
One-Pot Production of RNA in High Yield and Purity Through Cleaving Tandem Transcripts


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
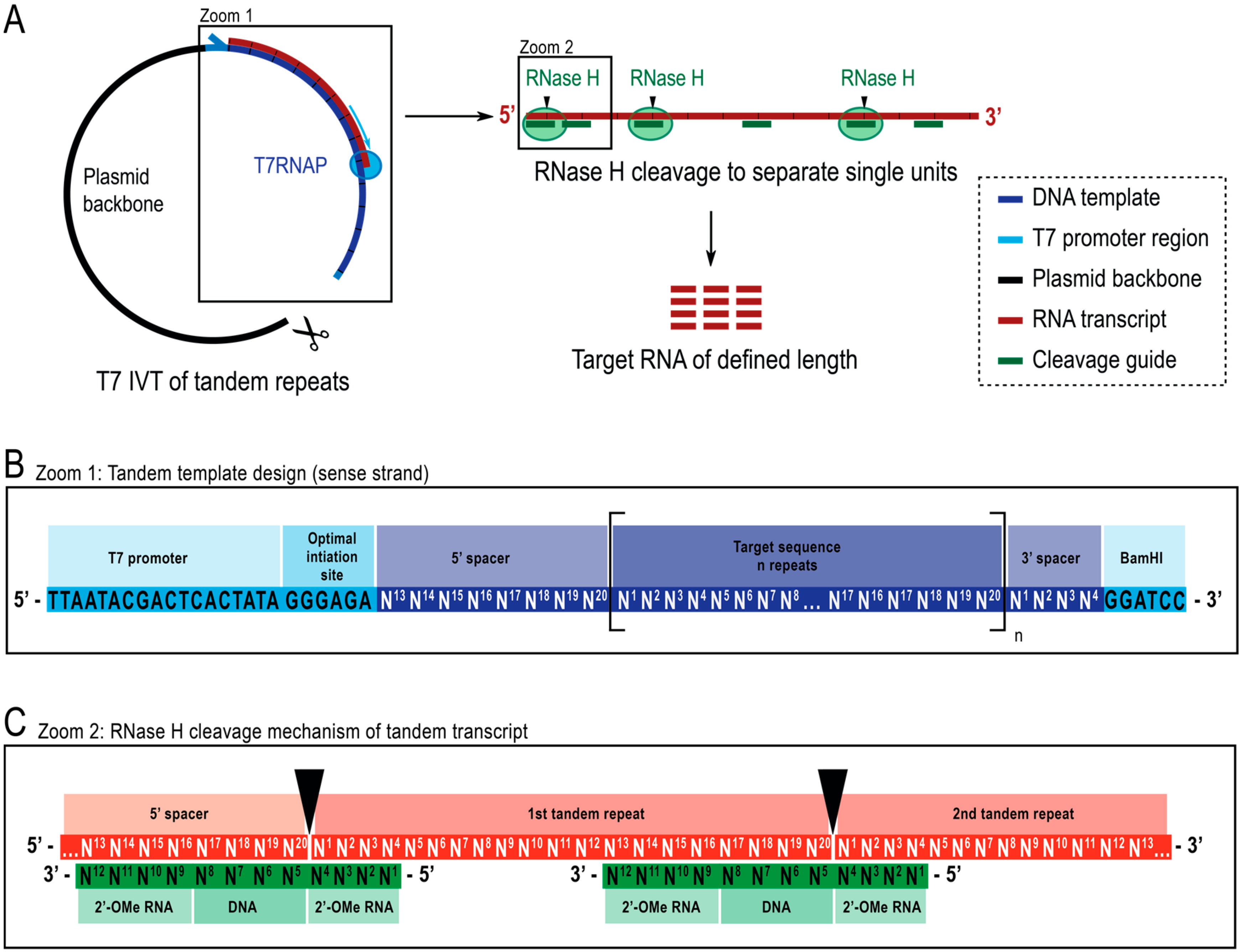
2.1. Template Design
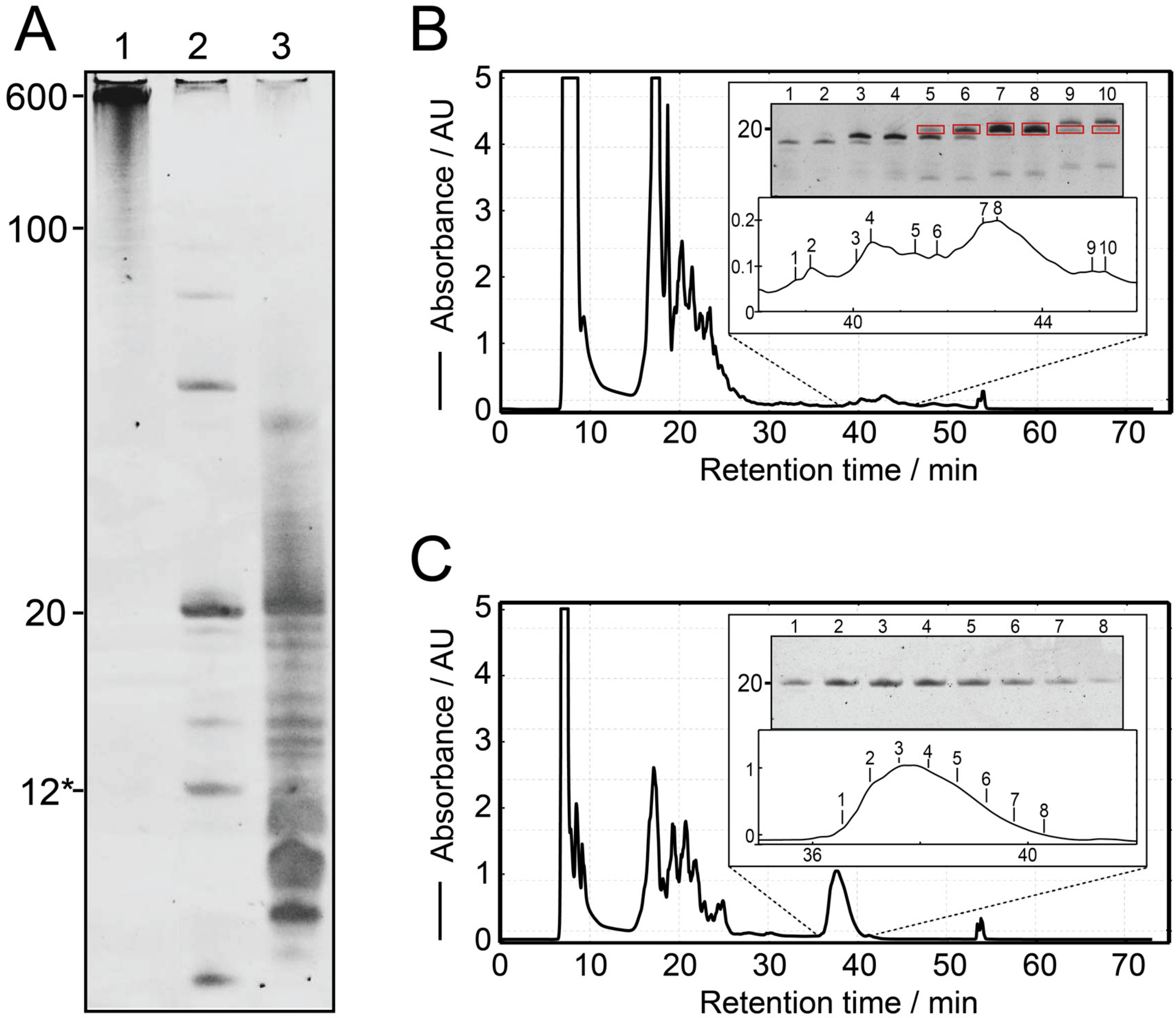
2.2. T7 In Vitro Transcription
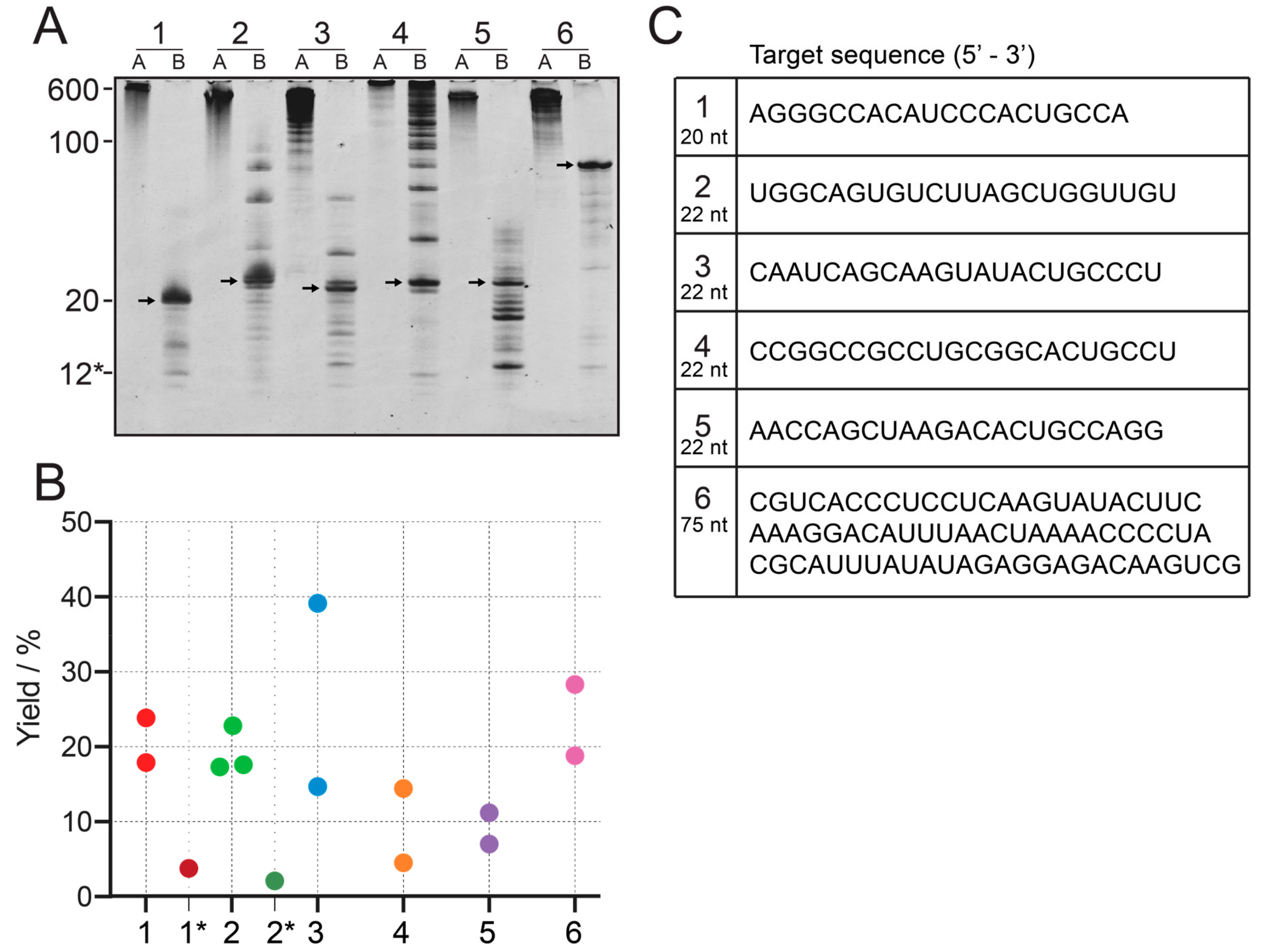
2.3. RNase H Cleavage Reaction
2.4. Ion-Exchange HPLC Purification
2.5. Yield Quantification
2.6. Differences Between Small Scale and Large Scale Reactions
3. Discussion
3.1. Large Scale Production of RNA from Repetitive Templates
3.2. Using Different Templates to Generate Tandem Transcripts
4. Materials and Methods
4.1. Plasmid Preparation
4.2. Gel Analysis
4.3. T7 In Vitro Transcription
4.4. RNase H Cleavage Reaction
4.5. Ion-Exchange HPLC Purification
4.6. UV Spectroscopy
4.7. Yield Calculation
4.8. Quantification with Ion-Exchange HPLC
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostankovitch, M.; Pyle, A.M. Noncoding RNAs: A story of networks and long-distance relationships. J. Mol. Biol. 2013, 425, 3577–3581. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Lui, P.Y.; Jin, D.Y.; Stevenson, N.J. MicroRNA: Master controllers of intracellular signaling pathways. Cell. Mol. Life Sci. 2015, 72, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, E.; Kang, Y.Y.; Song, J.; Mok, H.; Lee, J.B. Enzymatically Produced miR-34a Nanoparticles for Enhanced Antiproliferation Activity. Adv. Biosyst. 2018, 2, 1700158. [Google Scholar] [CrossRef]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinforma. 2017, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Cech, T.R. The chemical repertoire of natural ribozymes. Nature 2002, 418, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef]
