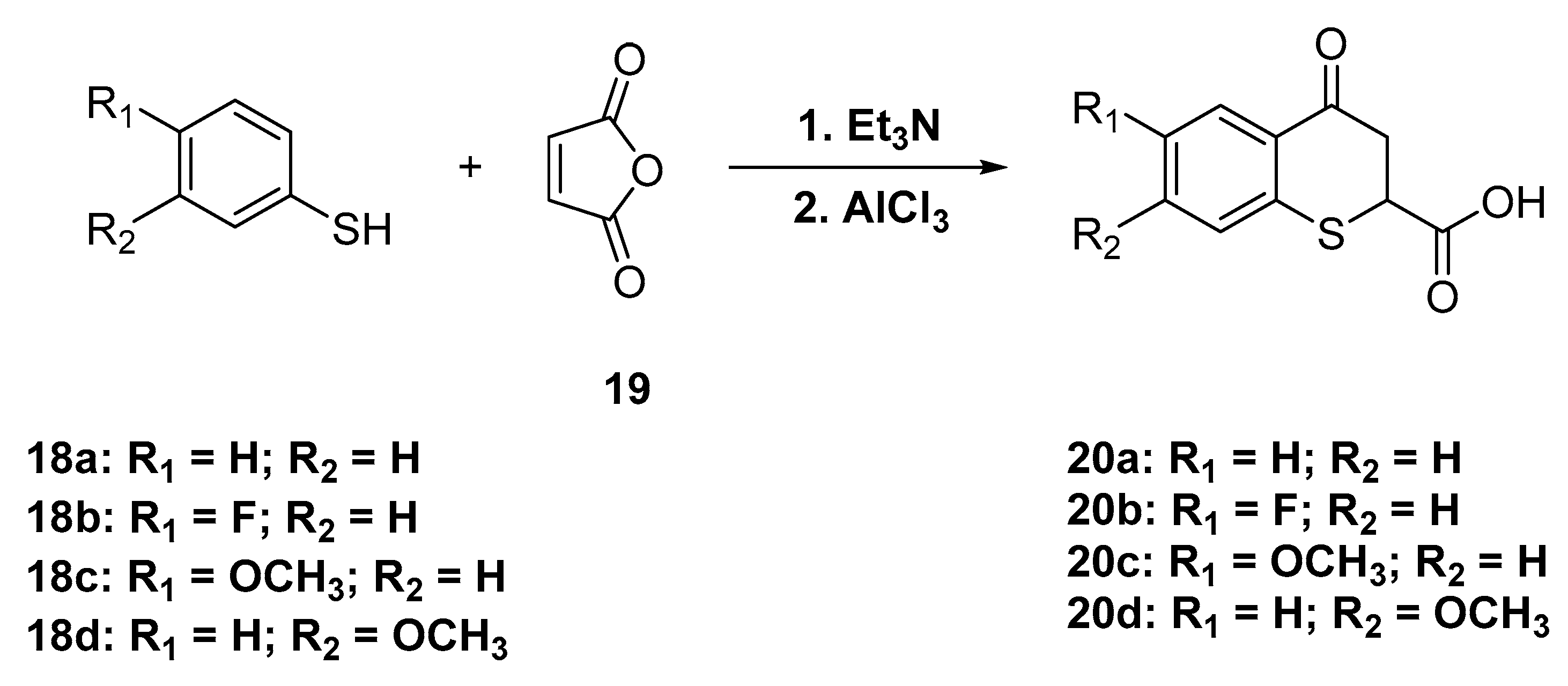
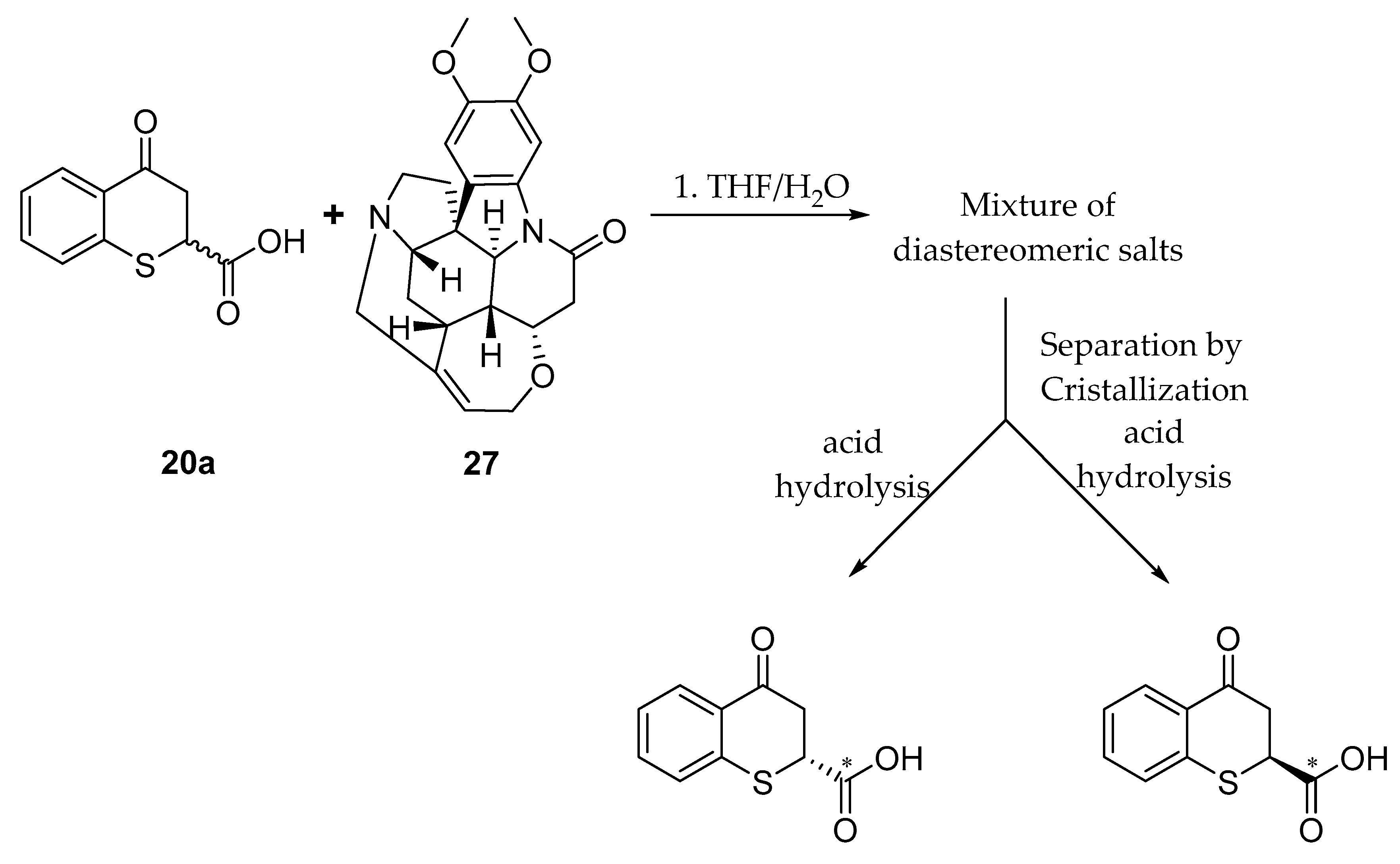
4.1.5. General Procedure for the Synthesis of the 4-oxo-thiochromane-2-carboxylic acids 20a–d
A round bottom flask equipped with a magnetic stirrer was loaded with a mixture of maleic anhydride (1.718 g, 17.5 mmol) and a slight excess of thiophenols (19.3 mmol, 1.1 equivalents) in acetonitrile dry and then triethylamine (10 drops) was slowly added. The reaction flask was closed with a glass-stopper and stirred at 50 °C for 2 h. The reaction was quenched at room temperature, the solvent was removed under reduced pressure, and the black oily residue was cooled at 0 °C in bath ice, and redissolved with dry DCM, after which a significant excess of AlCl3 was added. The mixture reaction was stirred at room temperature overnight. After the reaction was completed as determined by TLC, the mixture reaction was treated with a cold solution of hydrochloric acid (5%) and extracted with CH2Cl2 (3 × 25 mL) three times. The combined organic layers were dried over anhydrous Na2SO4, the residue after solvent evaporation the mixture was filtered through a silica gel column using as mobile phase hexanes/ethyl acetate with 5 % of acetic acid as an additive (80:20 v/v) to give pure compounds 20a–d in 55–70% global yield.
4-Oxothiochromane-2-carboxylic acid (20a)
White solid, m.p.: = 151–152 °C. 1H-NMR (300 MHz, DMSO-d6) δ 7.95 (dd, J = 7.9, 1.5 Hz, 1H), 7.49 (td, J = 7.6, 1.5 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.25 (t, J = 7.6 Hz, 1H), 4.39 (dd, J = 6.1, 4.3 Hz, 1H), 3.19–2.97 (m, 2H). 13C-NMR (75 MHz, DMSO) δ 192.63, 172.14, 138.99, 134.25, 130.52, 128.37, 127.82, 125.94, 41.71, 41.26. IR ν (cm−1) = 2918.37, 1695.44, 1681.12, 881.15, 768.35. HRMS-ESI (m/z): calcd. for C10H8O3S [M + H]+ 209.0267, found 209.0282.
