
Interplay between Polymorphism and Isostructurality in the 2-Fur- and 2-Thenaldehyde Semi- and Thiosemicarbazones

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
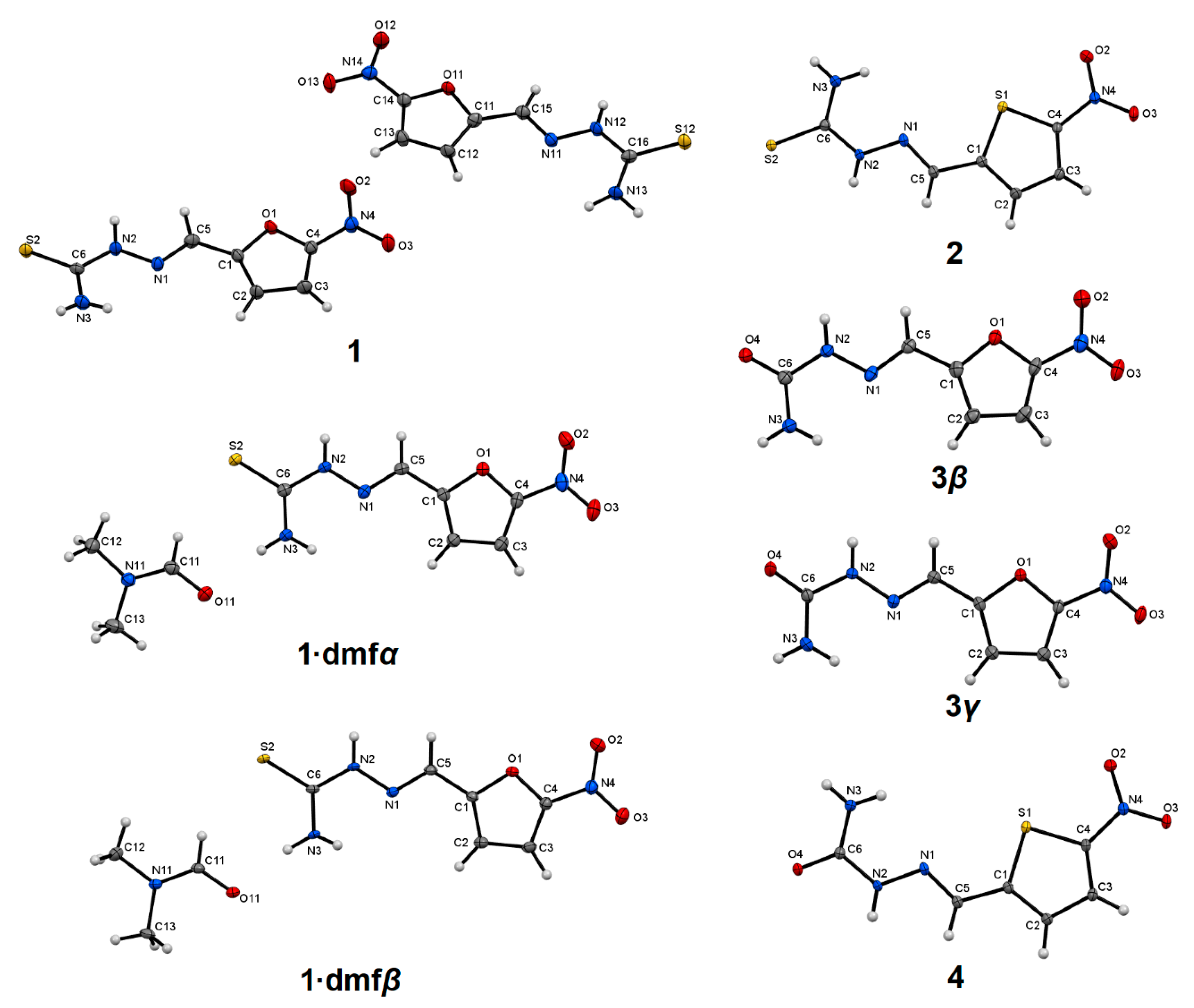
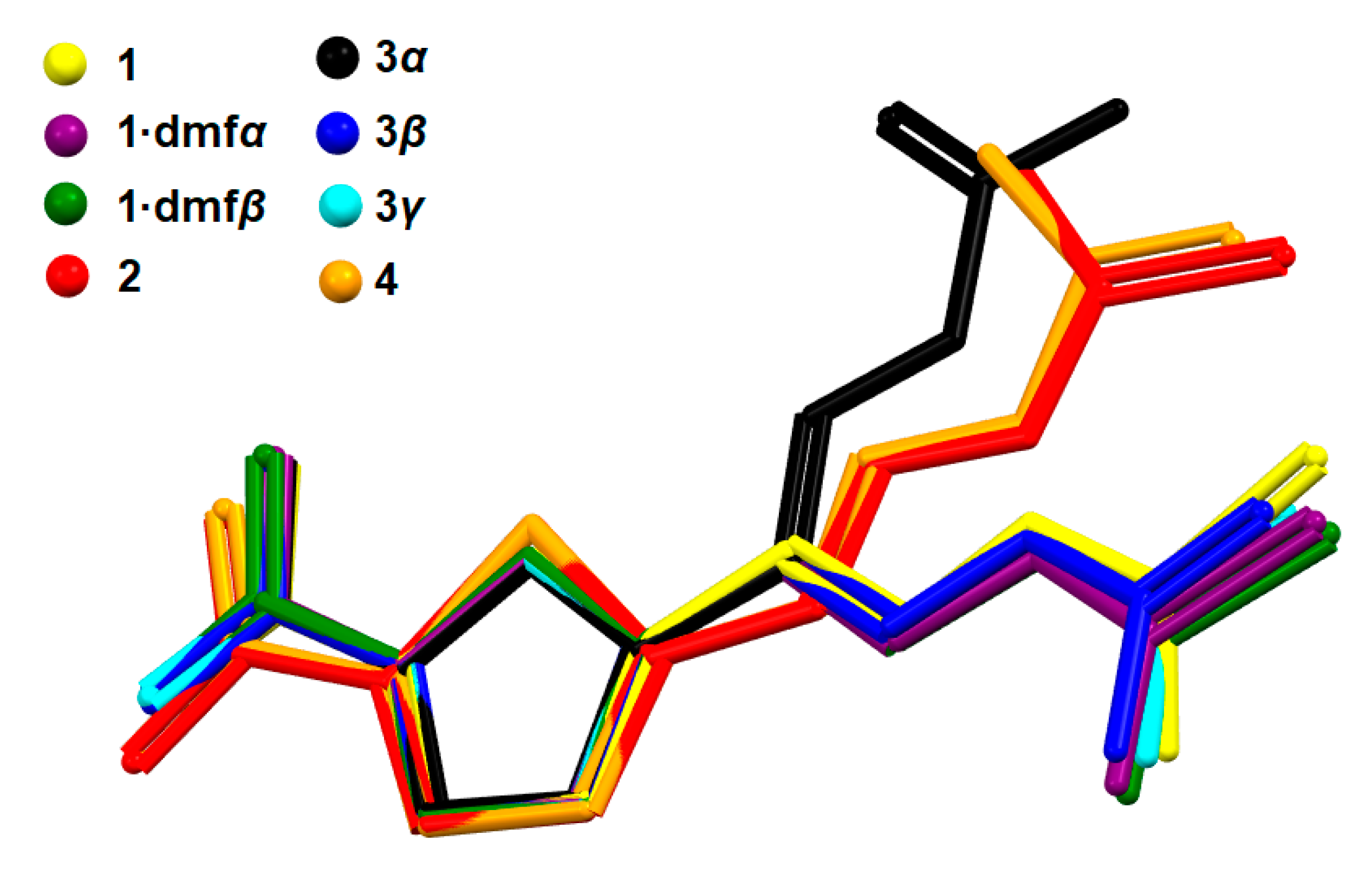
2.2. Molecular Conformation
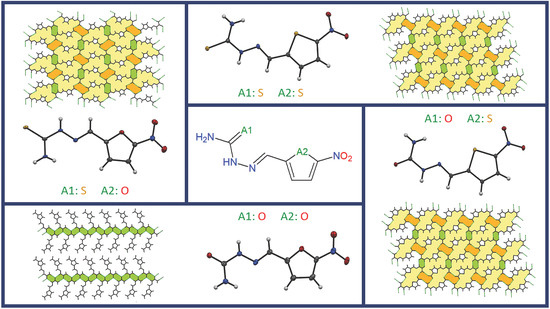
2.3. Crystal Packing
2.4. Polymorph Stability
2.5. IR Spectra
3. Materials and Methods
3.1. Synthesis
3.2. Crystal Structure Determination
3.3. IR Spectrometry and Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Michalczyk, M.; Zierkiewicz, W.; Wysokinski, R.; Scheiner, S. Theoretical studies of IR and NMR spectral changes induced by sigma-hole hydrogen, halogen, chalcogen, pnicogen, and tetrel bonds in a model protein environment. Molecules 2019, 24, 3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Weinhold, F. What Is the Nature of Supramolecular Bonding? Comprehensive NBO/NRT Picture of Halogen and Pnicogen Bonding in RPH2···IF/FI Complexes (R = CH3, OH, CF3, CN, NO2). Molecules 2019, 24, 2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, C.; Alkorta, I.; Elguero, J.; Sanchez-Sanz, G. Cooperative effects in weak interactions: Enhancement of tetrel bonds by intramolecular hydrogen bonds. Molecules 2019, 24, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieranski, T. The intricacies of the stacking interaction in a pyrrole–pyrrole system. Struct. Chem. 2016, 27, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Sieranski, T. Discovering the stacking landscape of a pyridine-pyridine system. J. Mol. Model. 2017, 23, 338. [Google Scholar] [CrossRef] [Green Version]
- Price, S.L. Control and prediction of the organic solid state: A challenge to theory and experiment. Proc. R. Soc. A 2018, 474, 20180351. [Google Scholar] [CrossRef] [Green Version]
- Iuzzolino, L.; Reilly, A.M.; McCabe, P.; Price, S.L. Use of Crystal Structure Informatics for Defining the Conformational Space Needed for Predicting Crystal Structures of Pharmaceutical Molecules. J. Chem. Theory Comput. 2017, 13, 5163–5171. [Google Scholar] [CrossRef]