- Zhu, L.; Jiang, H.; Sheong, F.K.; Cui, X.; Wang, Y.; Gao, X.; Huang, X. Understanding the core of RNA interference: The dynamic aspects of Argonaute-mediated processes. Prog. Biophys. Mol. Biol. 2016, 128, 39–46. [Google Scholar] [CrossRef]
- Koo, T.; Kim, J.S. Therapeutic applications of CRISPR RNA-guided genome editing. Brief. Funct. Genom. 2017, 16, 38–45. [Google Scholar] [CrossRef]
- Hoehn, B.; Rossi, J.J. Nucleic Acid-Based Therapies. Chem. Biol. Nucleic Acids 2010, 233–260. [Google Scholar] [CrossRef]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Lucchino, M. Current Challenges in Delivery and Cytosolic Translocation of Therapeutic RNAs. Nucleic Acid Ther. 2018, 28, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Flamme, M.; McKenzie, L.K.; Sarac, I.; Hollenstein, M. Chemical methods for the modification of RNA. Methods 2019, 161, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Behlke, M.A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Jian, C.; Yu, A.H.; Tu, M.J. RNA therapy: Are we using the right molecules? Pharmacol. Ther. 2019, 196, 91–104. [Google Scholar] [CrossRef]
- Borkotoky, S.; Murali, A. The highly efficient T7 RNA polymerase: A wonder macromolecule in biological realm. Int. J. Biol. Macromol. 2018, 118, 49–56. [Google Scholar] [CrossRef]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef]
- Price, S.R.; Ito, N.; Oubridge, C.; Avis, J.M.; Nagai, K. Crystallization of RNA-protein complexes I. Methods for the large-scale preparation of RNA suitable for crystallographic studies. J. Mol. Biol. 1995, 249, 398–408. [Google Scholar] [CrossRef]
- Helm, M.; Brule, H.; Giege, R.; Florentz, C. More mistakes by T7 RNA polymerase at the 5′ ends of in vitro- transcribed RNAs. RNA 1999, 5, 618–621. [Google Scholar] [CrossRef]
