
Phage Display-Based Nanotechnology Applications in Cancer Immunotherapy




Abstract
:1. Introduction
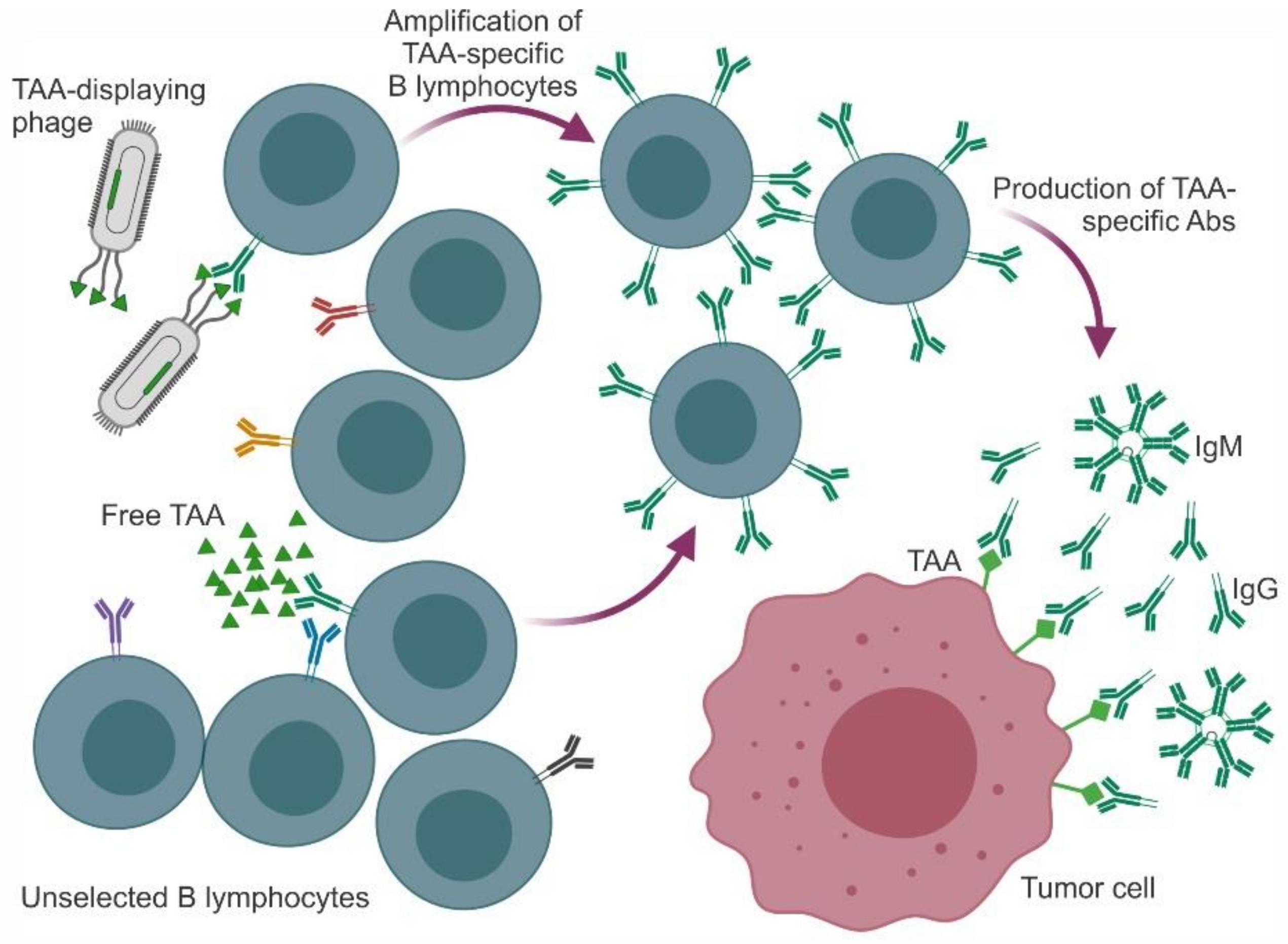
2. Mimotopes of Tumor-Associated Antigens
2.1. Mimotopes of CD20
2.2. Mimotopes of the Epidermal Growth Factor Receptor
2.3. Mimotopes of HER2
2.4. Miscellaneous Mimotopes
2.5. Mimotopes of Unknown Antigens
2.6. Mimotopes of Tumor-Associated Carbohydrate Antigens
2.7. Conclusions for this Section
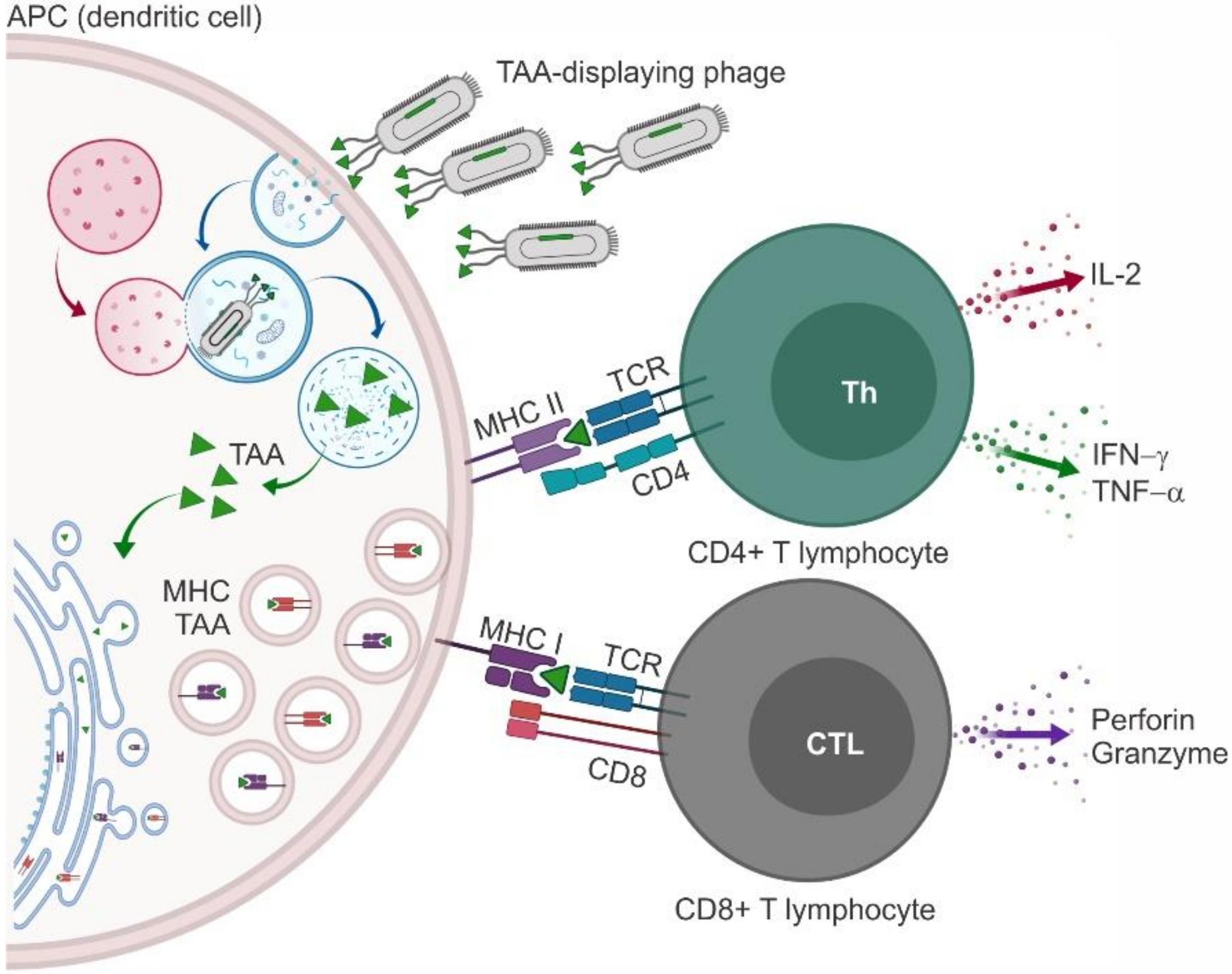
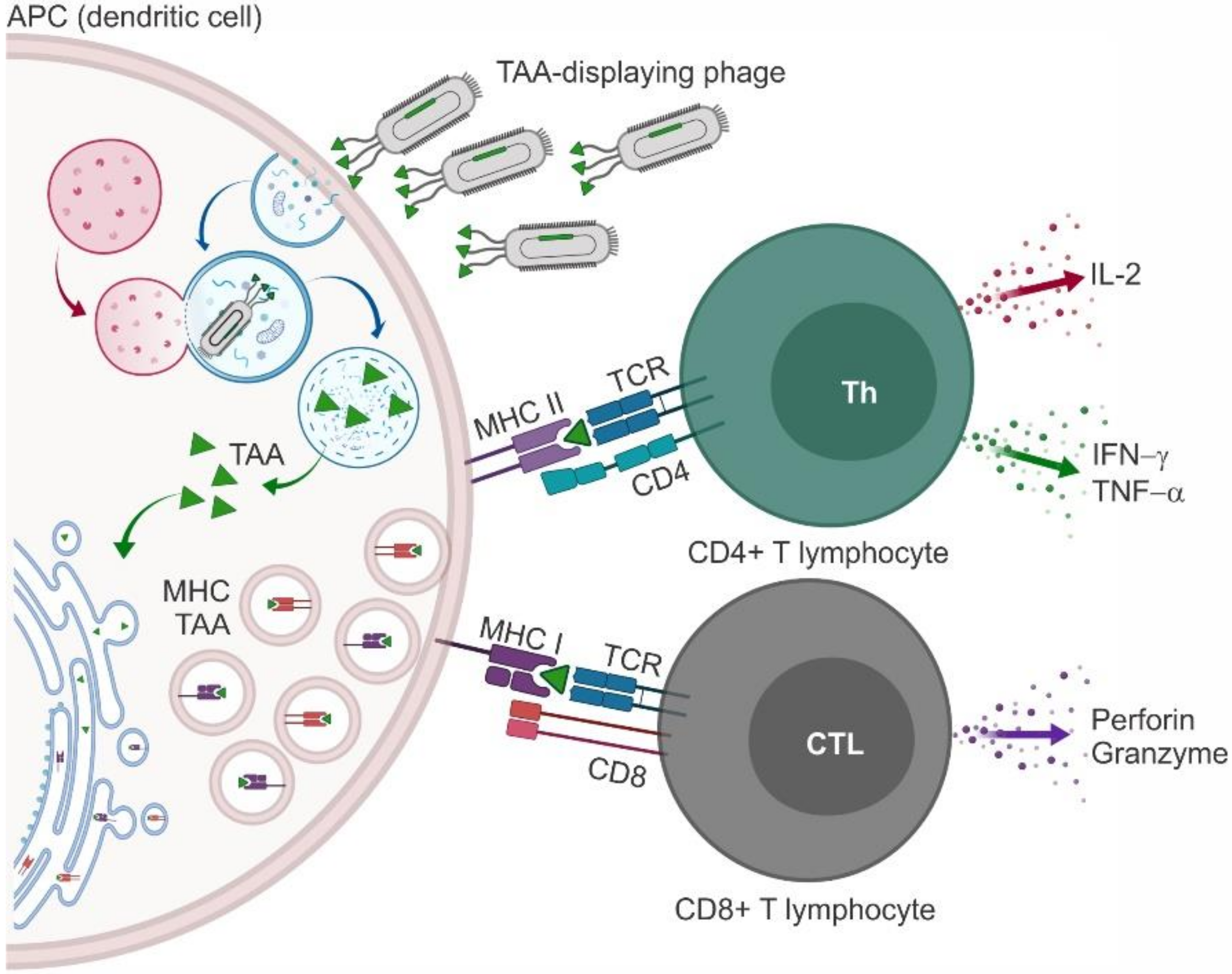
3. Phages as Nanocarriers for Anticancer Vaccines
3.1. Phage Particles Displaying Antigenic Portions of Melanoma Antigen Gene and Related Proteins
3.2. Phage Particles Displaying Antigenic Portions of HER2
3.3. Phage Particles Displaying Antigenic Portions of Mucin 1
3.4. Phage Particles Displaying Antigenic Portions of VEGFR2
3.5. Phage Particles Exposing Phage Display-Derived TAA Mimotopes (Miscellaneous)
3.6. Clinical Trials with Phage-Based Anticancer Vaccines
3.7. Conclusions for this Section
4. Phage Display-Derived Peptides as Nanomodulators of the Immune Response
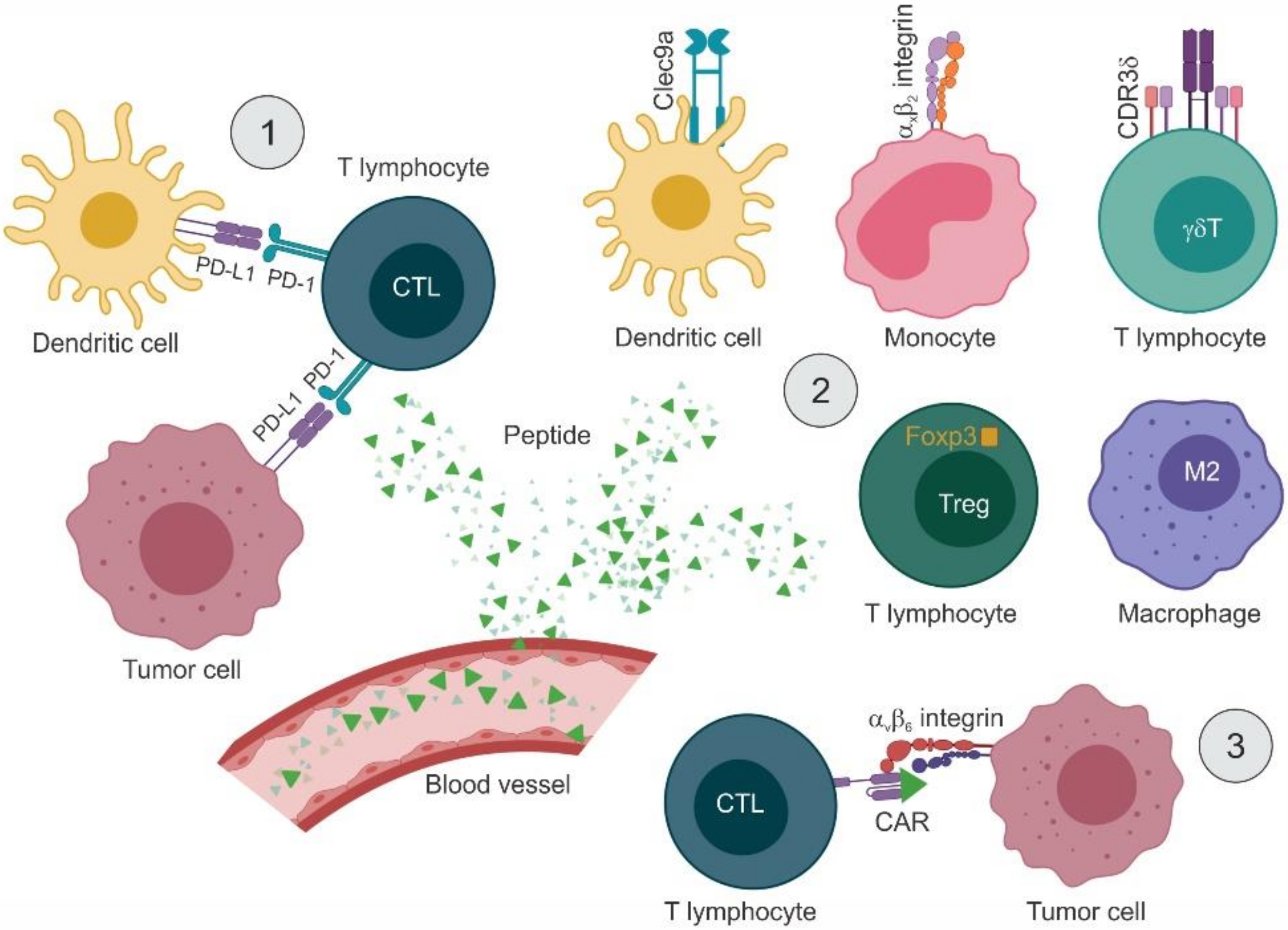
4.1. Targeting the Immune System Indirectly
4.2. Targeting the PD-1/PD-L1 Axis with Peptide Inhibitors
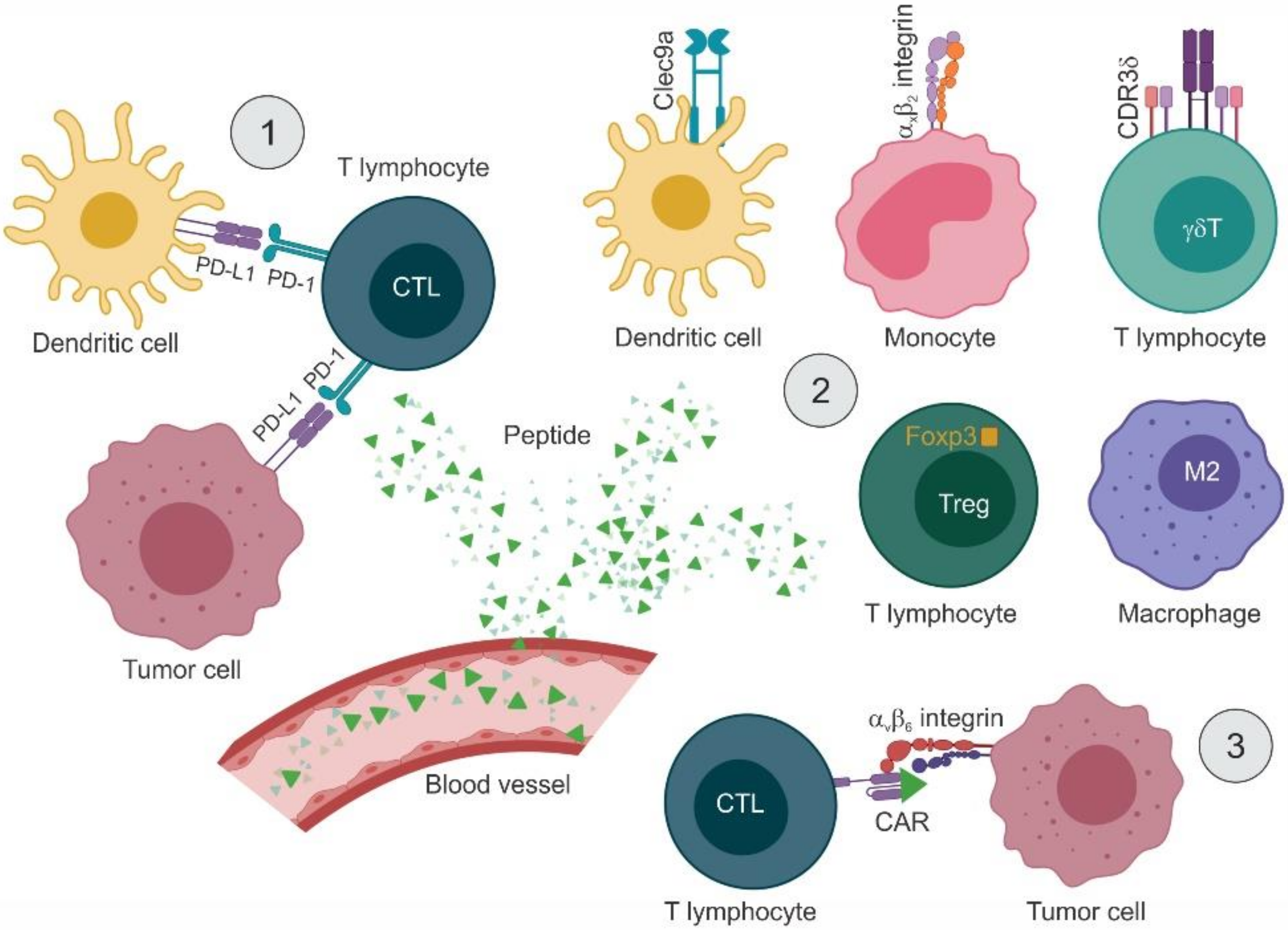
4.3. Modulating the Activity of Immune Cells
4.3.1. Inducing Immune Responses by Targeting APCs and T Lymphocytes
4.3.2. Reducing Immunosuppression by Targeting Tumor-Associated Macrophages and Treg Cells
4.4. Conclusions for this Section
5. Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Teicher, B.A. Molecular targets and cancer therapeutics: Discovery, development and clinical validation. Drug Resist. Updat. 2000, 3, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Thotathil, Z.; Jameson, M.B. Early experience with novel immunomodulators for cancer treatment. Expert Opin. Investig. Drugs 2007, 16, 1391–1403. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.L.; Jewell, C.M. Phage display as a tool for vaccine and immunotherapy development. Bioeng. Transl. Med. 2019, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunology 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spodzieja, M.; Lach, S.; Iwaszkiewicz, J.; Cesson, V.; Kalejta, K.; Olive, D.; Michielin, O.; Speiser, D.E.; Zoete, V.; Derré, L.; et al. Design of short peptides to block BTLA/HVEM interactions for promoting anticancer T-cell responses. PLoS ONE 2017, 12, e0179201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genome Res. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, S.; Lv, J.; Lin, Y.; Zhou, L.; Han, L. Phage Display Technology and its Applications in Cancer Immunotherapy. Anti-Cancer Agents Med. Chem. 2019, 19, 229–235. [Google Scholar] [CrossRef]
- Curdy, N.; Lanvin, O.; Laurent, C.; Fournié, J.-J.; Franchini, D.-M. Regulatory Mechanisms of Inhibitory Immune Checkpoint Receptors Expression. Trends Cell Boil. 2019, 29, 777–790. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Choi, G.; de Bruyn, M.; Wiersma, V.R.; Bremer, E. Antibody-Based Cancer Therapy: Successful Agents and Novel Approaches. Int. Rev. Cell Mol. Biol. 2017, 331, 289–383. [Google Scholar] [CrossRef] [PubMed]
- Kijanka, M.; Dorresteijn, B.; Oliveira, S.; Henegouwen, P.M.V.B.E. Nanobody-based cancer therapy of solid tumors. Nanomedicine 2015, 10, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-H.; Kim, M.R.; Jang, J.H.; Na, H.-J.; Lee, S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254. [Google Scholar] [CrossRef]
- Yonezawa, A.; Dutt, S.; Chester, C.; Kim, J.; Kohrt, H.E. Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin. Cancer Res. 2015, 21, 3113–3120. [Google Scholar] [CrossRef] [Green Version]
- Brämswig, K.H.; Knittelfelder, R.; Gruber, S.; Untersmayr, E.; Riemer, A.B.; Szalai, K.; Horvat, R.; Kammerer, R.; Zimmermann, W.; Zielinski, C.C.; et al. Immunization with Mimotopes Prevents Growth of Carcinoembryonic Antigen Positive Tumors in BALB/c Mice. Clin. Cancer Res. 2007, 13, 6501–6508. [Google Scholar] [CrossRef] [Green Version]
- Knittelfelder, R.; Riemer, A.B.; Jensen-Jarolim, E. Mimotope vaccination--from allergy to cancer. Expert Opin. Boil. Ther. 2009, 9, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Rafii, S. Vaccination against tumor neovascularization: Promise and reality. Cancer Cell 2002, 2, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Riemer, A.B.; Jensen-Jarolim, E. Mimotope vaccines: Epitope mimics induce anti-cancer antibodies. Immunol. Lett. 2007, 113, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Szalai, K.; Jensen-Jarolim, E.; Pali-Schöll, I. Vaccination strategies based on the mimotope concept. G. Ital. di Dermatol. e Venereol. 2008, 143, 95. [Google Scholar]