- Ether, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Ether, M.C. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Sarma, J.A.R.P. The chloro-methyl exchange rule and its violations in the packing of organic molecular solids. Proc. Indian Acad. Sci. (Chem. Sci.) 1986, 96, 599–605. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Guo, R.; Tocher, D.A.; Buanz, A.B.M.; Gaisford, S.; Price, S.L.; Bučar, D.-K. Are Oxygen and Sulfur Atoms Structurally Equivalent in Organic Crystals. Cryst. Growth Des. 2017, 17, 827–833. [Google Scholar] [CrossRef]
- Bis, J.A.; Zaworotko, M.J. The 2-Aminopyridinium-carboxylate Supramolecular Heterosynthon: A Robust Motif for Generation of Multiple-Component Crystals. Cryst. Growth Des. 2005, 5, 1169–1179. [Google Scholar] [CrossRef]
- Shattock, T.R.; Arora, K.K.; Vishweshwar, P.; Zaworotko, M.J. Hierarchy of Supramolecular Synthons: Persistent Carboxylic Acid···Pyridine Hydrogen Bonds in Cocrystals That also Contain a Hydroxyl Moiety. Cryst. Growth Des. 2008, 8, 4533–4545. [Google Scholar] [CrossRef]
- Kavuru, P.; Aboarayes, D.; Arora, K.K.; Clarke, H.D.; Kennedy, A.; Marshall, L.; Ong, T.T.; Perman, J.; Pujari, T.; Wojtas, Ł.; et al. Hierarchy of Supramolecular Synthons: Persistent Hydrogen Bonds Between Carboxylates and Weakly Acidic Hydroxyl Moieties in Cocrystals of Zwitterions. Cryst. Growth Des. 2010, 10, 3568–3584. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Wood, G.P.F.; Fischer, A.; Wojtas, Ł.; Perry, M.L.; Zaworotko, M.J. Hydrogen Bond Hierarchy: Persistent Phenol···Chloride Hydrogen Bonds in the Presence of Carboxylic Acid Moieties. Cryst. Growth Des. 2015, 15, 4341–4354. [Google Scholar] [CrossRef]
- Budziak, I.; Arczewska, M.; Kamiński, D.M. Formation of prenylated chalcone xanthohumol cocrystals: Single crystal X-Ray diffraction, vibrational spectroscopic study coupled with multivariate analysis. Molecules 2019, 24, 4245. [Google Scholar] [CrossRef] [Green Version]
- Wuest, J.D. Engineering crystals by the strategy of molecular tectonics. Chem. Commun. 2005, 5830–5837. [Google Scholar] [CrossRef]
- Hosseini, M.W. Molecular Tectonics: From Simple Tectons to Complex Molecular Networks. Acc. Chem. Res. 2005, 38, 313–323. [Google Scholar] [CrossRef]