6-Fluoro-4-oxo-thiochromane-2-carboxylic acid (20b)
White solid, m.p.: = 135–137 °C. 1H-NMR (300 MHz, DMSO-d6) δ 7.66 (d, J = 11.0 Hz, 1H), 7.44 (s, 1H), 7.23 (t, J = 8.8 Hz, 1H), 4.40 (t, J = 5.1 Hz, 1H), 3.10 (t, J = 4.9 Hz, 2H).13C-NMR (75 MHz, DMSO) δ 191.92, 172.33, 162.10, 158.86, 134.47, 127.97, 122.07, 116.53, 114.27, 41.62, 40.92. IR ν (cm−1) = 3498.15, 2895.35, 1723.95, 1684.65, 805.21, 238.57. HRMS-ESI (m/z): calcd. for C10H7O3FS [M + H]+ 227.0173, found 227.0173.
6-Methoxy-4-oxo-thiochromane-2-carboxylic acid (20c)
White solid, m.p.: = 165–166 °C 1H-NMR (300 MHz, DMSO-d6) δ 7.45 (d, J = 2.8 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 7.12 (d, J = 8.7 Hz, 1H), 3.77 (s, 3H), 3.15–2.96 (m, 2H). 13C-NMR (75 MHz, DMSO) δ 192.60, 172.21, 157.64, 131.43, 129.80, 129.28, 122.05, 111.46, 55.83, 41.70, 40.44. IR ν (cm−1) = 2888.72, 1714.93, 1626.73, 881.75, 807.73. HRMS-ESI (m/z): calcd. for C11H10O4S [M + H]+ 239.0373, found 239.0387.
7-Methoxy-4-oxo-thiochromane-2-carboxylic acid (20d)
White solid, m.p.: = 162–164 °C 1H-NMR (300 MHz, acetone-d6) δ 7.99 (d, J = 8.6 Hz, 1H), 6.83 (d, J = 2.4 Hz, 1H), 6.83–6.78 (m, 1H), 4.36 (dd, J = 6.4, 4.7 Hz, 1H), 3.88 (s, 3H), 3.08 (dd, J = 5.6, 3.5 Hz, 2H). 