- Zhao, L.; Liu, Z.; Fan, D. Overview of mimotopes and related strategies in tumor vaccine development. Expert Rev. Vaccines 2008, 7, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Kochin, V.; Nishikawa, H. Meddling with meddlers: Curbing regulatory T cells and augmenting antitumor immunity. Nagoya J. Med. Sci. 2019, 81, 1–18. [Google Scholar] [PubMed]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef]
- Arab, A.; Behravan, N.; Razazn, A.; Barati, N.; Mosaffa, F.; Nicastro, J.; Slavcev, R.; Behravan, J. The viral approach to breast cancer immunotherapy. J. Cell Physiol. 2019, 234, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ganesan, A.; Okoye, I.; Arutyunova, E.; Elahi, S.; Lemieux, M.J.; Barakat, K. Targeting B7-1 in immunotherapy. Med. Res. Rev. 2019. [Google Scholar] [CrossRef]
- Yao, V.J.; D’Angelo, S.; Butler, K.S.; Theron, C.; Smith, T.L.; Marchio’, S.; Gelovani, J.G.; Sidman, R.L.; Dobroff, A.S.; Brinker, C.J.; et al. Ligand-targeted theranostic nanomedicines against cancer. J. Control. Release 2016, 240, 267–286. [Google Scholar] [CrossRef] [Green Version]
- Hardy, B.; Raiter, A. A mimotope peptide-based anti-cancer vaccine selected by BAT monoclonal antibody. Vaccine 2005, 23, 4283–4291. [Google Scholar] [CrossRef]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Dammacco, F. CD20 Mimicry by a mAb Rituximab-Specific Linear Peptide: A Potential Tool for Active Immunotherapy of Autoimmune Diseases. Ann. N. Y. Acad. Sci. 2005, 1051, 672–683. [Google Scholar] [CrossRef]
- Li, M.; Yan, Z.; Han, W.; Zhang, Y. Mimotope vaccination for epitope-specific induction of anti-CD20 antibodies. Cell. Immunol. 2006, 239, 136–143. [Google Scholar] [CrossRef]
- Perosa, F.; Favoino, E.; Caragnano, M.A.; Dammacco, F. Generation of biologically active linear and cyclic peptides has revealed a unique fine specificity of rituximab and its possible cross-reactivity with acid sphingomyelinase-like phosphodiesterase 3b precursor. Blood 2006, 107, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Perosa, F.; Favoino, E.; Vicenti, C.; Merchionne, F.; Dammacco, F. Identification of an antigenic and immunogenic motif expressed by two 7-mer rituximab-specific cyclic peptide mimotopes: Implication for peptide-based active immunotherapy. J. Immunol. 2007, 179, 7967–7974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perosa, F.; Favoino, E.; Vicenti, C.; Guarnera, A.; Racanelli, V.; De Pinto, V.; Dammacco, F. Two structurally different rituximab-specific CD20 mimotope peptides reveal that rituximab recognizes two different CD20-associated epitopes. J. Immunol. 2009, 182, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favoino, E.; Prete, M.; Catacchio, G.; Conteduca, G.; Perosa, F. CD20-Mimotope Peptides: A Model to Define the Molecular Basis of Epitope Spreading. Int. J. Mol. Sci. 2019, 20, 1920. [Google Scholar] [CrossRef] [Green Version]
- Riemer, A.B.; Hantusch, B.; Sponer, B.; Kraml, G.; Hafner, C.; Zielinski, C.C.; Scheiner, O.; Pehamberger, H.; Jensen-Jarolim, E. High-molecular-weight melanoma-associated antigen mimotope immunizations induce antibodies recognizing melanoma cells. Cancer Immunol. Immunother. 2005, 54, 677–684. [Google Scholar] [CrossRef]
- Benhar, I.; Azriel, R.; Nahary, L.; Shaky, S.; Berdichevsky, Y.; Tamarkin, A.; Wels, W. Highly efficient selection of phage antibodies mediated by display of antigen as Lpp-OmpA′ fusions on live bacteria. J. Mol. Boil. 2000, 301, 893–904. [Google Scholar] [CrossRef]
- Hartmann, C.; Müller, N.; Blaukat, A.; Koch, J.; Benhar, I.; Wels, W.S. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-EGFR immune response. Oncogene 2010, 29, 4517–4527. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jiang, H.; Shi, B.; Wang, H.; Li, J.; Wang, H.; Yao, M.; Li, Z. Identification and characterization of Ch806 mimotopes. Cancer Immunol. Immunother. 2010, 59, 1481–1487. [Google Scholar] [CrossRef]
- Navari, M.; Zare, M.; Javanmardi, M.; Asadi-Ghalehni, M.; Modjtahedi, H.; Rasaee, M.J. Epitope mapping of epidermal growth factor receptor (EGFR) monoclonal antibody and induction of growth-inhibitory polyclonal antibodies by vaccination with EGFR mimotope. Immunopharmacol. Immunotoxicol. 2014, 36, 309–315. [Google Scholar] [CrossRef]
- Asadi-Ghalehni, M.; Ghaemmaghami, M.; Klimka, A.; Javanmardi, M.; Navari, M.; Rasaee, M.J. Cancer immunotherapy by a recombinant phage vaccine displaying EGFR mimotope: An in vivo study. Immunopharmacol. Immunotoxicol. 2015, 37, 1–6. [Google Scholar] [CrossRef]
- Wang, A.; Cui, M.; Qu, H.; Di, J.; Wang, Z.; Xing, J.; Wu, F.; Wu, W.; Wang, X.; Shen, L.; et al. Induction of anti-EGFR immune response with mimotopes identified from a phage display peptide library by panitumumab. Oncotarget 2016, 7, 75293–75306. [Google Scholar] [CrossRef] [Green Version]