- Kuzmine, I.; Gottlieb, P.A.; Martin, C.T. Binding of the priming nucleotide in the initiation of transcription by T7 RNA polymerase. J. Biol. Chem. 2003, 278, 2819–2823. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J.A.; Derrick, M.L.; Uhlenbeck, O.C. T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Gholamalipour, Y.; Mudiyanselage, A.K.; Martin, C.T. 3′ end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character. Nucleic Acids Res. 2018, 46, 9253–9263. [Google Scholar] [CrossRef] [PubMed]
- Easton, L.E.; Shibata, Y.; Lukavsky, P.J. Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA 2010, 16, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lv, H.; Wang, L.; Chen, M.; Li, F.; Liang, C.; Yu, Y.; Jiang, F.; Lu, A.; Zhang, G. Recent methods for purification and structure determination of oligonucleotides. Int. J. Mol. Sci. 2016, 17, 2134. [Google Scholar] [CrossRef]
- Baronti, L.; Karlsson, H.; Marušič, M.; Petzold, K. A guide to large-scale RNA sample preparation. Anal. Bioanal. Chem. 2018, 410, 3239–3252. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Kao, C.; Zheng, M.; Rüdisser, S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA 1999, 5, 1268–1272. [Google Scholar] [CrossRef]
- Becker, B.; Masquida, B. Synthesis of RNA by In Vitro Transcription. RNA Methods Protoc. Methods Mol. Biol. 2011, 703, 298–299. [Google Scholar]
- Guillerez, J.; Lopez, P.J.; Proux, F.; Launay, H.; Dreyfus, M. A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc. Natl. Acad. Sci. USA 2005, 102, 5958–5963. [Google Scholar] [CrossRef]
- Ferré-D’Amaré, A.R.; Doudna, J.A. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res. 1996, 24, 977–978. [Google Scholar] [CrossRef] [PubMed]