- Laliberté, D.; Maris, T.; Wuest, J.D. Molecular tectonics. Use of urethanes and ureas derived from tetraphenylmethane and tetraphenylsilane to build porous chiral hydrogen-bonded networks. Can. J. Chem. 2004, 82, 386–398. [Google Scholar] [CrossRef]
- Cole, J.C.; Groom, C.R.; Read, M.G.; Giangreco, I.; McCabe, P.; Reilly, A.M.; Shields, G.P. Generation of crystal structures using known crystal structures as analogues. Acta Cryst. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Thompson, H.P.G.; Day, G.M. Which conformations make stable crystal structures? Mapping crystalline molecular geometries to the conformational energy landscape. Chem. Sci. 2014, 5, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Olszak, T.A.; Peeters, O.M.; Blaton, N.M.; De Ranter, C.J. 5-Nitro-2-furaldehyde semicarbazone. Acta Crystallogr., Sect. C Cryst. Struct. Commun. 1994, 50, 948–950. [Google Scholar] [CrossRef]
- Pogoda, D.; Janczak, J.; Videnova-Adrabinska, V. New polymorphs of an old drug: Conformational and synthon polymorphism of 5-nitrofurazone. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mat. 2016, 72, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Sharara, K.N.; Nyamayaro, K.; Wicht, M.M.; Venter, G.A.; Bathori, N.B. Multicomponent crystals of nitrofurazone – when more is less. CrystEngComm 2019, 21, 1091–1096. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. Cambridge Structural Database, CSD v 5.39, August 2018. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M. The nature of.pi.-.pi. interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Huber, R.G.; Margreiter, M.A.; Fuchs, J.E.; von Grafenstein, S.; Tautermann, C.S.; Liedl, K.R.; Fox, T. Heteroaromatic π-Stacking Energy Landscapes. J. Chem. Inf. Model. 2014, 54, 1371–1379. [Google Scholar] [CrossRef]
- Kruszynski, R.; Sieranski, T. Can Stacking Interactions Exist beyond the Commonly Accepted Limits? Cryst. Growth Des. 2016, 12, 587–595. [Google Scholar] [CrossRef]
- Hayashi, N.; Higuchi, H.; Ninomiya, N. X/π Interactions in Aromatic Heterocycles: Basic Principles and Recent Advances. Top. Heterocycl. Chem. 2009, 18, 1–35. [Google Scholar]