- Riemer, A.B.; Klinger, M.; Wagner, S.; Bernhaus, A.; Mazzucchelli, L.; Pehamberger, H.; Scheiner, O.; Zielinski, C.C.; Jensen-Jarolim, E. Generation of Peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J. Immunol. 2004, 173, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Singer, J.; Manzano-Szalai, K.; Fazekas, J.; Thell, K.; Bentley-Lukschal, A.; Stremnitzer, C.; Roth-Walter, F.; Weghofer, M.; Ritter, M.; Tossi, K.P.; et al. Proof of concept study with an HER-2 mimotope anticancer vaccine deduced from a novel AAV-mimotope library platform. Onco. Immunol. 2016, 5, e1171446. [Google Scholar] [CrossRef] [Green Version]
- Vaisman, N.; Nissim, A.; Klapper, L.N.; Tirosh, B.; Yarden, Y.; Sela, M. Specific inhibition of the reaction between a tumor-inhibitory antibody and the ErbB-2 receptor by a mimotope derived from a phage display library. Immunol. Lett. 2000, 75, 61–67. [Google Scholar] [CrossRef]
- Itoh, K.; Inoue, K.; Tezuka, T.; Tada, H.; Hashimoto, Y.; Masuko, T.; Suzuki, T. Molecular structural and functional characterization of tumor suppressive anti-ErbB-2 monoclonal antibody by phage display system. J. Biochem. 2003, 133, 239–245. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Zhong, C.P.; Xu, W.F.; Lin, G.M.; Ye, G.Q.; Ji, Y.Y.; Sun, B.; Yeh, M. PSMA mimotope isolated from phage displayed peptide library can induce PSMA specific immune response. Cell Res. 1999, 9, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, A.; Suriano, R.; Chaudhuri, D.; Rajoria, S.; George, A.; Mittelman, A.; Tiwari, R.K. Identification of PSA peptide mimotopes using phage display peptide library. Peptide 2011, 32, 1097–1102. [Google Scholar] [CrossRef]
- Li, W.; Ran, Y.; Li, M.; Zhang, K.; Qin, X.; Xue, X.; Zhang, C.; Hao, Q.; Zhang, W.; Zhang, Y. Mimotope vaccination for epitope-specific induction of anti-VEGF antibodies. BMC Biotechnol. 2013, 13, 77. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.-Y.; Wu, K.-C.; He, F.-T.; Han, Q.-L.; Nie, Y.-Z.; Han, Y.; Liu, X.-N.; Zheng, J.-Y.; Xu, M.-H.; Lin, T.; et al. Screening and identification of mimotope of gastric cancer associated antigen MGb1-Ag. World J. Gastroenterol. 2003, 9, 1920–1924. [Google Scholar] [CrossRef]
- Xu, L.; Jin, B.-Q.; Fan, D.-M. Selection and identification of mimic epitopes for gastric cancer-associated antigen MG7 Ag. Mol. Cancer Ther. 2003, 2, 301–306. [Google Scholar]
- Hang, Q.-L.; Ding, J.; Gong, A.-C.; Yu, Z.-C.; Qiao, T.-D.; Chen, B.-J.; Zhang, X.-Y.; Fan, D.-M. Screening of bioactive peptide that mimic the epitope of gastric cancer associated antigen. Chin. J. Cell. Mol. Immunol. 2003, 19, 308–310. [Google Scholar]
- Shi, R.; Hong, L.; Wu, D.; Ning, X.; Chen, Y.; Lin, T.; Fan, D.; Wu, K. Enhanced immune response to gastric cancer specific antigen Peptide by coencapsulation with CpG oligodeoxynucleotides in nanoemulsion. Cancer Boil. Ther. 2005, 4, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, K.; Guo, C.; Liu, C.; Han, S.; Lin, T.; Ning, X.; Shi, R.; Shi, Y.; Fan, D. A novel DNA vaccine containing 4 mimicry epitopes for gastric cancer. Cancer Boil. Ther. 2005, 4, 308–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popkov, M.; Sidrac-Ghali, S.; Alakhov, V.; Mandeville, R. Epitope-specific antibody response to HT-1080 fibrosarcoma cells by mimotope immunization. Clin. Cancer Res. 2000, 6, 3629–3635. [Google Scholar] [PubMed]
- Hardy, B.; Yampolski, I.; Kovjazin, R.; Galli, M.; Novogrodsky, A. A monoclonal antibody against a human B lymphoblastoid cell line induces tumor regression in mice. Cancer Res. 1994, 54, 5793–5796. [Google Scholar] [PubMed]
- Feinmesser, M.; Raiter, A.; Hardy, B. Prevention of melanoma metastases in lungs of BAT treated and peptide immunized mice. Int. J. Oncol. 2006, 29, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Kieber-Emmons, T.; Saha, S.; Pashov, A.; Monzavi-Karbassi, B.; Murali, R. Carbohydrate-Mimetic Peptides for Pan Anti-Tumor Responses. Front. Immunol. 2014, 5, 308. [Google Scholar] [CrossRef] [Green Version]
- Taki, T.; Ishikawa, D.; Hamasaki, H.; Handa, S. Preparation of peptides which mimic glycosphingolipids by using phage peptide library and their modulation on β-galactosidase activity. FEBS Lett. 1997, 418, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, D.; Kikkawa, H.; Ogino, K.; Hirabayashi, Y.; Oku, N.; Taki, T. GD1α-replica peptides functionally mimic GD1α, an adhesion molecule of metastatic tumor cells, and suppress the tumor metastasis. FEBS Lett. 1998, 441, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Bolesta, E.; Kowalczyk, A.; Wierzbicki, A.; Rotkiewicz, P.; Bambach, B.; Tsao, C.-Y.; Horwacik, I.; Kolinski, A.; Rokita, H.; Brecher, M.; et al. DNA vaccine expressing the mimotope of GD2 ganglioside induces protective GD2 cross-reactive antibody responses. Cancer Res. 2005, 65, 3410–3418. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.; Wierzbicki, A.; Gil, M.; Bambach, B.; Kaneko, Y.; Rokita, H.; Repasky, E.; Fenstermaker, R.; Brecher, M.; Ciesielski, M.; et al. Induction of protective immune responses against NXS2 neuroblastoma challenge in mice by immunotherapy with GD2 mimotope vaccine and IL-15 and IL-21 gene delivery. Cancer Immunol. Immunother. 2007, 56, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.; Gil, M.; Ciesielski, M.; Fenstermaker, R.A.; Kaneko, Y.; Rokita, H.; Lau, J.T.; Kozbor, D. Immunization with a mimotope of GD2 ganglioside induces CD8+ T cells that recognize cell adhesion molecules on tumor cells. J. Immunol. 2008, 181, 6644–6653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozbor, D. Cancer vaccine with mimotopes of tumor-associated carbohydrate antigens. Immunol. Res. 2010, 46, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwacik, I.; Czaplicki, D.; Talarek, K.; Kowalczyk, A.; Bolesta, E.; Kozbor, D.; Rokita, H. Selection of novel peptide mimics of the GD2 ganglioside from a constrained phage-displayed peptide library. Int. J. Mol. Med. 2007, 19, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förster-Waldl, E.; Riemer, A.B.; Dehof, A.K.; Neumann, D.; Brämswig, K.; Boltz-Nitulescu, G.; Pehamberger, H.; Zielinski, C.C.; Scheiner, O.; Pollak, A.; et al. Isolation and structural analysis of peptide mimotopes for the disialoganglioside GD2, a neuroblastoma tumor antigen. Mol. Immunol. 2005, 42, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Fest, S.; Huebener, N.; Weixler, S.; Bleeke, M.; Zeng, Y.; Strandsby, A.; Volkmer-Engert, R.; Landgraf, C.; Gaedicke, G.; Riemer, A.B.; et al. Characterization of GD2 Peptide Mimotope DNA Vaccines Effective against Spontaneous Neuroblastoma Metastases. Cancer Res. 2006, 66, 10567–10575. [Google Scholar] [CrossRef] [Green Version]
- Bleeke, M.; Fest, S.; Huebener, N.; Landgraf, C.; Schraven, B.; Gaedicke, G.; Volkmer, R.; Lode, H.N. Systematic amino acid substitutions improved efficiency of GD2-peptide mimotope vaccination against neuroblastoma. Eur. J. Cancer 2009, 45, 2915–2921. [Google Scholar] [CrossRef]
- Wondimu, A.; Zhang, T.; Kieber-Emmons, T.; Gimotty, P.; Sproesser, K.; Somasundaram, R.; Ferrone, S.; Tsao, C.-Y.; Herlyn, R. Peptides mimicking GD2 ganglioside elicit cellular, humoral and tumor-protective immune responses in mice. Cancer Immunol. Immunother. 2008, 57, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Popa, I.; Ishikawa, D.; Tanaka, M.; Ogino, K.; Portoukalian, J.; Taki, T. GD3-replica peptides selected from a phage peptide library induce a GD3 ganglioside antibody response. FEBS Lett. 2006, 580, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Heimburg-Molinaro, J.; Almogren, A.; Morey, S.; Glinskii, O.V.; Roy, R.; Wilding, G.E.; Cheng, R.P.; Glinsky, V.V.; Rittenhouse-Olson, K. Development, Characterization, and Immunotherapeutic Use of Peptide Mimics of the Thomsen-Friedenreich Carbohydrate Antigen. Neoplasia 2009, 11, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Tuccillo, F.M.; Palmieri, C.; Fiume, G.; de Laurentiis, A.; Schiavone, M.; Falcone, C.; Iaccino, E.; Galandrini, R.; Capuano, C.; Santoni, A.; et al. Cancer-associated CD43 glycoforms as target of immunotherapy. Mol. Cancer Ther. 2014, 13, 752–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhoul, I.; Hutchins, L.; Emanuel, P.D.; Pennisi, A.; Siegel, E.; Jousheghany, F.; Monzavi-Karbassi, B.; Kieber-Emmons, T. Moving a Carbohydrate Mimetic Peptide into the clinic. Hum. Vaccin. Immunother. 2015, 11, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, L.F.; Makhoul, I.; Emanuel, P.D.; Pennisi, A.; Siegel, E.R.; Jousheghany, F.; Guo, X.; Pashov, A.D.; Monzavi-Karbassi, B.; Kieber-Emmons, T. Targeting tumor-associated carbohydrate antigens: A phase I study of a carbohydrate mimetic-peptide vaccine in stage IV breast cancer subjects. Oncotarget 2017, 8, 99161–99178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Houten, N.E.; Henry, K.A.; Smith, G.P.; Scott, J.K. Engineering filamentous phage carriers to improve focusing of antibody responses against peptides. Vaccine 2010, 28, 2174–2185. [Google Scholar] [CrossRef] [Green Version]
- Garg, P. Filamentous bacteriophage: A prospective platform for targeting drugs in phage-mediated cancer therapy. J. Cancer Res. Ther. 2019, 15, S1–S10. [Google Scholar] [CrossRef]
- Gaubin, M.; Fanutti, C.; Mishal, Z.; Durrbach, A.; De Berardinis, P.; Sartorius, R.; Del Pozzo, G.; Guardiola, J.; Perham, R.N.; Piatier-Tonneau, D. Processing of Filamentous Bacteriophage Virions in Antigen-Presenting Cells Targets Both HLA Class I and Class II Peptide Loading Compartments. DNA Cell Boil. 2003, 22, 11–18. [Google Scholar] [CrossRef]
- Hashemi, H.; Pouyanfard, S.; Bandehpour, M.; Noroozbabaei, Z.; Kazemi, B.; Saelens, X.; Mokhtari-Azad, T. Immunization with M2e-Displaying T7 Bacteriophage Nanoparticles Protects against Influenza A Virus Challenge. PLoS ONE 2012, 7, e45765. [Google Scholar] [CrossRef]