- Salzman, D.W.; Nakamura, K.; Nallur, S.; Dookwah, M.T.; Metheetrairut, C.; Slack, F.J.; Weidhaas, J.B. miR-34 activity is modulated through 5′-end phosphorylation in response to DNA damage. Nat. Commun. 2016, 7, 10954. [Google Scholar] [CrossRef] [PubMed]
- Duss, O.; Maris, C.; von Schroetter, C.; Allain, F.H.T. A fast, efficient and sequence-independent method for flexible multiple segmental isotope labeling of RNA using ribozyme and RNase H cleavage. Nucleic Acids Res. 2010, 38, e188. [Google Scholar] [CrossRef] [PubMed]
- Lapham, J.; Crothers, D.M. RNase H cleavage for processing of in vitro transcribed RNA for NMR studies and RNA ligation. RNA 1996, 2, 289–296. [Google Scholar]
- Miller, H.I.; Riggs, A.D.; Gill, G.N. Ribonuclease H (Hybrid) in Escherichia coli. J. Biol. Chem. 1973, 248, 2621–2624. [Google Scholar]
- Inoue, H.; Hayase, Y.; Iwai, S.; Ohtsuka, E. Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H. FEBS Lett. 1987, 215, 327–330. [Google Scholar] [CrossRef]
- Wang, X.; Li, C.; Gao, X.; Wang, J.; Liang, X. Preparation of small RNAs using rolling circle transcription and site-specific RNA disconnection. Mol. Ther. Nucleic Acids 2015, 4, e215. [Google Scholar] [CrossRef]
- Cheng, H.; Hong, S.; Wang, Z.; Sun, N.; Wang, T.; Zhang, Y.; Chen, H.; Pei, R. Self-assembled RNAi nanoflowers via rolling circle transcription for aptamer-targeted siRNA delivery. J. Mater. Chem. B 2018, 6, 4638–4644. [Google Scholar] [CrossRef]
- Brunelle, J.L.; Green, R. In vitro transcription from plasmid or PCR-amplified DNA. In Methods in Enzymology, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 530, pp. 101–114. [Google Scholar]
