
Solvent-Free Iron(III) Chloride-Catalyzed Direct Amidation of Esters

 and
and
Abstract
:1. Introduction
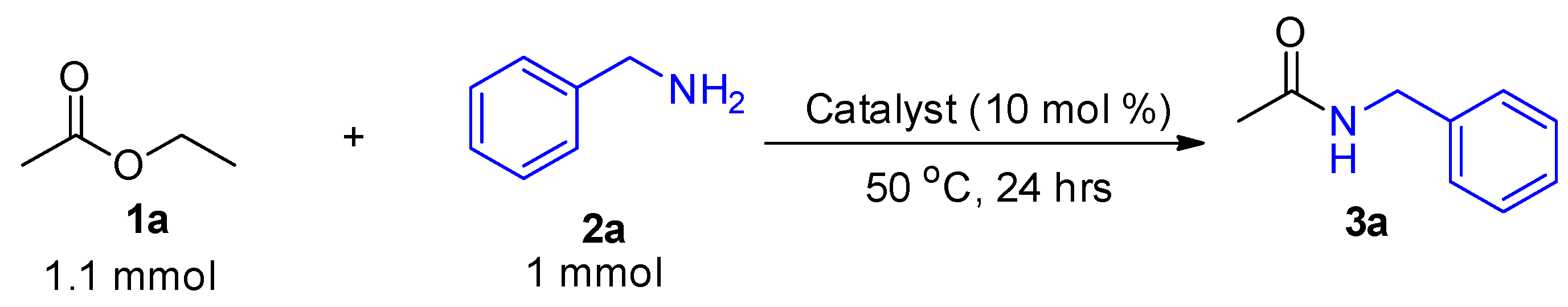
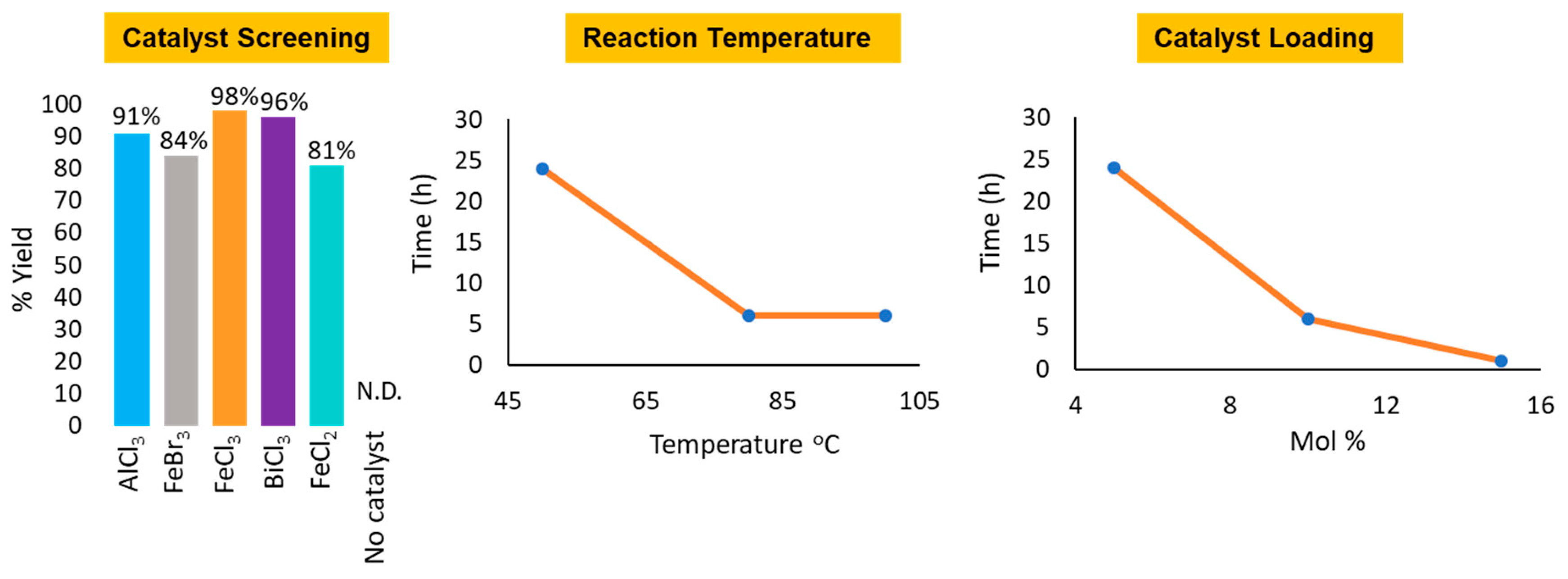
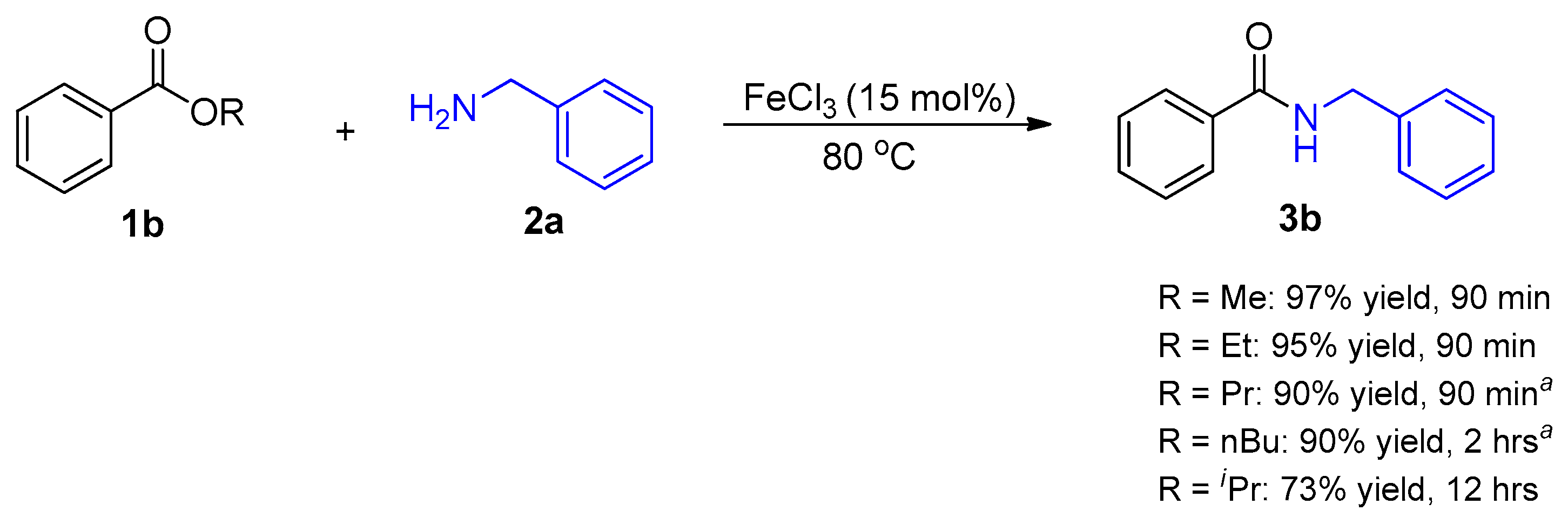
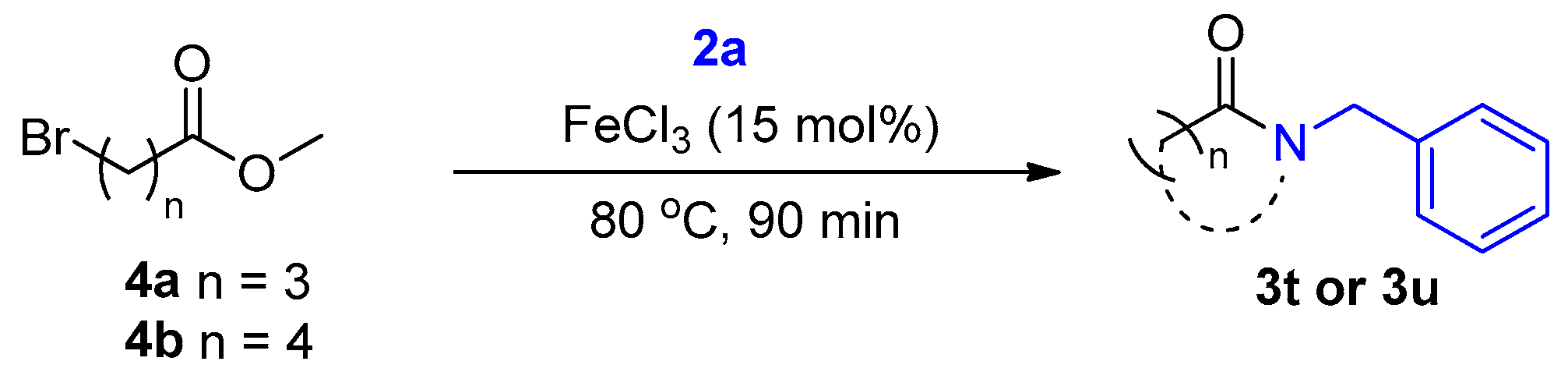
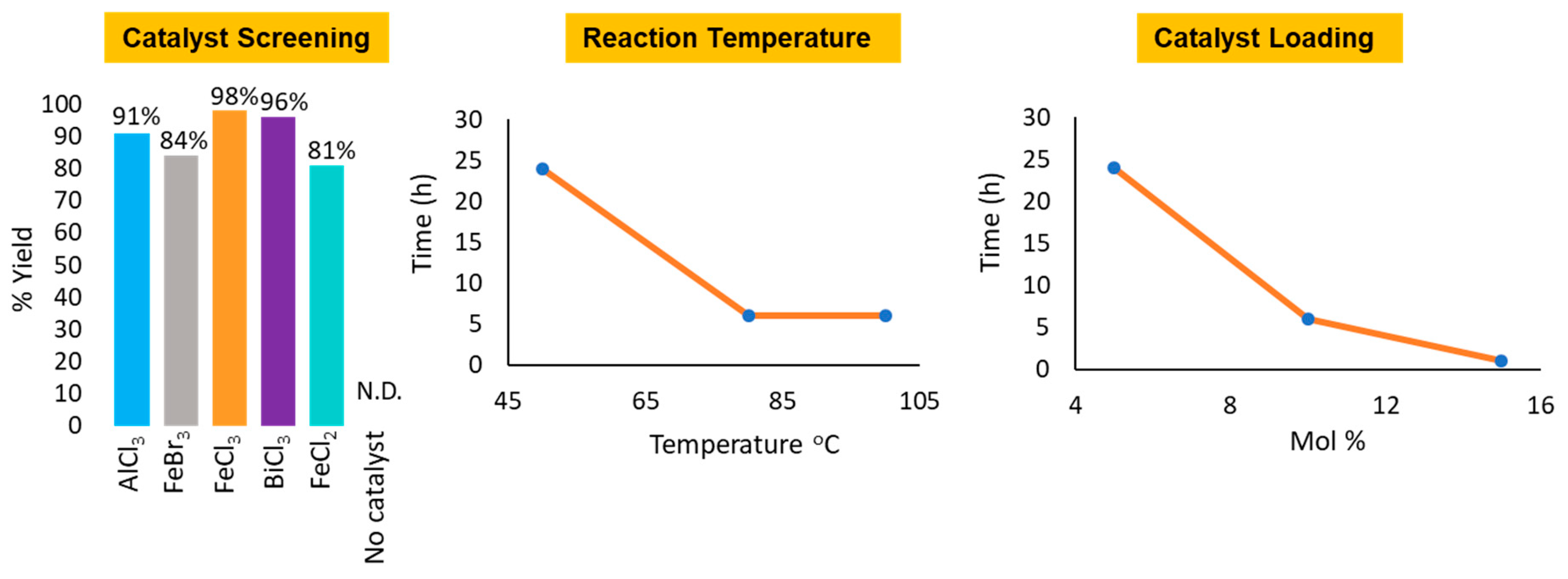
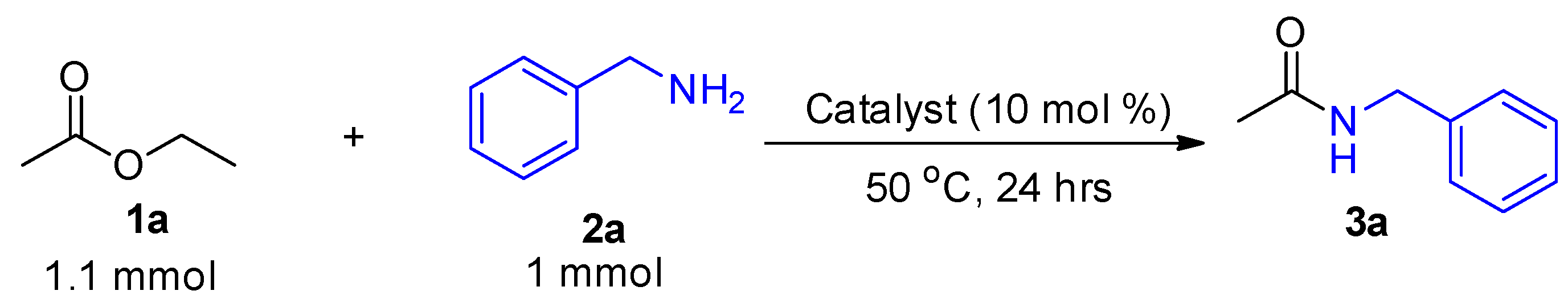
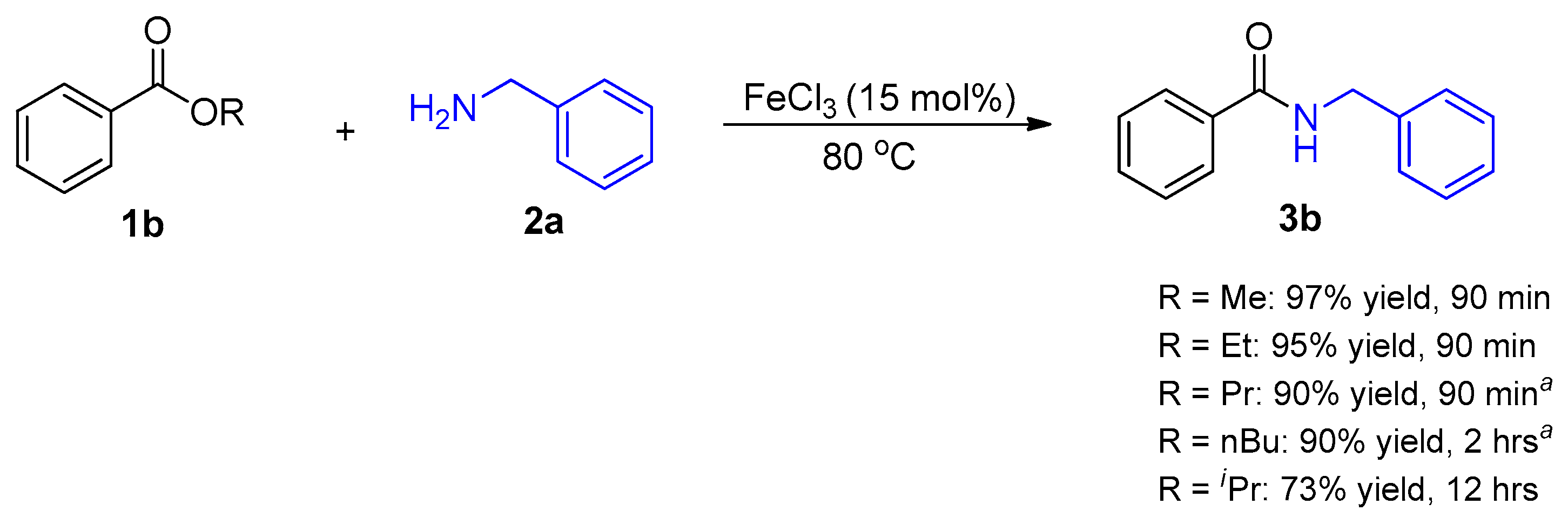
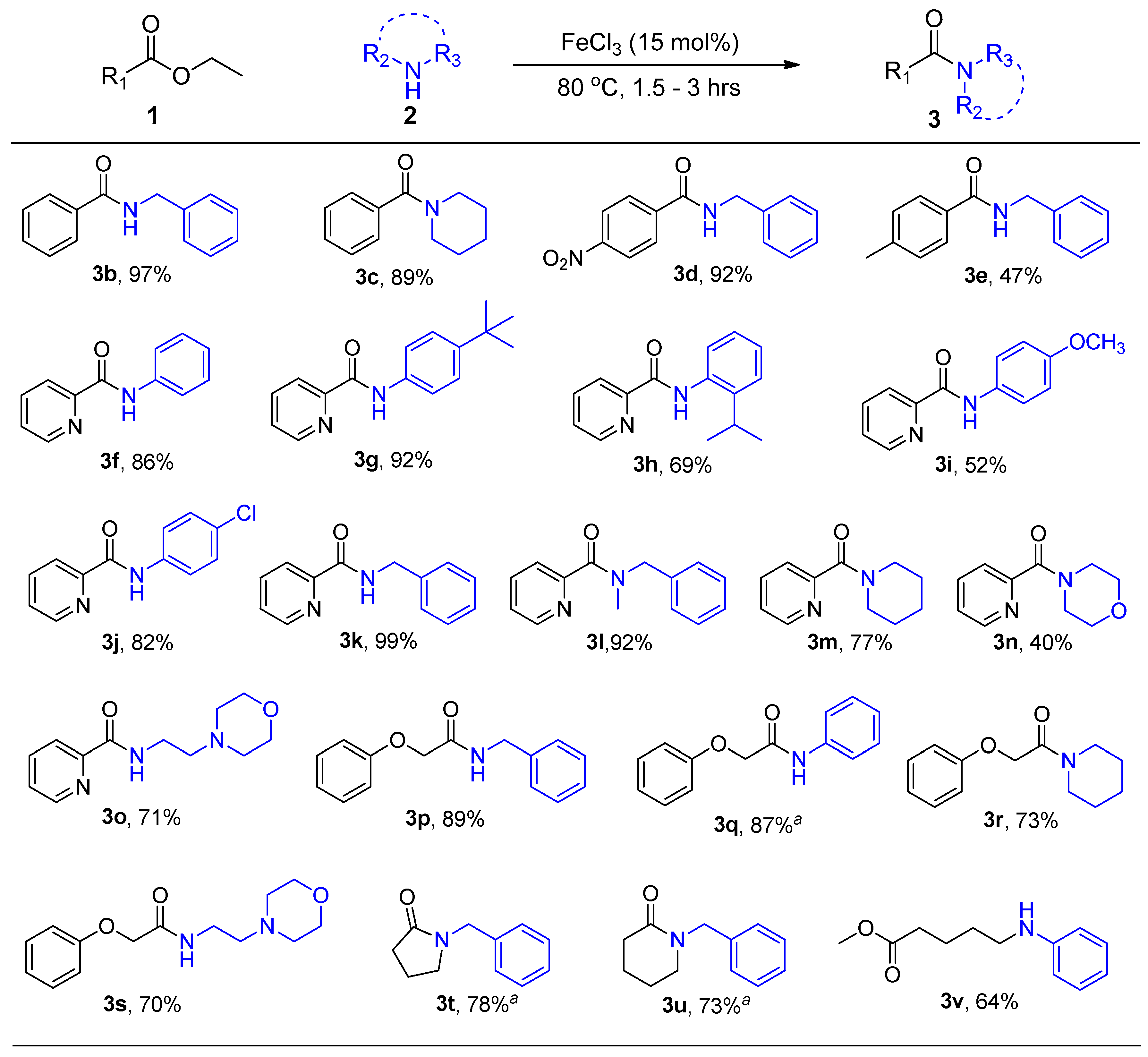
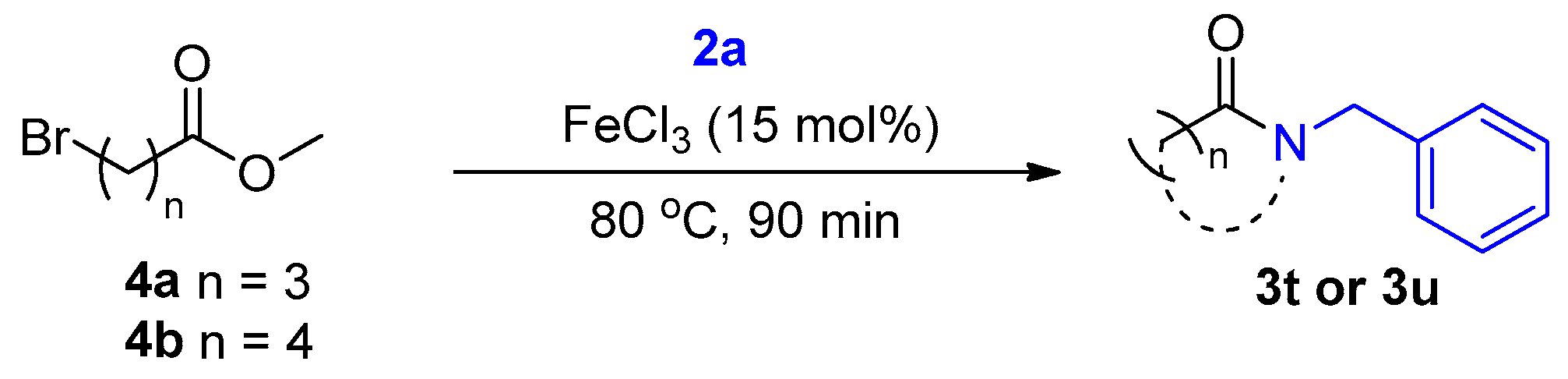
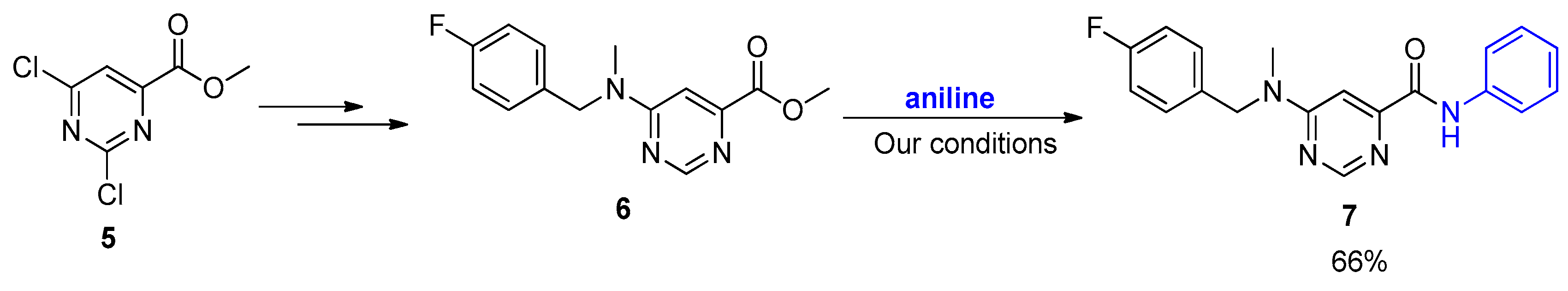
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Humphrey, J.M.; Chamberlin, A.R. Chemical Synthesis of Natural Product Peptides: Coupling Methods for the Incorporation of Noncoded Amino Acids into Peptides. Chem. Rev. 1997, 97, 2243–2266. [Google Scholar] [CrossRef]
- Agrawal, R.; Jain, P.; Dikshit, S.N. Apixaban: A New Player in the Anticoagulant Class. Curr. Drug Targets 2012, 13, 863–875. [Google Scholar] [CrossRef]
- Ponomaryov, Y.; Krasikova, V.; Lebedev, A.; Chernyak, D.; Varacheva, L.; Chernobroviy, A. Scalable and green process for the synthesis of anticancer drug lenalidomide. Chem. Heterocycl. Comp. 2015, 51, 133–138. [Google Scholar] [CrossRef]
