Primary Human Hepatocytes, But not HepG2 or Balb/c 3T3 Cells, Efficiently Metabolize Salinomycin and Are Resistant to Its Cytotoxicity


Abstract
:1. Introduction
2. Results
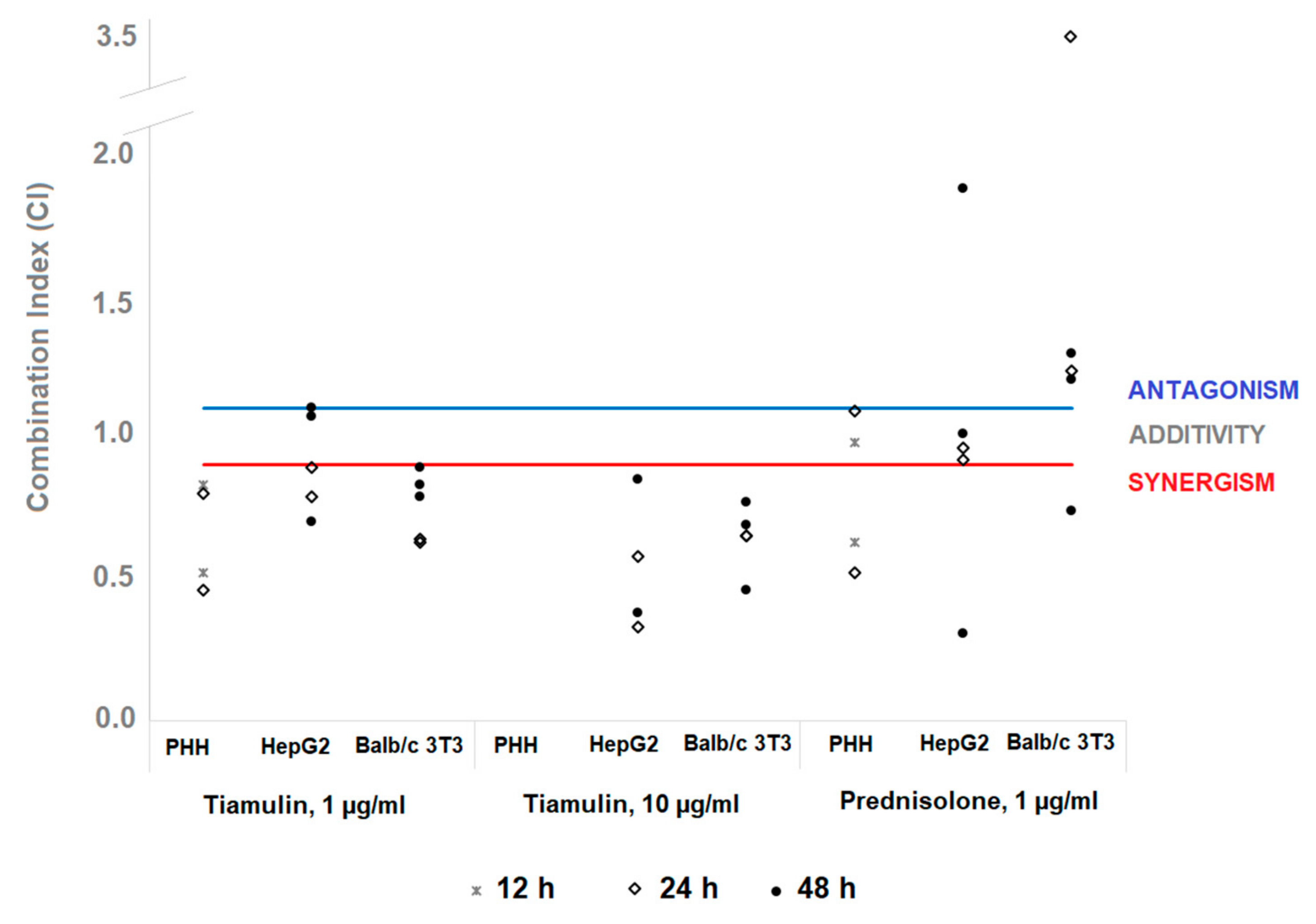
2.1. Cytotoxicity of Salinomycin and its Combinations with Tiamulin or Prednisolone
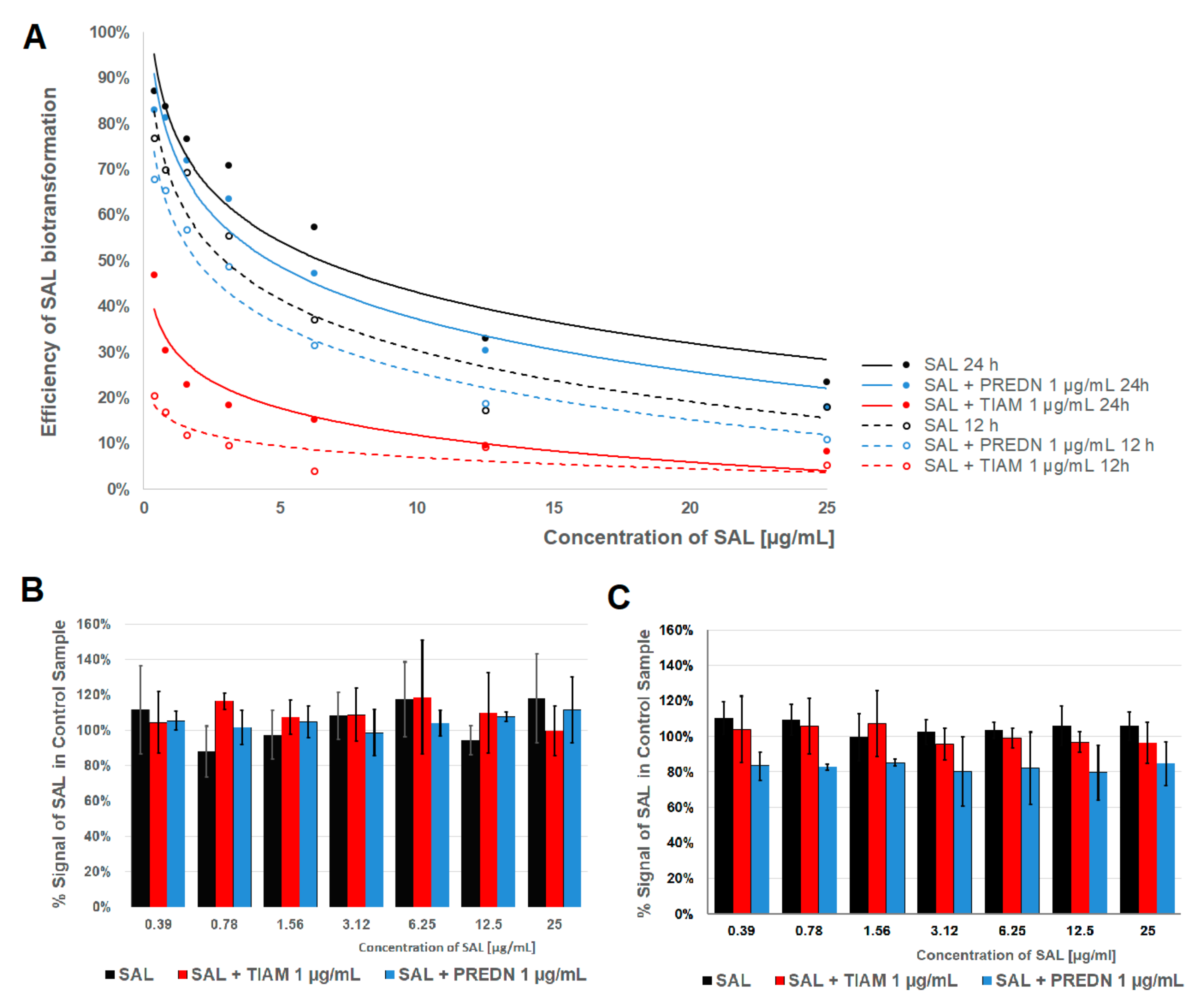
2.2. Metabolism of SAL
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures
4.2.1. PHH and Culture Conditions
4.2.2. Cell lines and Culture Conditions
4.3. Exposure to Drugs
4.4. Cytotoxicity Assessment
4.4.1. MTT Assay
4.4.2. NRU Assay
4.4.3. TPC Assay
4.4.4. LDH Leakage Assay
4.5. Analysis of Drug Interactions
4.6. Determination of SAL Metabolites
4.6.1. Sample Preparation
4.6.2. LC-MS/MS Determination
4.6.3. Data Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, A.-K.; Hermawan, A.; Mickler, F.M.; Ljepoja, B.; Knyazev, P.; Bräuchle, C.; Ullrich, A.; Wagner, E.; Roidl, A. Salinomycin co-treatment enhances tamoxifen cytotoxicity in luminal A breast tumor cells by facilitating lysosomal degradation of receptor tyrosine kinases. Oncotarget 2016, 7, 50461–50476. [Google Scholar] [CrossRef] [Green Version]
- Huczyński, A. Polyether ionophores-promising bioactive molecules for cancer therapy. Bioorg. Med. Chem. Lett. 2012, 22, 7002–7010. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Li, H.; Qaed, E.; Zhang, J.; Song, Y.; Wu, R.; Bu, X.; Wang, Q.; Tang, Z. Salinomycin, as an autophagy modulator—A new avenue to anticancer: A review. J. Exp. Clin. Cancer Res. 2018, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Story, P.; Doube, A. A case of human poisoning by salinomycin, an agricultural antibiotic. New Zeal. Med. J. 2004, 117, 1–4. [Google Scholar]
- Dorne, J.L.C.M.; Fernández-Cruz, M.L.; Bertelsen, U.; Renshaw, D.W.; Peltonen, K.; Anadon, A.; Feil, A.; Sanders, P.; Wester, P.; Fink-Gremmels, J. Risk assessment of coccidostatics during feed cross-contamination: Animal and human health aspects. Toxicol. Appl. Pharmacol. 2013, 270, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Oehme, F.W.; Pickrell, J.A. An analysis of the chronic oral toxicity of polyether ionophore antibiotics in animals. Vet. Hum. Toxicol. 1999, 41, 251–257. [Google Scholar] [PubMed]