- Narayan, V.; Mishra, H.N.; Prasad, O.; Sinha, L. Electronic structure, electric moments and vibrational analysis of 5-nitro-2-furaldehyde semicarbazone: A D.F.T. study. Comput. Theor. Chem. 2011, 973, 20–27. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Adv. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables, 3rd ed.; Prince, E. (Ed.) Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Gavezzotti, A. Efficient computer modeling of organic materials. The atom–atom, Coulomb–London–Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies. New J. Chem. 2011, 35, 1360–1368. [Google Scholar] [CrossRef]
- Maschio, L.; Civalleri, B.; Ugliengo, P.; Gavezzotti, A. Intermolecular Interaction Energies in Molecular Crystals: Comparison and Agreement of Localized Møller–Plesset 2, Dispersion-Corrected Density Functional, and Classical Empirical Two-Body Calculations. J. Phys. Chem. A 2011, 115, 11179–11186. [Google Scholar] [CrossRef]
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of molecular crystal structures using Full Interaction Maps. CrystEngComm 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Fábián, L.; Motherwell, W.D.S.; Allen, F.H.; Feeder, N. Knowledge-based model of hydrogen-bonding propensity in organic crystals. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2007, 63, 768–782. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0– new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Lommerse, J.P.M.; Rowland, R.S.; Taylor, R.; Verdonk, M.L.J. IsoStar: A library of information about nonbonded interactions. Comput. Aided Mol. Des. 1997, 11, 525–537. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1, 1·dmfα, 1·dmfβ, 2, 3β, 3γ and 4 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 1·dmfα | 1·dmfβ | 2 | 4 |
|---|---|---|---|---|---|
| CCDC number | 1976784 | 1976786 | 1976789 | 1976785 | 1976790 |
| Empirical formula | C6H6N4O3S | C9H13N5O4S | C9H13N5O4S | C6H6N4O2S2 | C6H6N4O3S |
| Formula weight | 214.21 | 287.30 | 287.30 | 230.27 | 214.21 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P21/n (No. 14) | P21/n (No. 14) | Cc (No. 9) | P-1 (No. 2) | P-1 (No. 2) |
| Temperature (K) | 100.0(1) | 100.0(1) | 100.0(1) | 100.0(1) | 100.0(1) |