- Ulivieri, C.; Citro, A.; Ivaldi, F.; Mascolo, D.; Ghittoni, R.; Fanigliulo, D.; Manca, F.; Baldari, C.T.; Pira, G.L.; Del Pozzo, G. Antigenic properties of HCMV peptides displayed by filamentous bacteriophages vs. synthetic peptides. Immunol. Lett. 2008, 119, 62–70. [Google Scholar] [CrossRef]
- Eriksson, F.; Culp, W.D.; Massey, R.; Egevad, L.; Garland, D.; Persson, M.A.; Pisa, P. Tumor specific phage particles promote tumor regression in a mouse melanoma model. Cancer Immunol. Immunother. 2007, 56, 677–687. [Google Scholar] [CrossRef]
- Bruttin, A.; Brüssow, H. Human Volunteers Receiving Escherichia coli Phage T4 Orally: A Safety Test of Phage Therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.R.; March, J.B. Bacteriophages and biotechnology: Vaccines, gene therapy and antibacterials. Trends Biotechnol. 2006, 24, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; De Aluja, A.S.; Martínez, J.J.; Hernández, M.; Rosas, G.; Villalobos, N.; Hernández, B.; Blancas, A.; Manoutcharian, K.; Gevorkian, G.; et al. Recombinant S3Pvac-phage anticysticercosis vaccine: Simultaneous protection against cysticercosis and hydatid disease in rural pigs. Veter. Parasitol. 2011, 176, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Prisco, A.; De Berardinis, P. Filamentous Bacteriophage Fd as an Antigen Delivery System in Vaccination. Int. J. Mol. Sci. 2012, 13, 5179–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, N.; Abediankenari, S. Phage Particles as Vaccine Delivery Vehicles: Concepts, Applications and Prospects. Asian Pac. J. Cancer Prev. 2015, 16, 8019–8029. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wang, G.; Yang, Q.; Song, J.; Wang, Y.; Wang, L. The potential of phage display virions expressing malignant tumor specific antigen MAGE-A1 epitope in murine model. Vaccine 2005, 23, 4860–4866. [Google Scholar] [CrossRef]
- Pascolo, S.; Bervas, N.; Ure, J.M.; Smith, A.G.; Lemonnier, F.A.; Perarnau, B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J. Exp. Med. 1997, 185, 2043–2051. [Google Scholar] [CrossRef] [Green Version]
- Sartorius, R.; Pisu, P.; D’Apice, L.; Pizzella, L.; Romano, C.; Cortese, G.; Giorgini, A.; Santoni, A.; Velotti, F.; De Berardinis, P. The use of filamentous bacteriophage fd to deliver MAGE-A10 or MAGE-A3 HLA-A2-restricted peptides and to induce strong antitumor CTL responses. J. Immunol. 2008, 180, 3719–3728. [Google Scholar] [CrossRef] [Green Version]
- Shadidi, M.; Sørensen, D.; Dybwad, A.; Furset, G.; Sioud, M. Mucosal vaccination with phage-displayed tumour antigens identified through proteomics-based strategy inhibits the growth and metastasis of 4T1 breast adenocarcinoma. Int. J. Oncol. 2008, 32, 241–247. [Google Scholar] [CrossRef]
- Wu, Y.; Wan, Y.; Bian, J.; Zhao, J.; Jia, Z.; Zhou, L.; Zhou, W.; Tan, Y. Phage display particles expressing tumor-specific antigens induce preventive and therapeutic anti-tumor immunity in murine p815 model. Int. J. Cancer 2002, 98, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Arab, A.; Nicastro, J.; Slavcev, R.; Razazan, A.; Barati, N.; Nikpoor, A.R.; Brojeni, A.A.M.; Mosaffa, F.; Badiee, A.; Jaafari, M.R.; et al. Lambda phage nanoparticles displaying HER2-derived E75 peptide induce effective E75-CD8(+) T response. Immunol. Res. 2018, 66, 200–206. [Google Scholar] [CrossRef]
- Barati, N.; Razazan, A.; Nicastro, J.; Slavcev, R.; Arab, A.; Mosaffa, F.; Nikpoor, A.R.; Badiee, A.; Jaafari, M.R.; Behravan, J. Immunogenicity and antitumor activity of the superlytic λF7 phage nanoparticles displaying a HER2/neu-derived peptide AE37 in a tumor model of BALB/c mice. Cancer Lett. 2018, 424, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasopoulou, E.A.; Voutsas, I.F.; Keramitsoglou, T.; Gouttefangeas, C.; Kalbacher, H.; Thanos, A.; Papamichail, M.; Perez, S.A.; Baxevanis, C.N. A pilot study in prostate cancer patients treated with the AE37 Ii-key-HER-2/neu polypeptide vaccine suggests that HLA-A*24 and HLA-DRB1*11 alleles may be prognostic and predictive biomarkers for clinical benefit. Cancer Immunol. Immunother. 2015, 64, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Chamani, R.; Ranji, P.; Hadji, M.; Nahvijou, A.; Esmati, E.; Alizadeh, A.M. Application of E75 peptide vaccine in breast cancer patients: A systematic review and meta-analysis. Eur. J. Pharmacol. 2018, 831, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Chianese-Bullock, K.A.; Irvin, W.P.; Petroni, G.R.; Murphy, C.; Smolkin, M.; Olson, W.C.; Coleman, E.; Boerner, S.A.; Nail, C.J.; Neese, P.Y.; et al. A Multipeptide Vaccine is Safe and Elicits T-cell Responses in Participants With Advanced Stage Ovarian Cancer. J. Immunother. 2008, 31, 420–430. [Google Scholar] [CrossRef]