- Novilla, M.N. Ionophores. In Veterinary Toxicology (Ed. R.C. Gupta); Elsevier Inc.: Cambridge, MA, USA, 2018; pp. 1073–1092. [Google Scholar]
- Szucs, G.; Tamási, V.; Laczay, P.; Monostory, K. Biochemical background of toxic interaction between tiamulin and monensin. Chem. Biol. Interact. 2004, 147, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Matoulkova, P.; Pavek, P.; Maly, J.; Vlcek, J. Cytochrome P450 enzyme regulation by glucocorticoids and consequences in terms of drug interaction. Expert Opin. Drug Metab. Toxicol. 2014, 10, 425–435. [Google Scholar] [CrossRef]
- Radko, L.; Olejnik, M. Cytotoxicity of anticancer candidate salinomycin and identification of its metabolites in rat cell cultures. Toxicol. in Vitro 2018, 52, 314–320. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Korzeniewski, C.; Callewaert, D.M. An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Zou, Z.-Z.; Nie, P.-P.; Li, Y.-W.; Hou, B.-X.; Li, R.; Shi, X.-P.; Ma, Z.-K.; Han, B.-W.; Luo, X.-Y. Synergistic induction of apoptosis by salinomycin and gefitinib through lysosomal and mitochondrial dependent pathway overcomes gefitinib resistance in colorectal cancer. Oncotarget 2017, 8, 22414–22432. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; He, L.; Dai, W.-Q.; Xu, Y.-P.; Wu, D.; Lin, C.-L.; Wu, S.-M.; Cheng, P.; Zhang, Y.; Shen, M.; et al. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS ONE 2012, 7, e50638. [Google Scholar] [CrossRef] [Green Version]
- Lieke, T.; Ramackers, W.; Bergmann, S.; Klempnauer, J.; Winkler, M.; Klose, J. Impact of Salinomycin on human cholangiocarcinoma: Induction of apoptosis and impairment of tumor cell proliferation in vitro. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Luo, Q.; Liu, L.; Yang, X.; Zhu, S.; Song, G. Salinomycin attenuates liver cancer stem cell motility by enhancing cell stiffness and increasing F-actin formation via the FAK-ERK1/2 signalling pathway. Toxicology 2017, 384, 1–10. [Google Scholar] [CrossRef]