13C NMR (75 MHz, acetone-d6) δ 190.56, 170.91, 163.51, 141.17, 130.44, 124.13, 112.67, 110.55, 55.29, 41.97, 40.89. IR ν (cm−1) = 3419.98, 2906.14, 1698.81, 1671.88, 870.22, 824.56. HRMS-ESI (m/z): calcd. for C11H10O4S [M + H]+ 239.0373, found = 239.0375.
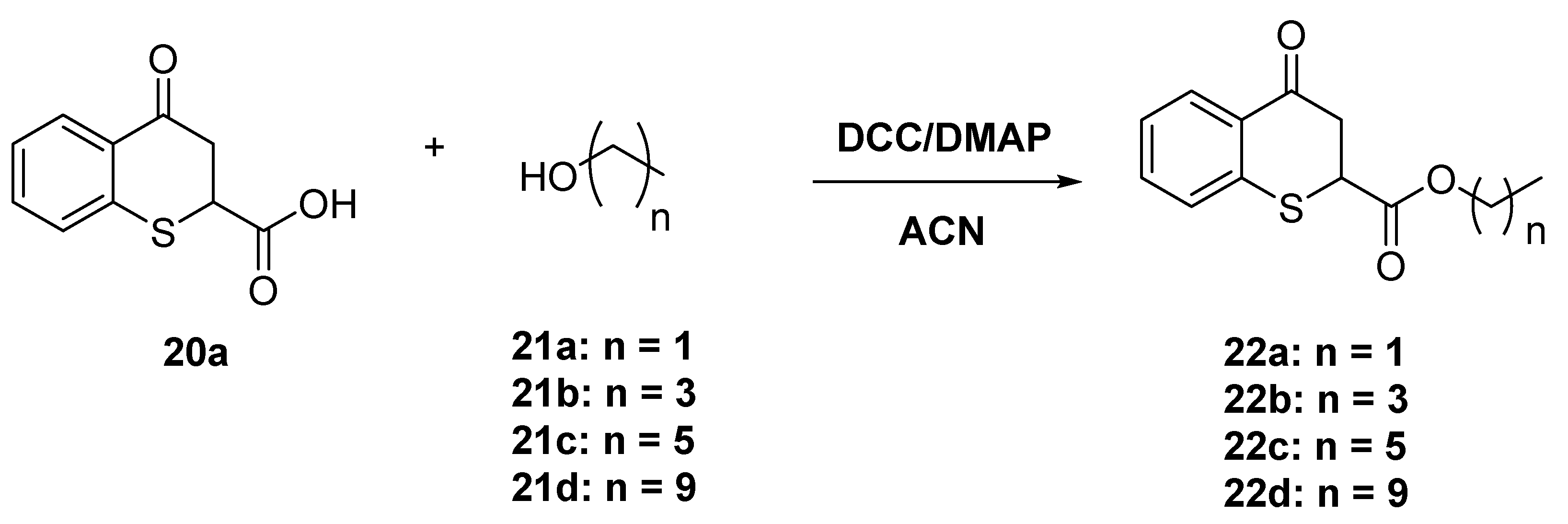
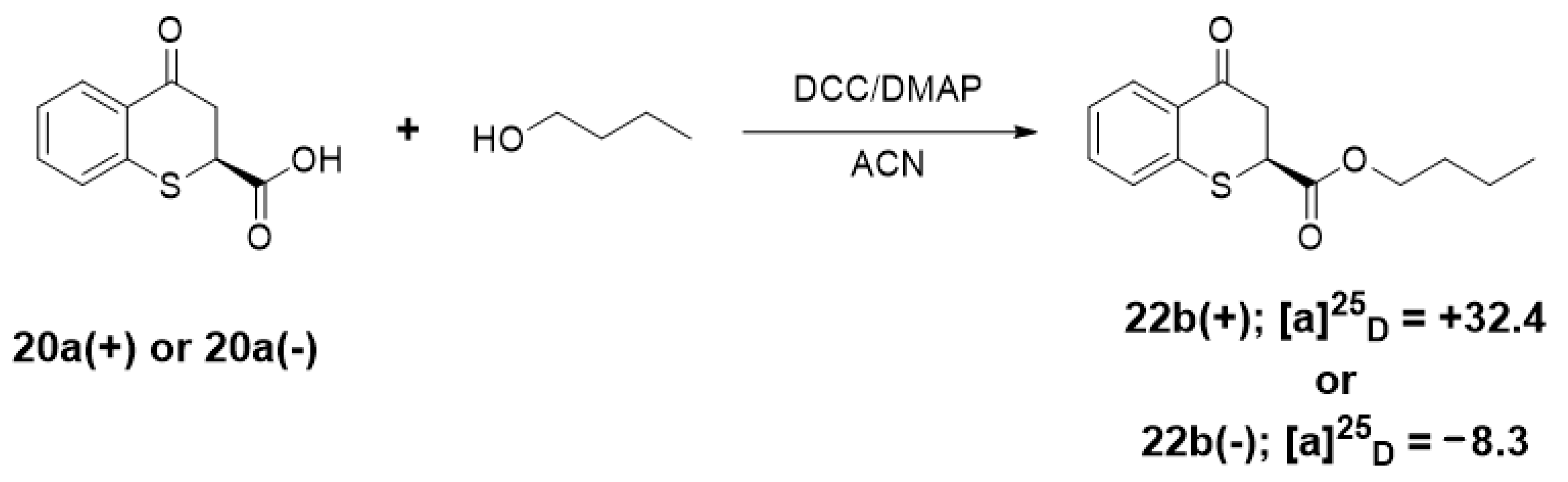
4.1.6. General Procedure for the Synthesis of the Esters from 4-oxo-thiochromane-2-carboxylic acids 22a–d
A mixture of 4-oxo-thiochromane-2-carboxylic acid (20a, 100 mg, 0.48 mmol), N,N´dicyclohexylcarbodiimide (DCC, 150 mg) and small amount of 4-dimethylaminopyridine (DMAP) were dissolved in acetonitrile (10 mL) and heated under microwave irradiation at 70 °C for 20 min, then 1.3 equivalents of the corresponding aliphatic alcohol were added. After that the reaction vial was sealed and heated to 70 °C for 40 min. When the reaction was completed as determined by TLC, the reaction mixture was diluted with ethyl acetate and washed with brine (2 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, and the residue after solvent evaporation was purified by flash chromatography using hexanes/ethyl acetate (80:20 v/v) to give 22a–d in 65–80% yield.