| Wavelength (Å) | 0.71073 λ(MoKα) | 1.54184 λ(CuKα) | 0.71073 λ(MoKα) | 0.71073 λ(MoKα) | 0.71073 λ(MoKα) |
| Unit cell dimensions | |||||
| a (Å) | 12.262(3) | 4.7031(1) | 5.9513(4) | 4.5364(1) | 4.5040(2) |
| b (Å) | 12.4011(14) | 24.9324(5) | 19.6479(15) | 8.7380(2) | 8.5940(3) |
| c (Å) | 12.670(3) | 11.1468(2) | 11.4607(7) | 12.2261(3) | 11.7247(4) |
| α (°) | 90.00 | 90.00 | 90.00 | 76.291(2) | 71.920(3) |
| β (°) | 113.84(3) | 98.552(1) | 104.431(5) | 87.644(2) | 86.928(3) |
| γ (°) | 90.00 | 90.00 | 90.00 | 79.401(2) | 77.680(3) |
| Volume (Å3) | 1762.3(6) | 1292.54(3) | 1297.82(16) | 462.791(19) | 421.45(3) |
| Z | 8 | 4 | 4 | 2 | 2 |
| Calculated density (Mg/m3) | 1.615 | 1.476 | 1.470 | 1.652 | 1.688 |
| Absorption coefficient (mm−1) | 0.354 | 2.435 | 0.269 | 0.553 | 0.370 |
| F(000) | 880 | 600 | 600 | 236 | 220 |
| Crystal size (mm) | 0.098 0.090 0.007 | 0.173 0.131 0.118 | 0.121 0.084 0.024 | 0.107 0.098 0.091 | 0.098 0.093 0.025 |
| θ Range for data collection (°) | 3.416 to 25.049 | 4.386 to 78.482 | 3.671 to 32.078 | 3.430 to 31.793 | 3.563 to 31.940 |
| Index ranges | −14 ≤ h ≤ 14, −14 ≤ k ≤ 14, −15 ≤ l ≤ 14 | −5 ≤ h ≤ 5, −31 ≤ k ≤ 31, −14 ≤ l ≤ 13 | −8 ≤ h ≤ 8, −28 ≤ k ≤ 28, −16 ≤ l ≤ 14 | −6 ≤ h ≤6, −12 ≤ k ≤ 12, −17 ≤ l ≤ 17 | −6 ≤ h ≤ 6, −12 ≤ k ≤ 12, −17 ≤ l ≤ 17 |
| Reflections collected/unique | 14539/3111 | 25641/2646 | 12573/3526 | 12959/2743 | 12046/2544 |
| Rint | 0.0647 | 0.0350 | 0.0464 | 0.0278 | 0.0338 |
| Completeness (%) to θ = 25°(Mo) and θ = 67°(Cu) | 99.8 | 100.0 | 99.8 | 99.8 | 99.8 |
| Min. and max. transmission | 0.48113 and 1.00000 | 0.63893 and 1.00000 | 0.86402 and 1.00000 | 0.94704 and 1.00000 | 0.96223 and 1.00000 |
| Data/restraints/parameters | 3111/1/271 | 2646/0/183 | 3526/2/183 | 2743/0/136 | 2544/0/136 |
| Goodness-of-fit on F2 | 1.044 | 1.043 | 1.084 | 1.078 | 1.050 |
| Final R indices [I > 2σ(I)] | R1 = 0.0632, wR2 = 0.1595 | R1 = 0.0271, wR2 = 0.0698 | R1 = 0.0357, wR2 = 0.0951 | R1 = 0.0249, wR2 = 0.0658 | R1 = 0.0282, wR2 = 0.0725 |
| R indices (all data) | R1 = 0.0834, wR2 = 0.1705 | R1 = 0.0289, wR2 = 0.0708 | R1 = 0.0366, wR2 = 0.0957 | R1 = 0.0290, wR2 = 0.0674 | R1 = 0.0337, wR2 = 0.0745 |