- Antoszczak, M.; Maj, E.; Napiórkowska, A.; Stefańska, J.; Augustynowicz-Kopeć, E.; Wietrzyk, J.; Janczak, J.; Brzezinski, B.; Huczyński, A. Synthesis, anticancer and antibacterial activity of salinomycin N-benzyl amides. Molecules 2014, 19, 19435–19459. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Ghavami, S.; Grabarek, J.; Kratz, G.; Wiechec, E.; Fredriksson, B.A.; Pariti, R.K.R.; Cieślar-Pobuda, A.; Panigrahi, S.; Łos, M.J. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: Differences between primary and cancer cells. Biochim. Biophys. Acta 2013, 1833, 2057–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehmerle, W.; Endres, M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011, 2, e168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lascala, A.; Martino, C.; Parafati, M.; Salerno, R.; Oliverio, M.; Pellegrino, D.; Mollace, V.; Janda, E. Analysis of proautophagic activities of Citrus flavonoids in liver cells reveals the superiority of a natural polyphenol mixture over pure flavones. J. Nutr. Biochem. 2018, 58, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Klose, J.; Stankov, M.V.; Kleine, M.; Ramackers, W.; Panayotova-Dimitrova, D.; Jäger, M.D.; Klempnauer, J.; Winkler, M.; Bektas, H.; Behrens, G.M.N.; et al. Inhibition of autophagic flux by salinomycin results in anti-cancer effect in hepatocellular carcinoma cells. PLoS ONE 2014, 9, e95970. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, W.; Radko, L.; Rzeski, W. Cytotoxicity of monensin, narasin and salinomycin and their interaction with silybin in HepG2, LMH and L6 cell cultures. Toxicol. In Vitro 2015, 29, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Scherzed, A.; Hackenberg, S.; Froelich, K.; Rak, K.; Technau, A.; Radeloff, A.; Nöth, U.; Koehler, C.; Hagen, R.; Kleinsasser, N. Effects of salinomycin on human bone marrow-derived mesenchymal stem cells in vitro. Toxicol. Lett. 2013, 218, 207–214. [Google Scholar] [CrossRef]