Ethyl 4-oxo-thiochromane-2-carboxylate (22a)
Yellowish oil. 1H-NMR (300 MHz, CDCl3) δ 8.14 (d, J = 9.2 Hz, 1H), 7.42 (d, J = 8.7 Hz, 1H), 7.31–7.19 (m, 2H), 4.27–4.12 (m, 3H), 3.21 (d, J = 6.5 Hz, 1H), 1.25 (t, J = 7.1 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 192.24, 169.85, 138.45, 133.71, 130.45, 128.85, 127.37, 126.22, 125.77, 77.55, 77.13, 76.70, 62.27, 42.34, 41.34, 13.97. IR ν (cm−1) = 2981.39, 1731.29, 1684.23, 761.18. HRMS-ESI (m/z): calcd. for C12H12O3S [M + H]+ 237.0580, found 237.0592.
Butyl 4-oxo-thiochromane-2-carboxylate (22b)
Yellowish oil. 1H-NMR (300 MHz, CDCl3) δ 8.14 (dd, J = 7.9, 1.6 Hz, 1H), 7.43 (ddd, J = 8.0, 7.2, 1.6 Hz, 1H), 7.34–7.18 (m, 2H), 4.24–4.04 (m, 3H), 3.21 (dd, J = 5.6, 1.6 Hz, 2H), 1.66–1.45 (m, 2H), 1.40–1.22 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 192.18, 169.94, 141.93, 138.49, 137.47, 133.68, 132.32, 130.47, 129.25, 129.14, 128.84, 128.69, 128.30, 127.36, 125.74, 77.55, 77.12, 76.70, 67.08, 66.06, 42.46, 41.34, 30.38, 19.16, 18.94, 13.71, 13.63. IR ν (cm−1) = 2960.28, 2933.24, 1731.63, 1683.60, 758.76. HRMS-ESI (m/z): calcd. for C14H16O3S [M + Na]+ 287.0712, found 287.0709.
Hexyl 4-oxo-thiochromane-2-carboxylate (22c)
Yellowish oil. 1H-NMR (300 MHz, CDCl3) δ 8.13 (d, J = 7.9 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 7.28–7.16 (m, 2H), 4.14 (q, J = 6.5, 5.8 Hz, 3H), 3.20 (d, J = 5.8 Hz, 2H), 1.57 (p, J = 6.4 Hz, 3H), 1.25 (s, 6H), 0.88 (t, J = 6.0 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 192.14, 169.95, 138.48, 133.67, 130.45, 128.81, 127.33, 125.72, 66.33, 42.42, 41.30, 31.31, 28.34, 25.35, 22.49, 14.01. IR ν (cm−1) = 3056.06, 2959.62, 1730.43, 1684.96, 738.80. HRMS-ESI (m/z): calcd. for C16H20O3S [M + Na]+ 315.1025, found 315.1005
Decyl 4-oxo-thiochromane-2-carboxylate (22d)