| Largest diff. peak and hole (e•Å−3) | 0.995 and −0.352 | 0.414 and −0.205 | 0.582 and −0.546 | 0.425 and −0.233 | 0.428 and −0.229 |
| Compound | Packing Index (%) | Density (g/cm3) | Elatt | ECoul | Epol | Edisp | Erep | EMP2 | EM06-2X |
|---|---|---|---|---|---|---|---|---|---|
| Elatt+EMP2 | Elatt+EM06-2X | ||||||||
| 3α | 69.9 | 1.571 | −178.6 | −148.4 | −60.3 | −117.3 | 147.3 | −1968944 | −1973692 |
| −1969123 | −1973871 | ||||||||
| 3β | 68.8 | 1.547 | −133.4 | −107.1 | −38.2 | −109.9 | 126.6 | −1968999 | −1973732 |
| −1969132 | −1973865 | ||||||||
| 3γ | 71.2 | 1.592 | −134.7 | −99.7 | −33.9 | −116.1 | 114.9 | −1969005 | −1973732 |
| −1969140 | −1973867 | ||||||||
| 1 | — | — | — | — | — | — | — | −2815948 | −2821619 |
| — | — | ||||||||
| 1·dmfα | 71.2 | 1.476 | −107.6 | −81.3 | −33.3 | −100.2 | 107.2 | −3466466 | — |
| −3466574 | — | ||||||||
| 1·dmfβ | 70.6 | 1.470 | −109.1 | −84.0 | −33.5 | −101.2 | 111.8 | −3466466 | — |
| −3466575 | — |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swiatkowski, M.; Trzesowska-Kruszynska, A.; Danielewicz, A.; Sobczak, P.; Kruszynski, R. Interplay between Polymorphism and Isostructurality in the 2-Fur- and 2-Thenaldehyde Semi- and Thiosemicarbazones. Molecules 2020, 25, 993. https://doi.org/10.3390/molecules25040993
Swiatkowski M, Trzesowska-Kruszynska A, Danielewicz A, Sobczak P, Kruszynski R. Interplay between Polymorphism and Isostructurality in the 2-Fur- and 2-Thenaldehyde Semi- and Thiosemicarbazones. Molecules. 2020; 25(4):993. https://doi.org/10.3390/molecules25040993
Chicago/Turabian StyleSwiatkowski, Marcin, Agata Trzesowska-Kruszynska, Agnieszka Danielewicz, Paulina Sobczak, and Rafal Kruszynski. 2020. "Interplay between Polymorphism and Isostructurality in the 2-Fur- and 2-Thenaldehyde Semi- and Thiosemicarbazones" Molecules 25, no. 4: 993. https://doi.org/10.3390/molecules25040993
APA StyleSwiatkowski, M., Trzesowska-Kruszynska, A., Danielewicz, A., Sobczak, P., & Kruszynski, R. (2020). Interplay between Polymorphism and Isostructurality in the 2-Fur- and 2-Thenaldehyde Semi- and Thiosemicarbazones. Molecules, 25(4), 993. https://doi.org/10.3390/molecules25040993