- Karlsson, H.; Baronti, L.; Petzold, K. A robust and versatile method for production and purification of large-scale RNA samples for structural biology. RNA, under Rev. 2020. [Google Scholar]
- Kern, J.A.; Davis, R.H. Application of solution equilibrium analysis to in vitro RNA transcription. Biotechnol. Prog. 1997, 13, 747–756. [Google Scholar] [CrossRef]
- AbouHaidar, M.G.; Ivanov, I.G. Non-enzymatic RNA hydrolysis promoted by the combined catalytic activity of buffers and magnesium ions. Z. Naturforsch. Sect. C J. Biosci. 1999, 54, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.A.; Chipperfield, R.; Birnboim, H.C. A procedure for the large-scale isolation of highly purified plasmid DNA using alkaline extraction and binding to glass powder. Anal. Biochem. 1982, 121, 382–387. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
Not available. |



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feyrer, H.; Munteanu, R.; Baronti, L.; Petzold, K. One-Pot Production of RNA in High Yield and Purity Through Cleaving Tandem Transcripts. Molecules 2020, 25, 1142. https://doi.org/10.3390/molecules25051142
Feyrer H, Munteanu R, Baronti L, Petzold K. One-Pot Production of RNA in High Yield and Purity Through Cleaving Tandem Transcripts. Molecules. 2020; 25(5):1142. https://doi.org/10.3390/molecules25051142
Chicago/Turabian StyleFeyrer, Hannes, Raluca Munteanu, Lorenzo Baronti, and Katja Petzold. 2020. "One-Pot Production of RNA in High Yield and Purity Through Cleaving Tandem Transcripts" Molecules 25, no. 5: 1142. https://doi.org/10.3390/molecules25051142
APA StyleFeyrer, H., Munteanu, R., Baronti, L., & Petzold, K. (2020). One-Pot Production of RNA in High Yield and Purity Through Cleaving Tandem Transcripts. Molecules, 25(5), 1142. https://doi.org/10.3390/molecules25051142