
1,2,4-Triazolo[1,5-a]pyrimidines as a Novel Class of Inhibitors of the HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity



 , ,
, ,  ,
,  and
and 
Abstract
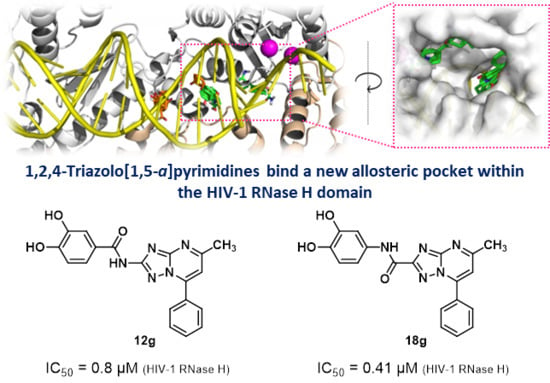
:
1. Introduction
2. Results and Discussion
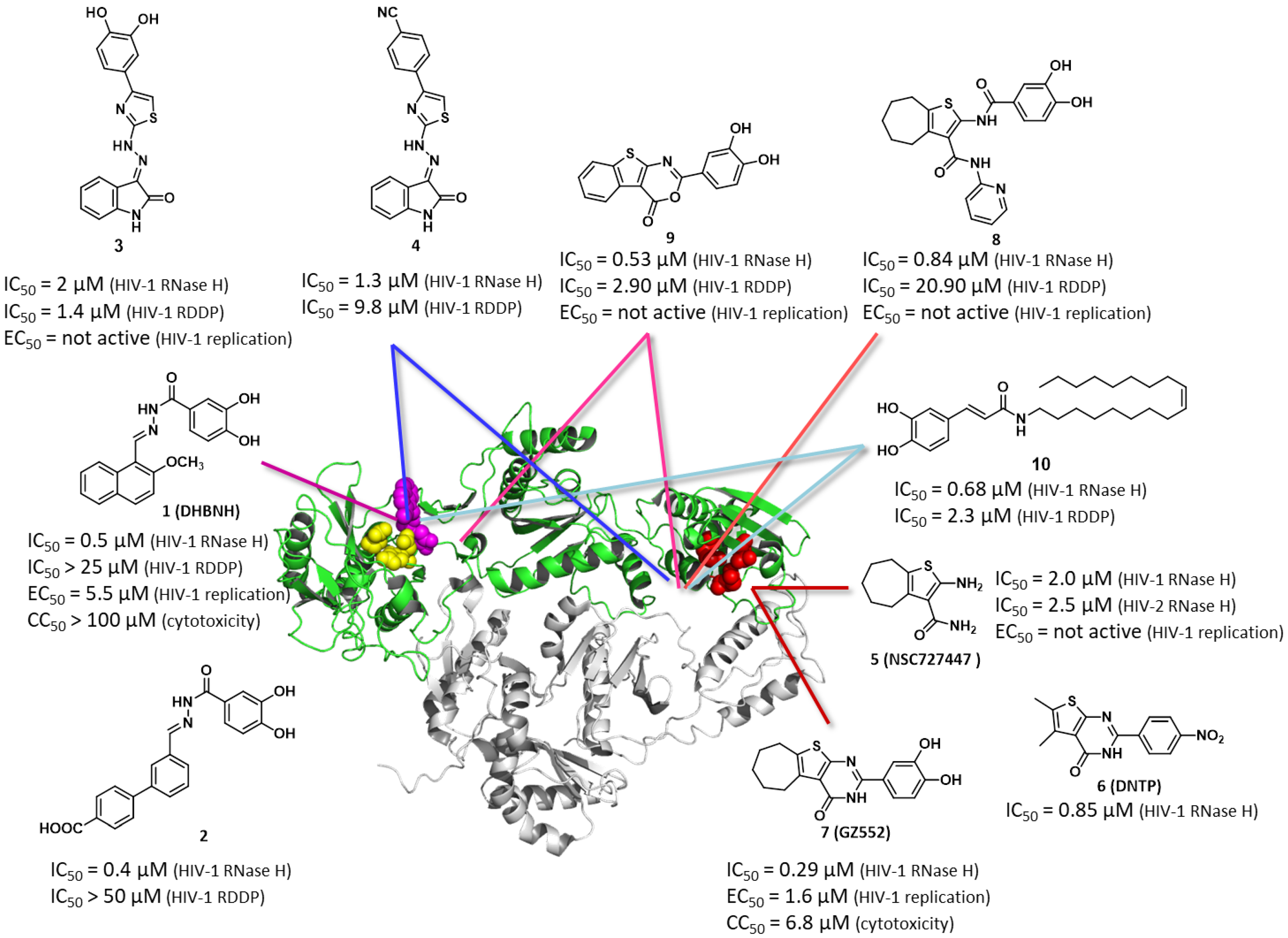
2.1. Structural Exploration of the 1,2,4-Triazolo[1,5-a]pyrimidine Scaffold