- Errico, A. Breast cancer: E75-a safe and effective vaccine for the prevention of disease recurrence. Nat. Rev. Clin. Oncol. 2014, 11, 440. [Google Scholar]
- Holmes, J.P.; Benavides, L.C.; Gates, J.D.; Carmichael, M.G.; Hueman, M.T.; Mittendorf, E.A.; Murray, J.L.; Amin, A.; Craig, D.; Von Hofe, E.; et al. Results of the First Phase I Clinical Trial of the Novel Ii-Key Hybrid Preventive HER-2/neu Peptide (AE37) Vaccine. J. Clin. Oncol. 2008, 26, 3426–3433. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Schneble, E.; Van Echo, D.; Ponniah, S.; Peoples, G.E. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann. Oncol. 2014, 25, 1735–1742. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Ardavanis, A.; Symanowski, J.; Murray, J.L.; Shumway, N.M.; Litton, J.K.; Hale, D.F.; Perez, S.A.; Anastasopoulou, E.A.; Pistamaltzian, N.F.; et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann. Oncol. 2016, 27, 1241–1248. [Google Scholar] [CrossRef]
- Perez, S.A.; Kallinteris, N.L.; Bisias, S.; Tzonis, P.K.; Georgakopoulou, K.; Varla-Leftherioti, M.; Papamichail, M.; Thanos, A.; Von Hofe, E.; Baxevanis, C.N. Results from a Phase I Clinical Study of the Novel Ii-Key/HER-2/neu(776–790) Hybrid Peptide Vaccine in Patients with Prostate Cancer. Clin. Cancer Res. 2010, 16, 3495–3506. [Google Scholar] [CrossRef] [Green Version]
- Sander, A.F.; Lollini, P.-L. Virus-like antigen display for cancer vaccine development, what is the potential? Expert Rev. Vaccines 2018, 17, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Wu, X.; Kaczanowska, K.; Sungsuwan, S.; Comellas-Aragonès, M.; Pett, C.; Yu, J.; Baniel, C.; Westerlind, U.; Finn, M.; et al. Antitumor Humoral and T Cell Responses by Mucin-1 Conjugates of Bacteriophage Qβ in Wild-type Mice. ACS Chem. Boil. 2018, 13, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, B.S.; Pestana, E.S.; Hidalgo, G.G.; García, T.H.; Rodríguez, R.P.; Ullrich, A.; Fernández, L.E. Active antimetastatic immunotherapy in Lewis lung carcinoma with self EGFR extracellular domain protein in VSSP adjuvant. Int. J. Cancer 2006, 119, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Yu, F.; Zuo, S.; Zhao, M.; Wang, X.; Wang, X.; Chen, Y.; Wu, Z.; Ren, Z. Inhibition of tumor angiogenesis in lung cancer by T4 phage surface displaying mVEGFR2 vaccine. Vaccine 2011, 29, 5802–5811. [Google Scholar] [CrossRef] [PubMed]
- Asadi-Ghalehni, M.; Rasaee, M.J.; Namvar Asl, N.; Khosravani, M.; Rajabibazl, M.; Khalili, S.; Modjtahedi, H.; Sadroddiny, E. Construction of a Recombinant Phage-vaccine Capable of Reducing the Growth Rate of an Established LL2 Tumor Model. Iran J. Allergy Asthma Immunol. 2018, 17, 240–249. [Google Scholar] [PubMed]
- Samoylov, A.; Cochran, A.; Schemera, B.; Kutzler, M.; Donovan, C.; Petrenko, V.; Bartol, F.; Samoylova, T. Humoral immune responses against gonadotropin releasing hormone elicited by immunization with phage-peptide constructs obtained via phage display. J. Biotechnol. 2015, 216, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Ali, Z.; Khan, M.; Bostan, N.; Naseem, S. The dawn of phage therapy. Rev. Med. Virol. 2019, 29, e2041. [Google Scholar] [CrossRef] [PubMed]
- Roehnisch, T.; Then, C.; Nagel, W.; Blumenthal, C.; Braciak, T.; Donzeau, M.; Böhm, T.; Flaig, M.; Bourquin, C.; Oduncu, F.S. Phage idiotype vaccination: First phase I/II clinical trial in patients with multiple myeloma. J. Transl. Med. 2014, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Roehnisch, T.; Then, C.; Nagel, W.; Blumenthal, C.; Braciak, T.; Donzeau, M.; Böhm, T.; Bourquin, C.; Oduncu, F. Chemically linked phage idiotype vaccination in the murine B cell lymphoma 1 model. J. Transl. Med. 2013, 11, 267. [Google Scholar] [CrossRef] [Green Version]
- Górski, A.; Ważna, E.; Dąbrowska, B.W.; Dąbrowska, K.; Świtała-Jeleń, K.; Międzybrodzki, R. Bacteriophage translocation. FEMS Immunol. Med. Microbiol. 2006, 46, 313–319. [Google Scholar] [CrossRef]
- Pajtasz-Piasecka, E.; Rossowska, J.; Dus, D.; Weber-Dąbrowska, B.; Zabłocka, A.; Górski, A. Bacteriophages support anti-tumor response initiated by DC-based vaccine against murine transplantable colon carcinoma. Immunol. Lett. 2008, 116, 24–32. [Google Scholar] [CrossRef]
- Sunderland, K.S.; Yang, M.; Mao, C. Phage-Enabled Nanomedicine: From Probes to Therapeutics in Precision Medicine. Angew. Chem. Int. Ed. 2017, 56, 1964–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankiewicz, E.; Liivak, M.; Mankiewicz, M.L.E. Mycobacteriophages isolated from Human Sources. Nature 1967, 216, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Parent, K.; Wilson, I.D. Mycobacteriophage in Crohn’s disease. Gut 1971, 12, 1019–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renschler, M.F.; Bhatt, R.R.; Dower, W.J.; Levy, R. Synthetic peptide ligands of the antigen binding receptor induce programmed cell death in a human B-cell lymphoma. Proc. Natl. Acad. Sci. USA 1994, 91, 3623–3627. [Google Scholar] [CrossRef] [Green Version]