- Anadón, A.; Arzo, M.A.; Bories, G.; Brantom, P.; de Barbera, J.B.; Chesson, A.; Sandro Cocconcelli, P.; de Knecht, J.; Dierick, N.; Flachowsky, G.; et al. Opinion of the Scientific Panel on Additives and Products or Substances used in Animal Feed on a request from the Commission on the safety and efficacy of product “BIO-COX 120G” as feed additive in accordance with Council Directive 70/524/EEC. EFSA J. 2004, 75, 1–51. [Google Scholar]
- Olejnik, M.; Radko, L.; Jedziniak, P. Identification of metabolites of anticancer candidate salinomycin using liquid chromatography coupled with Q-TOF and hybrid QQQ-linear ion trap mass spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 629–634. [Google Scholar] [CrossRef]
- Wilk-Zasadna, I.; Bernasconi, C.; Pelkonen, O.; Coecke, S. Biotransformation in vitro: An essential consideration in the quantitative in vitro-to-in vivo extrapolation (QIVIVE) of toxicity data. Toxicology 2015, 332, 8–19. [Google Scholar] [CrossRef]
- Rychen, G.; Aquilina, G.; Azimonti, G.; Bampidis, V.; de Lourdes Bastos, M.; Bories, G.; Chesson, A.; Cocconcelli, P.S.; Flachowsky, G.; Kolar, B.; et al. Scientific opinion on the safety and efficacy of Sacox® microGranulate (salinomycin sodium) for chickens for fattening and chickens reared for laying. EFSA J. 2017. [Google Scholar] [CrossRef] [Green Version]
- Nebbia, C.; Ceppa, L.; Dacasto, M.; Nachtmann, C.; Carletti, M. Oxidative monensin metabolism and cytochrome P450 3A content and functions in liver microsomes from horses, pigs, broiler chicks, cattle and rats. J. Vet. Pharmacol. Ther. 2001, 24, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Pascussi, J.M.; Drocourt, L.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dexamethasone induces pregnane X receptor and retinoid X receptor-α expression in human hepatocytes: Synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol. Pharmacol. 2000, 58, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, A.; Stolz, D.B.; Ellis, E.C.; Strom, S.C.; Michalopoulos, G.K.; Hengstler, J.G. Dieter Runge Long term cultures of primary human hepatocytes as an alternative to drug testing in animals. ALTEX 2009, 4, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