Yellowish oil. 1H-NMR (300 MHz, CDCl3) δ 8.14 (dd, J = 7.9, 1.5 Hz, 1H), 7.43 (td, J = 7.7, 1.6 Hz, 1H), 7.31–7.20 (m, 2H), 4.19–4.11 (m, 3H), 3.25–3.18 (m, 2H), 1.58 (d, J = 6.6 Hz, 2H), 1.27 (d, J = 5.7 Hz, 16H), 0.95–0.87 (m, 3H).13C-NMR (75 MHz, CDCl3) δ 192.15, 169.94, 138.50, 133.67, 130.47, 128.85, 127.34, 125.73, 77.53, 77.11, 76.68, 66.36, 42.45, 41.32, 31.91, 29.53, 29.48, 29.33, 29.17, 28.40, 25.70, 22.71, 14.16. IR ν (cm−1) = 2954.83, 2925.62, 1734.22, 1686.36, 759.28. HRMS-ESI (m/z): calcd. for C20H28O3S [M + H]+ 349.1832, found 349.1864
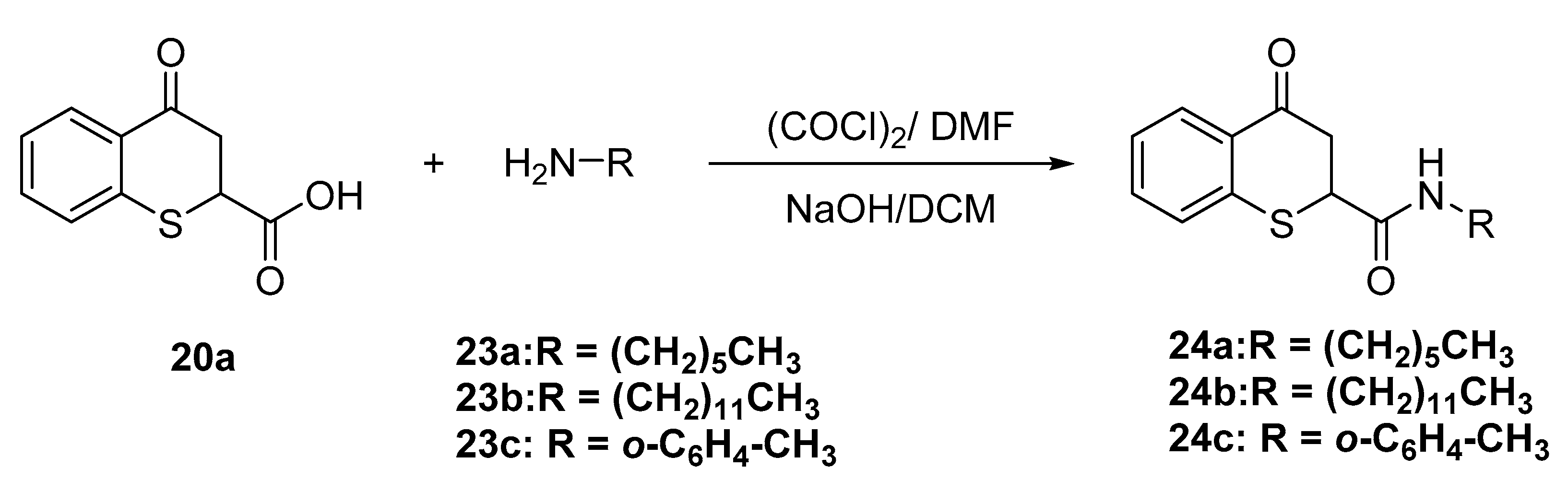
4.1.7. General Procedure for the Synthesis of the -4-oxo-thiochromane-2-carboxamides 24a–c
To a mixture of 4-oxo-thiochromane-2-carboxylic acid (20a, 1.0 g, 4.8 mmol) in anhydrous DCM (5 mL), at 0 °C was added a small amount of DMF, followed by oxalyl chloride (10.0 mmol). Once the effervescence stopped, the solution was quenched to room temperature for 2 h. FT-IR analyses determined that the acid chloride was formed as a brownish oil since Fermi resonance was observed. Next, a solution of amine (10.0 mmol) in DCM (5 mL) and NaOH (2.0 M, 5 mL) was slowly added to the initial mixture, under Schotten-Baumann conditions and stirred to room temperature overnight to give to corresponding amides. When the reaction was completed as determined by TLC, the reaction mixture was diluted with ethyl acetate and washed with brine (2 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, and the residue after solvent evaporation was purified by flash chromatography using hexanes/ethyl acetate (80:20 v/v) as eluent to give pure molecules 3i–k in 30–35% yield.
N-hexyl-4-oxo-thiochromane-2-carboxamide (24a)
White solid. m.p.: 128–129 °C. 1H-NMR (300 MHz, CDCl3) δ 8.13 (d, J = 7.9 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.33–7.22 (m, 2H), 6.56 (t, J = 5.9 Hz, 1H), 4.05 (t, J = 5.4 Hz, 1H), 3.48 (dd, J = 16.8, 6.5 Hz, 1H), 3.36–3.05 (m, 3H), 1.41 (p, J = 7.1 Hz, 2H), 1.23 (td, J = 12.1, 10.4, 5.6 Hz, 6H), 0.87 (t, J = 6.6 Hz, 3H). 13C-NMR (75 MHz, CDCl3) δ 192.52, 168.50, 138.06, 133.66, 131.04, 129.19, 127.47, 126.00, 77.53, 77.11, 76.68, 43.57, 41.75, 40.07, 31.39, 29.29, 26.31, 22.52, 14.04. IR ν (cm−1) = 3331.59, 2926.30, 2859.39, 1658.69, 755.77. HRMS-ESI (m/z): calcd. for C16H21NO2S [M + H]+ 292.1366, found 292.1393.