- Renschler, M.F.; Wada, H.G.; Fok, K.S.; Levy, R. B-lymphoma cells are activated by peptide ligands of the antigen binding receptor or by anti-idiotypic antibody to induce extracellular acidification. Cancer Res. 1995, 55, 5642–5647. [Google Scholar]
- Howell, R.C.; Revskaya, E.; Pazo, V.; Nosanchuk, J.D.; Casadevall, A.; Dadachova, E. Phage Display Library Derived Peptides that Bind to Human Tumor Melanin as Potential Vehicles for Targeted Radionuclide Therapy of Metastatic Melanoma. Bioconjugate Chem. 2007, 18, 1739–1748. [Google Scholar] [CrossRef]
- Chang, H.-N.; Liu, B.-Y.; Qi, Y.-K.; Zhou, Y.; Chen, Y.-P.; Pan, K.-M.; Li, W.-W.; Zhou, X.-M.; Ma, W.-W.; Fu, C.-Y.; et al. Blocking of the PD-1/PD-L1 Interaction by aD-Peptide Antagonist for Cancer Immunotherapy. Angew. Chem. Int. Ed. 2015, 54, 11760–11764. [Google Scholar] [CrossRef]
- Li, C.; Zhang, N.; Zhou, J.; Ding, C.; Jin, Y.; Cui, X.; Pu, K.; Zhu, Y. Peptide Blocking of PD-1/PD-L1 Interaction for Cancer Immunotherapy. Cancer Immunol. Res. 2018, 6, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liang, Z.; Tian, Y.; Cai, W.; Weng, Z.; Chen, L.; Zhang, H.; Bao, Y.; Zheng, H.; Zeng, S.; et al. High-affinity PD-1 molecules deliver improved interaction with PD-L1 and PD-L2. Cancer Sci. 2018, 109, 2435–2445. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhao, Z.; Li, Y.; Cheng, K. Discovery of small anti-PD-L1 peptides for cancer immunotherapy. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Basran, A.; Jenkins, E.; Adam, E.; Laurent, F.; Writer, M.; Oumie, A.; Sivula, J.; West, M.; Stanley, E.; Hillman, J.; et al. Preclinical evaluation of half-life extended Affimer® biotherapeutics targeting the PD-L1 pathway. Canc. Res. 2019, 79. [Google Scholar] [CrossRef]
- Son, S.; Park, J.; Seo, H.; Lee, H.T.; Heo, Y.S.; Kim, H.S. A small-sized protein binder specific for human PD-1 effectively suppresses the tumor growth in tumor mouse model. J. Drug Target 2019, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Frick, C.; Odermatt, A.; Zen, K.; Mandell, K.J.; Edens, H.; Portmann, R.; Mazzucchelli, L.; Jaye, D.L.; Parkos, C.A. Interaction of ICAM-1 with β2-integrin CD11c/CD18: Characterization of a peptide ligand that mimics a putative binding site on domain D4 of ICAM-1. Eur. J. Immunol. 2005, 35, 3610–3621. [Google Scholar] [CrossRef] [PubMed]
- Faham, A.; Altin, J.G. Ag-bearing liposomes engrafted with peptides that interact with CD11c/CD18 induce potent Ag-specific and antitumor immunity. Int. J. Cancer 2011, 129, 1391–1403. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wu, Y.; Du, J.; Li, G.; Wang, S.; Cao, W.; Zhou, X.; Wu, C.; Zhang, D.; Jing, X.; et al. A novel peptide targeting Clec9a on dendritic cell for cancer immunotherapy. Oncotarget 2016, 7, 40437–40450. [Google Scholar] [CrossRef] [Green Version]
- Kraft, S.; Diefenbach, B.; Mehta, R.; Jonczyk, A.; Luckenbach, G.A.; Goodman, S.L. Definition of an Unexpected Ligand Recognition Motif for v 6 Integrin. J. Boil. Chem. 1999, 274, 1979–1985. [Google Scholar] [CrossRef] [Green Version]
- Pameijer, C.R.; Navanjo, A.; Meechoovet, B.; Wagner, J.R.; Aguilar, B.; Wright, C.L.; Chang, W.C.; Brown, C.E.; Jensen, M.C. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Cancer Gene Ther. 2007, 14, 91–97. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Chen, H.; Wu, D.; Cui, L.; He, W. Tandem-epitope peptide: A novel stimulator for γδT cells in tumor immunotherapy. Cancer Lett. 2010, 288, 86–93. [Google Scholar] [CrossRef]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Del Rio, G.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Ruoslahti, E.; et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038. [Google Scholar] [CrossRef]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zavaljevski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [Green Version]
- Casares, N.; Rudilla, F.; Arribillaga, L.; Llopiz, D.; Riezu-Boj, J.-I.; Lozano, T.; López-Sagaseta, J.; Guembe, L.; Sarobe, P.; Prieto, J.; et al. A Peptide Inhibitor of FOXP3 Impairs Regulatory T Cell Activity and Improves Vaccine Efficacy in Mice. J. Immunol. 2010, 185, 5150–5159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, T.; Soldevilla, M.M.; Casares, N.; Villanueva, H.; Bendandi, M.; Lasarte, J.J.; Pastor, F. Targeting inhibition of Foxp3 by a CD28 2′-Fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials 2016, 91, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, Y.; Miyata, H.; Komiyama, M.; Nogami, M.; Ozawa, K.; Oshita, C.; Kume, A.; Ashizawa, T.; Sakura, N.; Mochizuki, T.; et al. The identification of affinity peptide ligands specific to the variable region of human antibodies. Biomed. Res. 2014, 35, 105–116. [Google Scholar] [CrossRef] [Green Version]
| 1 | CD20 |


{kind=link}