Sample Availability: Not available. |

{kind=link}
{kind=link}
| Cell Model | Assay | Time | SAL | SAL + T1 | SAL + T10 | SAL + P |
|---|---|---|---|---|---|---|
| PHH | MTT | 12 h | >25 | >25 | N/A* | >25 |
| 24 h | >25 | >25 | N/A | >25 | ||
| NRU | 12 h | 20.4 ± 1.9 a | 4.4 ± 2.3 b | N/A | 8.2 ± 2.5 c | |
| 24 h | 19.8 ± 1.6 a | 2.2 ± 0.6 b | N/A | 5.2 ± 1.2 c | ||
| TPC | 12 h | >25 | >25 | N/A | >25 | |
| 24 h | >25 | >25 | N/A | >25 | ||
| LDH | 12 h | 12.9 ± 3.2 a | 8.7 ± 1.3 b | N/A | 12.5 ± 0.7 a | |
| 24 h | 11.3 ± 0.7 a | 5.9 ± 2.4 b | N/A | 11.5 ± 2.2 a | ||
| HepG2 | MTT | 24 h | 15.4 ± 1.7 a | 13.5 ± 1.1 a | 11.0 ± 1.9 b | >25 |
| 48 h | 12.9 ± 1.4 abc | 12.8 ± 0.2 a | 10.5 ± 1.7 b | 14.9 ± 1.5 c | ||
| NRU | 24 h | 0.6 ± 0.2 a | <0.39 | <0.39 | 0.7 ± 0.1 a | |
| 48 h | <0.39 | <0.39 | <0.39 | <0.39 | ||
| TPC | 24 h | 13.3 ± 2.5 a | 12.6 ± 1.7 a | 4.6 ± 0.6 b | 10.5 ± 1.5 a | |
| 48 h | 11.3 ± 1.3 a | 9.6 ± 0.7 a | 3.0 ± 0.1 b | 4.9 ± 1.2 c | ||
| LDH | 24 h | >25 | 18.5 ± 1.1 b | 18.4 ± 1.1 b | >25 | |
| 48 h | 13.8 ± 0.2 a | 12.1 ± 1.3 ac | 5.6 ± 0.4 b | 11.4 ± 1.1 c | ||
| Balb/c 3T3 | MTT | 24 h | 10.4 ± 1.4 a | 4.5 ± 0.7 b | 4.9 ± 0.7 b | 16.4 ± 1.5 c |
| 48 h | 4.3 ± 0.9 a | 3.7 ± 0.7 ac | 2.3 ± 0.4 b | 2.6 ± 0.5 bc | ||
| NRU | 24 h | 0.6 ± 0.2 | <0.39 | <0.39 | <0.39 | |
| 48 h | <0.39 | <0.39 | <0.39 | <0.39 | ||
| TPC | 24 h | 7.9 ± 1.1 a | 3.9 ± 0.7 b | 3.1 ± 0.7 b | 12.3 ± 2.0 c | |
| 48 h | 1.4 ± 0.5 ac | 0.9 ± 0.2 a | 0.5 ± 0.1 b | 1.8 ± 0.3 c | ||
| LDH | 24 h | >25 | >25 | >25 | >25 | |
| 48 h | 23.1 ± 0.8 a | 22.6 ± 1.5 a | 19.1 ± 1.6 a | 24.8 ± 1.2 a |
| ID | Relative RT a | Biotransformation Route | Ratio to Control b | % of Signal of All Metabolites in PHH | Detected In | ||||
|---|---|---|---|---|---|---|---|---|---|
| SAL | SAL + P | SAL + T | HepG2 | Balb/c 3T3 | |||||
| M1 | 0.501 | Trihydroxylation | 1424 | 1.64 | 1.40 | 0.61 | |||