- Zhou, A.-N.; Li, B.; Ruan, L.; Wang, Y.; Duan, G.; Li, J. An improved and practical route for the synthesis of enzalutamide and potential impurities study. Chin. Chem. Lett. 2017, 28, 426–430. [Google Scholar] [CrossRef]
- Volovych, I.; Neumann, M.; Schmidt, M.; Buchner, G.; Yang, J.-Y.; Wölk, J.; Sottmann, T.; Strey, R.; Schomäcker, R.; Schwarze, M. A novel process concept for the three step Boscalid® synthesis. RSC Adv. 2016, 6, 58279–58287. [Google Scholar] [CrossRef]
- Jarvis, L.M. The new drugs of 2018. C&EN 2019, 97, 33–37. [Google Scholar]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, Jr.J.L.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas—a perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef]
- Sabatini, M.T.; Boulton, L.T.; Sheppard, T.D. Borate esters: Simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 2017, 3, e1701028. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.A. An assessment of boric acid and borax using the IEHR evaluative process for assessing human developmental and reproductive toxicity of agents. Reprod. Toxicol. 1997, 11, 123–160. [Google Scholar] [CrossRef]
- Sabatini, M.T.; Boulton, L.T.; Sneddon, H.F.; Sheppard, T.D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10–17. [Google Scholar] [CrossRef]
- Allen, C.L.; Williams, J.M.J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 2011, 40, 3405–3415. [Google Scholar] [CrossRef]
- de Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical routes for amide bond formation. Chem. Rev. 2016, 116, 19–12029. [Google Scholar] [CrossRef]
- Ojeda-Porras, A.; Gamba-Sánchez, D. Recent developments in amide synthesis using nonactivated starting materials. J. Org. Chem. 2016, 81, 11548–11555. [Google Scholar] [CrossRef] [PubMed]
- Ranu, B.C.; Dutta, P. A simple and convenient procedure for the conversion of esters to secondary amides. Synth. Commun. 2003, 33, 297–301. [Google Scholar] [CrossRef]
- Price, K.E.; Larriveé-Aboussafy, C.; Lillie, B.M.; McLaughlin, R.W.; Mustakis, J.; Hettenbach, K.W.; Hawkins, J.M.; Vaidyanathan, R. Mild and efficient DBU-catalyzed amidation of ayanoacetates. Org. Lett. 2009, 11, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Lee, H.-G.; Kang, S.-B.; Sung, G.H.; Kim, J.-J.; Park, J.K.; Lee, S.-G.; Yoon, Y.-J. tert-Butoxide-Assisted Amidation of Esters under Green Conditions. Synthesis 2012, 44, 42–50. [Google Scholar]