{kind=link}
{kind=link}
| Target | Ab | Peptide Motif | Reference |
|---|---|---|---|
| CD20 | Rituximab | ITPWPHWLERSS | [29] |
| QDKLTQWPKWLE | [30] | ||
| WPxWLE (A/S)NPS | [31] | ||
| WAANPS PYANPSL | [33] | ||
| WPKWLE PYANPSL | [34] | ||
| EGFR | Cetuximab | CQFDLSTRRLKC CQYNLSSRALKC | [35] |
| Cetuximab, Matuzumab | KTL YPLG | [37] | |
| 12H23, Ch806 | WHTEILKSYPHE LPAFFVTNQTQD | [38] | |
| ICR-62 | QHYNIVNTQSRV | [39] | |
| Panitumumab | DTDWVRMRDSAR VPGWSQAFMALA | [41] | |
| ErbB2 | Trastuzumab | CQWMAPQWGPDC | [42] |
| WxxGxAxGS | [43] | ||
| L26, N12, L288 | ALVRYKDPLFVWGFL | [44] | |
| SER4 | INNEYVESPLYM | [45] | |
| PSA | 4G5 | VDPGKYNKY EGPAKGFKL GCYEAPSKAAKC | [46] |
| RRSHPCRTCTTHTP HRKTTCTRCPATSP HRRGECRACPLLPA RRPAHCHHCPRNP | [47] | ||
| CEA | Col-1 | DRGGLWKTP | [17] |
| VEGF | Bevacizumab | DHTLYTPYHTHP | [48] |
| Gastric antigen MGb1 | MGb1 | HxQ LxS | [49] |
| Gastric antigen MG7 | MG7 | NAIYARNAQ TCHLRVYAQ SWAPVYARN | [50] |
| KPHVHTK | [51] | ||
| KPHLHFH KPHSHLH SWAPVYARAN | [53] | ||
| Human fibrosarcoma cell line | BCD-9 | GRRPGGWWMR | [54] |
| Human B lymphoblastoid cell line | BAT | PRRIKPRKIMLG QKILQQINLPRI | [28,55,56] |
| Lc4Cer, nLc4Cer | AD117m, H11 | RNVPPTFNDVYWIAF | [58] |
| GD1α | KA17 | WHWRHRIPLQLAAGR | [59] |
| GD2 | 14G2a | EDPSHSLGLDVALFM | [60,61] |
| NCDLLTGPMLCV SCQSTRMDPNCW VCNPLTGALLCS RCNPNMEPPRCF GCDALSGHLLCS | [64] | ||
| ch14.18 | CDGGWLSKGSWC CGRLKMVPDLEC | [65,66,67] | |
| ME361 | LDVVLAWRDGLSGAS GVVWRYTAPVHLGDG | [68] | |
| GD3 | 4F6 | LAPPRPRSELVFLSV PHFDSLLYPCELLGC GLAPPDYAERFFLLS RHAYRSMAEWGFLYS | [69] |
| TF-Ag | JAA-F11 | HIHGWKSPLSSL HHSHKTNLATTP GHPHYITHKPNR YPSLPVYHSLRS MHKPWSGHMQVP | [70] |
| Tumor-specific CD43 glycoforms | UN1 | TCKLLDECVPLW SFAATPHTCKLLDEC-VPLWPAEG | [71] |
| Antigen | Epitope | Phage | Peptide Motif | Model | Reference |
|---|---|---|---|---|---|
| MAGE | MAGE-A1161–169 | pfd8wf (fd derivative) | EADPTGFSY | B16-F10 melanoma | [85] |
| MAGE-A10254–262 | fd23 (fd derivative) | GLYDGMEHL | EL-4-HHD lymphoma | [87] | |
| MAGE-A3271–279 | fd23 (fd derivative) | FLWGPRALV | EL-4-HHD lymphoma | [87] | |
| MAGE-1 | T7 | Entire antigen | 4T1 breast carcinoma | [88] | |
| P1A | P1A35–43 | pc89 (fd derivative) | LPYLGWLVF | P815 mastocytoma | [89] |
| HER2 | HER2369-377 | λ | KIFGSLAFL | TUBO breast carcinoma | [90] |
| HER2776-790 | λF7 | GVGSPYVSRLLGICL | TUBO breast carcinoma | [91] | |
| MUC1 | MUC1 | Qß | PDTRPAPGSTAPPAHGVTSA | Non tumoral | [101] |
| VEGFR2 | VEGFR2 | T4 | Extracellular portion | Lewis lung carcinoma | [103] |
| EGFR | EGFR | M13-pAK8-VIII (fd derivative) | QHYNIVNTQSRV | Lewis lung carcinoma | [104] |
| Cell line | Unknown | M13KE (fd derivative) | TRTKLPRLHLQS | B16/A2Kb melanoma | [79] |
| GnRH | GnRH | f8-8 (fd derivative) | EHPSYGLA | Non-tumoral | [105] |
| Tumor idiotype | B-cell receptor | M13K07 (fd derivative) | Entire antigen | BCL1 lymphoma | [108] |
| Different myeloma idiotypes | M13K07 (fd derivative) | Entire antigen | Clinical trial in patients | [107] |
| Target | Effect | Peptide Motif | Reference |
|---|---|---|---|
| IgM λ receptor | Activation of B-lymphoma cells | KP--xRV -W--WxR Y--EDLRRR --PVDHGL | [114,115] |
| Melanin | Reversal of melanin-induced immune response | HTTHHRN TTHQFPF NPNWGPR | [116] |
| PD-L1 and PD-1 | Competitive inhibition of PD-1/PD-L1 binding | NYSKPTDRQYHFKHAHHTHNLRLP | [117] |
| SGQYASYHCWCWRDPGRSGGSK | [118] | ||
| VNYMSNQTKAPPGLSAILPYIQIE | [119] | ||
| CLP002 | [120] | ||
| Affimer® antagonists | [121] | ||
| r_G9 repeabdy | [122] | ||
| CD11c/CD18 integrin αXβ2 | Targeting of APCs and induction of CTLs | CGRWSGWPADLC | [123,124] |
| Clec9a | WPRFHSSVFHTH | [125] | |
| αvβ6 integrin | CAR | RTDLDSLRTYTL | [127] |
| CDR3δ | Activation of γδT cells | WPHNWWPHFKVK PLLPMHPMKVSH KPPTQKRRRQTM RPRTRLHTHRNR YPWHWWHSVSPW FHWSWYTPSRPS WHHPWWYPRPGV | [128] |
| M2 TAMs | Reduction of infiltration by M2 TAMs | YEQDPWGVKWWY | [130] |
| Foxp3 | Inhibition of Treg activity | RDFQSFRKMWPFFAM | [131,132] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goracci, M.; Pignochino, Y.; Marchiò, S. Phage Display-Based Nanotechnology Applications in Cancer Immunotherapy. Molecules 2020, 25, 843. https://doi.org/10.3390/molecules25040843
Goracci M, Pignochino Y, Marchiò S. Phage Display-Based Nanotechnology Applications in Cancer Immunotherapy. Molecules. 2020; 25(4):843. https://doi.org/10.3390/molecules25040843
Chicago/Turabian StyleGoracci, Martina, Ymera Pignochino, and Serena Marchiò. 2020. "Phage Display-Based Nanotechnology Applications in Cancer Immunotherapy" Molecules 25, no. 4: 843. https://doi.org/10.3390/molecules25040843
APA StyleGoracci, M., Pignochino, Y., & Marchiò, S. (2020). Phage Display-Based Nanotechnology Applications in Cancer Immunotherapy. Molecules, 25(4), 843. https://doi.org/10.3390/molecules25040843