| M2 | 0.552 | Dihydroxylation | 5357 | 4.00 | 4.00 | 2.59 | Yes, P c | ||
| M3 | 0.563 | Dihydroxylation | 468 | 1.09 | 1.05 | 0.59 | |||
| M4 | 0.573 | Dihydroxylation | 414 | 0.83 | 0.40 | 0.42 | |||
| M5 | 0.617 | Dihydroxylation | ∞ d | 0.20 | 0.20 | 0.13 | |||
| M6 | 0.621 | Dihydroxylation | 6167 | 15.36 | 14.18 | 9.59 | Yes, P | ||
| M7 | 0.644 | Dihydroxylation | ∞ | 0.27 | 0.25 | 0.16 | |||
| M8 | 0.656 | Dihydroxylation | 408 | 1.08 | 1.12 | 0.97 | |||
| M9 | 0.670 | Dihydroxylation | 490 | 1.10 | 1.10 | 1.02 | |||
| M10 | 0.682 | Hydroxylation + Demethylathion | ∞ | 0.05 | 0.05 | 0.05 | |||
| M11 | 0.691 | Demethylathion | 11.3 | 0.04 | 0.04 | 0.03 | |||
| M12 | 0.703 | Hydroxylation | 190 | 48.1 | 49.9 | 52.0 | Yes | ||
| M13 | 0.709 | Dihydroxylation | 155 | 0.22 | 0.22 | 0.31 | |||
| M14 | 0.722 | Hydroxylation | ∞ | 23.9 | 23.6 | 28.4 | Yes | ||
| M15 | 0.733 | Dihydroxylation | 155 | 0.17 | 0.18 | 0.28 | |||
| M16 | 0.789 | Dihydroxylation | 29.6 | 0.19 | 0.22 | 0.24 | |||
| M17 | 0.801 | Hydroxylation | 59.4 | 1.33 | 1.39 | 1.86 | |||
| M18 | 0.851 | Demethylathion | 48.5 | 0.04 | 0.22 | 0.32 | |||
| M19 | 0.866 | Dihydroxylation | 14.0 | 0.29 | 0.33 | 0.28 | |||
| M20 | 0.956 | Dihydroxylation | 3.00 | 0.12 | 0.13 | 0.12 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radko, L.; Olejnik, M.; Posyniak, A. Primary Human Hepatocytes, But not HepG2 or Balb/c 3T3 Cells, Efficiently Metabolize Salinomycin and Are Resistant to Its Cytotoxicity. Molecules 2020, 25, 1174. https://doi.org/10.3390/molecules25051174
Radko L, Olejnik M, Posyniak A. Primary Human Hepatocytes, But not HepG2 or Balb/c 3T3 Cells, Efficiently Metabolize Salinomycin and Are Resistant to Its Cytotoxicity. Molecules. 2020; 25(5):1174. https://doi.org/10.3390/molecules25051174
Chicago/Turabian StyleRadko, Lidia, Małgorzata Olejnik, and Andrzej Posyniak. 2020. "Primary Human Hepatocytes, But not HepG2 or Balb/c 3T3 Cells, Efficiently Metabolize Salinomycin and Are Resistant to Its Cytotoxicity" Molecules 25, no. 5: 1174. https://doi.org/10.3390/molecules25051174