N-dodecyl-4-oxo-thiochromane-2-carboxamide (24b)
White solid, m.p.: 125–127 °C. 1H-NMR (300 MHz, CDCl3) δ 8.14 (d, J = 7.9 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.34–7.22 (m, 2H), 6.50 (d, J = 7.2 Hz, 1H), 4.05 (t, J = 5.4 Hz, 1H), 3.49 (dd, J = 16.7, 6.5 Hz, 1H), 3.37–3.08 (m, 3H), 1.49–1.14 (m, 19H), 0.97–0.86 (m, 3H). 13C-NMR (75 MHz, CDCl3) δ 192.44, 168.41, 138.00, 133.66, 131.08, 129.24, 127.48, 126.04, 77.52, 77.09, 76.67, 43.59, 41.75, 40.09, 31.96, 29.68, 29.68, 29.61, 29.53, 29.40, 29.40, 29.34, 29.24, 29.24, 26.66, 22.74, 14.18. IR ν (cm−1) = 3365.75, 2979.88, 2913.42, 1686.63, 779.84. HRMS-ESI (m/z): calcd. for C22H33NO2S [M + H]+ 376.2305, found 376.2314.
4-Oxo-N-(o-tolyl)thiochromane-2-carboxamide (24c)
Yellowish solid, m.p.: 144–146 °C. 1H-NMR (300 MHz, CDCl3) δ 7.61 (s, 1H), 7.40 (d, J = 7.8 Hz, 1H), 6.95 (d, J = 7.9 Hz, 1H), 6.71 (t, J = 7.7 Hz, 1H), 6.64–6.36 (m, 5H), 6.33 (d, J = 7.3 Hz, 1H), 3.48 (d, J = 5.5 Hz, 1H), 2.88 (dd, J = 17.1, 4.9 Hz, 1H), 2.45 (dd, J = 16.9, 4.0 Hz, 1H), 1.33 (s, 3H). 13C- NMR (75 MHz, CDCl3) δ 192.00, 166.95, 137.28, 134.97, 133.87, 131.26, 130.54, 129.57, 129.51, 129.41, 127.64, 126.82, 126.64, 126.46, 125.71, 122.97, 77.54, 77.11, 76.69, 44.11, 41.51, 17.43. IR ν (cm−1) = 3261.48, 1673.35, 1650.48, 748.51. HRMS-ESI (m/z): calcd. for C17H15NO2S [M + H]+ 298.0896, found 298.0919
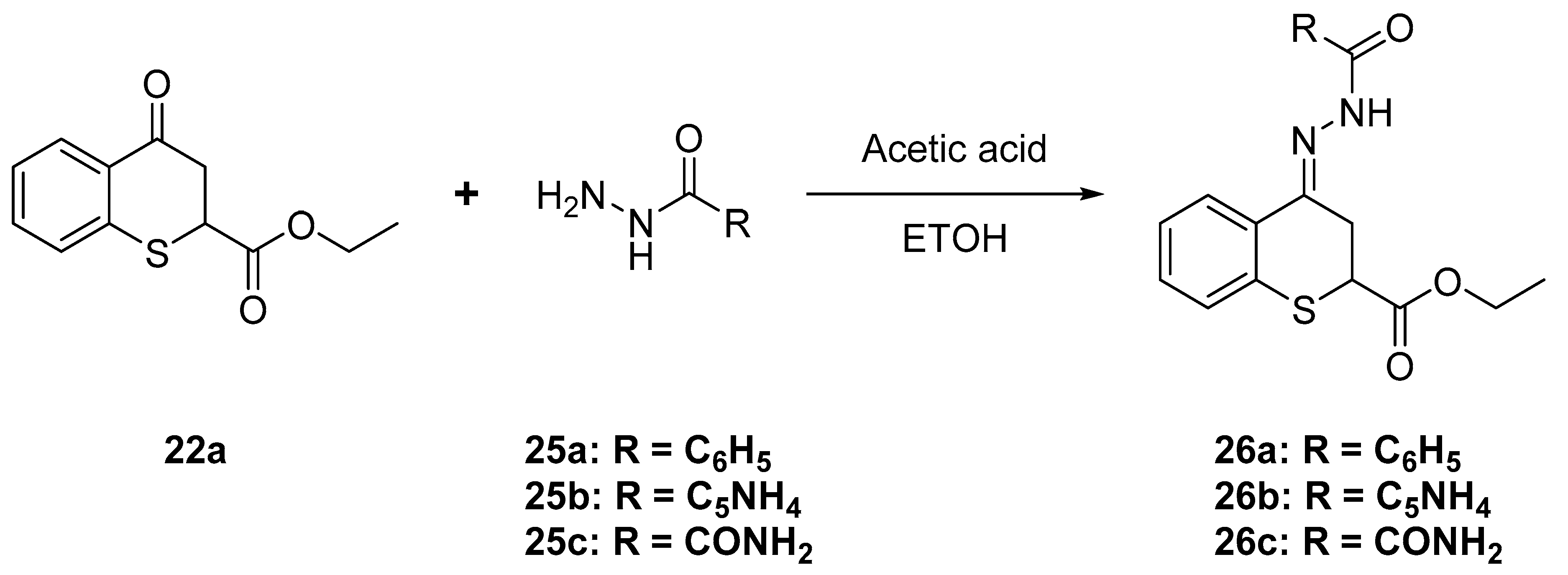
4.1.8. General Procedure for the Synthesis of Thiochromane Hydrazones
Ethyl 4-oxo-thiochromane-2-carboxylate (22a, 500 mg) was dissolved in anhydrous methanol (25 mL). The mixture was heated at reflux for 12 h with the corresponding hydrazide (1.6 mmol) and glacial acetic acid (100 µL). When the reaction was completed, as determined by the resulting precipitate, the acyl hydrazones were collected by filtration and washed with methanol to give pure products 26a–c as white solids in 90–98% yield.