- Ohshima, T.; Hayashi, Y.; Agura, K.; Fujii, Y.; Yoshiyama, A.; Mashima, K. Sodium methoxide: A simple but highly efficient catalyst for the direct amidation of esters. Chem. Commun. 2012, 48, 5434–5436. [Google Scholar] [CrossRef]
- Rzhevskiy, S.A.; Ageshina, A.A.; Chesnokov, G.A.; Gribanov, P.S.; Topchiy, M.A.; Nechaev, M.S.; Asachenko, A.F. Solvent- and transition metal-free amide synthesis from phenyl esters and aryl amines. RSC Adv. 2019, 9, 1536–1540. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, H.; Fujiwara, R.; Shimizu, Y.; Morisaki, K.; Ohshima, T. Lanthanum(III) triflate catalyzed direct amidation of esters. Org. Lett. 2014, 16, 2018–2021. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.W.; Ploeger, M.L.; Hu, X. Direct amidation of esters with nitroarenes. Nat. Commun. 2017, 8, 14878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, L.; Chen, C.; Luo, M.; Zeng, X. Chromium-catalyzed activation of acyl C−O Bonds with magnesium for amidation of esters with nitroarenes. Org Lett. 2019, 21, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Ben Halima, T.; Masson-Makdissi, J.; Newman, S.G. Nickel catalysed amide bond formation from methyl esters. Angew. Chem. Int. Ed. 2018, 57, 12925–12929. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Szostak, M. Highly selective transition-metal-free transamidation of amides and amidation of esters at room temperature. Nat Commun. 2018, 9, 4165. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, N.; Campbell, P.S.; Jamieson, C.; Potjewyd, F.; Simpson, I.; Watson, A.J.B. Amidation of esters with amino alcohols using organobase catalysis. J. Org. Chem. 2014, 79, 9347–9354. [Google Scholar] [CrossRef] [Green Version]
- Correa, A.; Mancheño, O.G.; Bolm, C. Iron-catalysed carbon–heteroatom and heteroatom–heteroatom bond forming processes. Chem. Soc. Rev. 2008, 37, 1108–1117. [Google Scholar] [CrossRef]
- Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Direct C−H Transformation via Iron Catalysis. Chem. Rev. 2011, 111, 1293–1314. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Wilson, C.R.; Gessner, R.K.; Moosa, A.; Seldon, R.; Warner, D.F.; Mizrahi, V.; de Melo, C.S.; Simelane, S.B.; Nchinda, A.; Abay, E.; et al. Novel antitubercular 6-dialkylaminopyrimidine carboxamides from phenotypic whole-cell high throughput screening of a softfocus library: Structure−activity relationship and target identification studies. J. Med. Chem. 2017, 60, 10118–10134. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.E.; Jones, M.D.; Williams, J.M.J.; Bull, S.D. N-Acyl DBN tetraphenylborate salts as N-acylating agents. J. Org. Chem. 2012, 77, 2808–2818. [Google Scholar] [CrossRef] [PubMed]
- Sawant, D.N.; Bagal, D.B.; Ogawa, S.; Selvam, K.; Saito, S. Diboron-Catalyzed Dehydrative Amidation of Aromatic Carboxylic Acids with Amines. Org. Lett. 2018, 20, 4397–4400. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Z.; Liebeskind, L.S. In Situ Carboxyl Activation Using a Silatropic Switch: A New Approach to Amide and Peptide Constructions. J. Am. Chem. Soc. 2011, 133, 14256–14259. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Fujita, K.-I.; Yamaguchi, R. Aerobic Oxidative Amidation of Aromatic and Cinnamic Aldehydes with Secondary Amines by CuI/2-Pyridonate Catalytic System. J. Org. Chem. 2012, 77, 9102–9109. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, H.; Liu, J.; Wan, X.; Xu, Q. Clean synthesis of primary to tertiary carboxamides by CsOH-catalyzed aminolysis of nitriles in water. Green Chem. 2016, 18, 4865–4870. [Google Scholar] [CrossRef]
- Kiselyov, A.S. Reaction of N-fluoropyridinium fluoride with isonitriles and diazo compounds: A one-pot synthesis of (pyridin-2-yl)-1H-1,2,3-triazoles. Tetrahedron Lett. 2006, 47, 2631–2634. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Adolfsson, H. Direct Amide Coupling of Non-activated Carboxylic Acids and Amines Catalysed by Zirconium(IV) Chloride. Chem. Eur. J. 2012, 18, 3822–3826. [Google Scholar] [CrossRef]
- Nagarajan, S.; Ran, P.; Shanmugavelan, P.; Sathishkumar, M.; Ponnuswamy, A.; Suk, N.K.; Gnana, K. The catalytic activity of titania nanostructures in the synthesis of amides under solvent-free conditions. New J. Chem. 2012, 36, 1312–1319. [Google Scholar] [CrossRef]
- Kim, K.; Hong, S.H. Iridium-Catalyzed Single-Step N-Substituted Lactam Synthesis from Lactones and Amines. J. Org. Chem. 2015, 80, 4152–4156. [Google Scholar] [CrossRef]
- Yuan, M.-L.; Xie, J.-H.; Zhou, Q.-L. Boron Lewis Acid Promoted Ruthenium-Catalyzed Hydrogenation of Amides: An Efficient Approach to Secondary Amines. ChemCatChem 2016, 8, 3036–3040. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the compounds 3a–3v and 7 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Product Yield |
|---|---|---|
| 1 | Toluene | 84% a |
| 2 | Acetonitrile | 96% |
| 3 | Tetrahydrofuran | 85% a |
| 4 | 1,2 Dichloroethane | 94% |
| 5 | No Solvent | 99% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mkhonazi, B.D.; Shandu, M.; Tshinavhe, R.; Simelane, S.B.; Moshapo, P.T. Solvent-Free Iron(III) Chloride-Catalyzed Direct Amidation of Esters. Molecules 2020, 25, 1040. https://doi.org/10.3390/molecules25051040
Mkhonazi BD, Shandu M, Tshinavhe R, Simelane SB, Moshapo PT. Solvent-Free Iron(III) Chloride-Catalyzed Direct Amidation of Esters. Molecules. 2020; 25(5):1040. https://doi.org/10.3390/molecules25051040
Chicago/Turabian StyleMkhonazi, Blessing D., Malibongwe Shandu, Ronewa Tshinavhe, Sandile B. Simelane, and Paseka T. Moshapo. 2020. "Solvent-Free Iron(III) Chloride-Catalyzed Direct Amidation of Esters" Molecules 25, no. 5: 1040. https://doi.org/10.3390/molecules25051040
APA StyleMkhonazi, B. D., Shandu, M., Tshinavhe, R., Simelane, S. B., & Moshapo, P. T. (2020). Solvent-Free Iron(III) Chloride-Catalyzed Direct Amidation of Esters. Molecules, 25(5), 1040. https://doi.org/10.3390/molecules25051040