Ethyl 4-(2-benzoylhydrazineylidene)thiochromane-2-carboxylate (26a)
White solid m.p.: 170–171 °C. 1H-NMR (300 MHz, DMSO-d6) δ 11.20 (s, 1H), 8.77 (d, J = 5.3 Hz, 3H), 8.15 (d, J = 7.9 Hz, 1H), 7.80 (d, J = 5.1 Hz, 2H), 7.29 (tt, J = 15.7, 7.4 Hz, 4H), 4.38 (dd, J = 6.7, 4.5 Hz, 1H), 4.08 (q, J = 7.1 Hz, 3H), 3.22 (dd, J = 17.5, 4.6 Hz, 1H), 1.13 (q, J = 8.2, 7.1 Hz, 4H). 13C-NMR (75 MHz, DMSO) δ 170.41, 163.11, 152.94, 150.62, 141.41, 133.54, 131.33, 130.66, 128.21, 127.05, 126.27, 122.39, 61.85, 40.72, 40.58, 40.44, 40.17, 39.89, 39.61, 39.34, 39.05, 30.28, 14.32. IR ν (cm−1) = 3173.40, 2978.57, 1718.42, 1652.27, 754.99, 734.77. HRMS-ESI (m/z): calcd. for C19H18N2O3S [M + H]+ 355.1111, found 355.1135.
Ethyl 4-(2-isonicotinoylhydrazineylidene)thiochromane-2-carboxylate (26b)
White solid, m.p.: 190–192 °C. 1H-NMR (300 MHz, DMSO-d6) δ 10.97 (s, 1H), 8.14 (s, 1H), 7.88 (d, J = 7.4 Hz, 2H), 7.55 (dt, J = 14.8, 7.2 Hz, 3H), 7.25 (d, J = 18.9 Hz, 3H), 4.38 (t, J = 5.5 Hz, 1H), 4.08 (q, J = 7.2 Hz, 2H), 1.12 (t, J = 7.1 Hz, 3H). 13C-NMR (75 MHz, DMSO) δ 170.43, 164.52, 134.38, 134.37, 133.29, 132.09, 131.61, 130.34, 128.79, 128.46, 128.18, 126.92, 126.21, 61.81, 30.11, 14.33. IR ν (cm−1) = 3145.28, 2947.51, 1717.64, 1651.30, 759.40, 708.68. HRMS-ESI (m/z): calcd. for C18H17N3O3S [M + H]+ 356.1063, found 356.1096.
4-(2-Carbamoylhydrazineylidene)thiochromane-2-carboxylic acid (26c)
White solid, m.p.: 238 °C. 1H-NMR (300 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.73 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.21 (d, J = 4.0 Hz, 2H), 7.14 (dq, J = 8.3, 4.3 Hz, 1H), 6.58 (s, 2H), 4.18 (dd, J = 6.8, 4.7 Hz, 1H), 3.12 (dd, J = 17.6, 7.0 Hz, 1H), 2.99 (dd, J = 17.6, 4.9 Hz, 1H).13C-NMR (75 MHz, DMSO-d6) δ 171.99, 157.60, 141.08, 132.46, 132.16, 129.08, 128.17, 126.80, 126.10, 41.16, 30.31. IR ν (cm−1) = 3469.94, 3237.11, 2906.14, 1713.23, 1645.36, 757.97, 736.96. HRMS-ESI (m/z): calcd. for C11H11N3O3S [M + H]+ 266.0594, found 266.0595.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