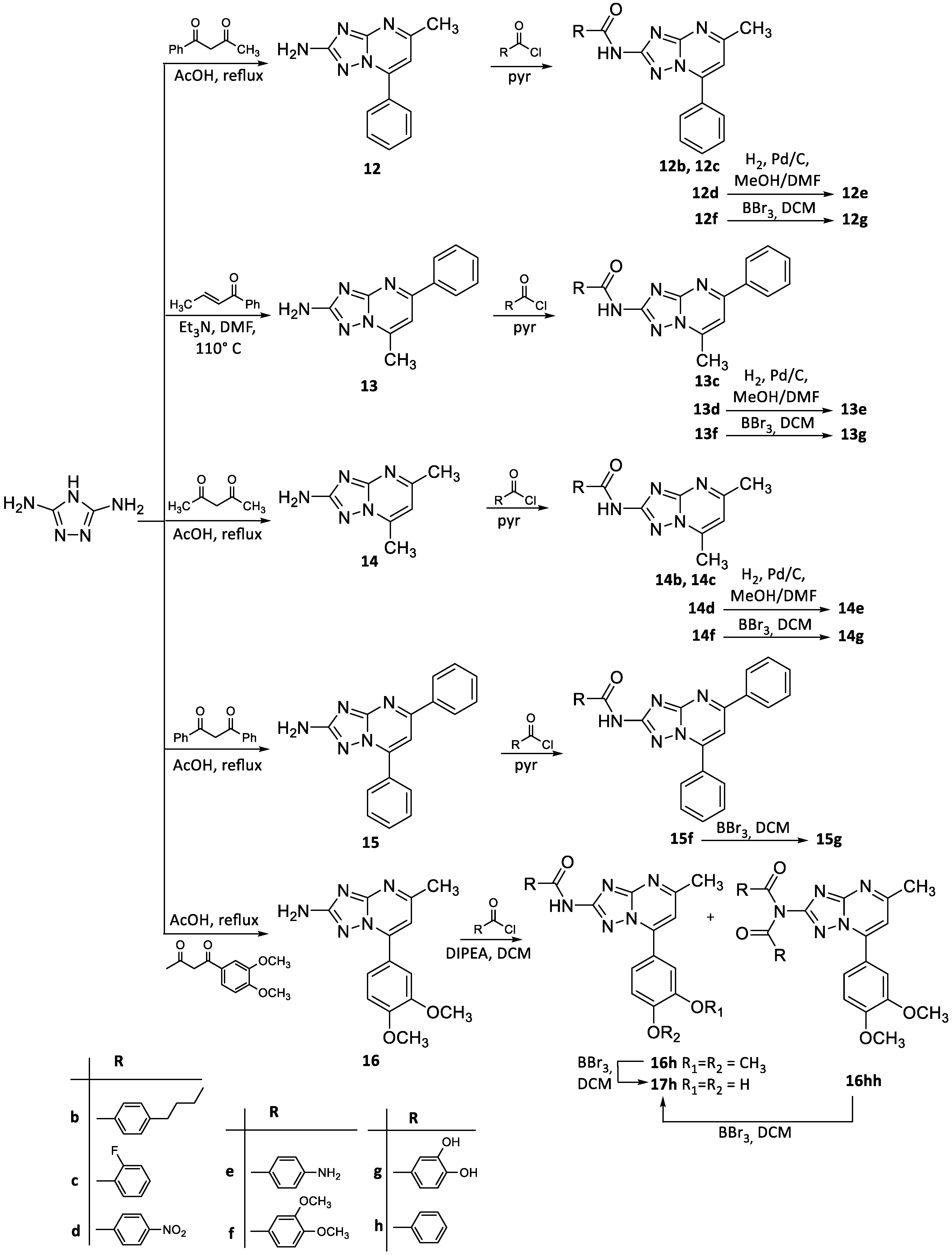
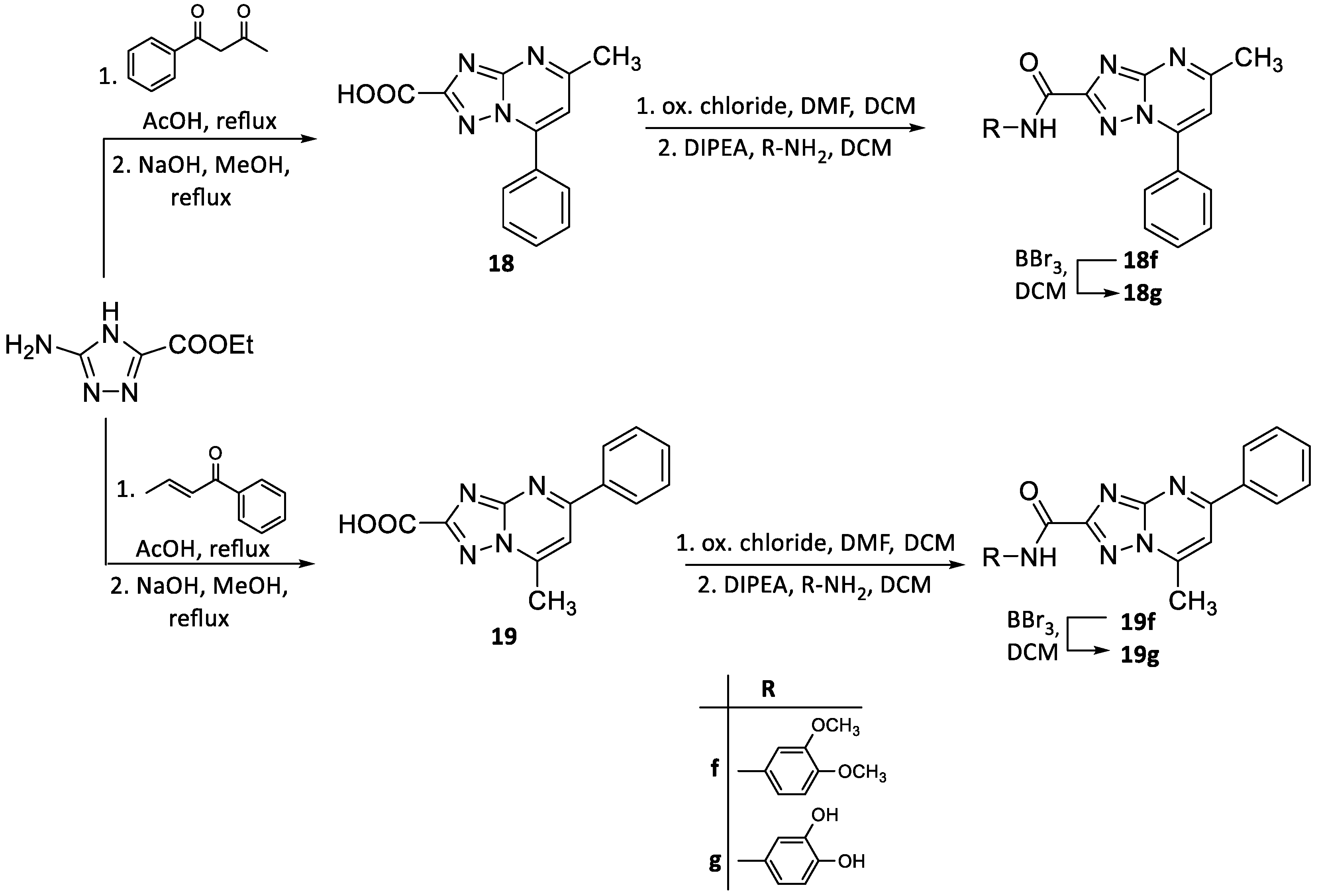
2.2. Chemistry
2.3. Evaluation of RNase H and RDDP Inhibitory Activity
2.4. Mg2+ Coordination Analysis
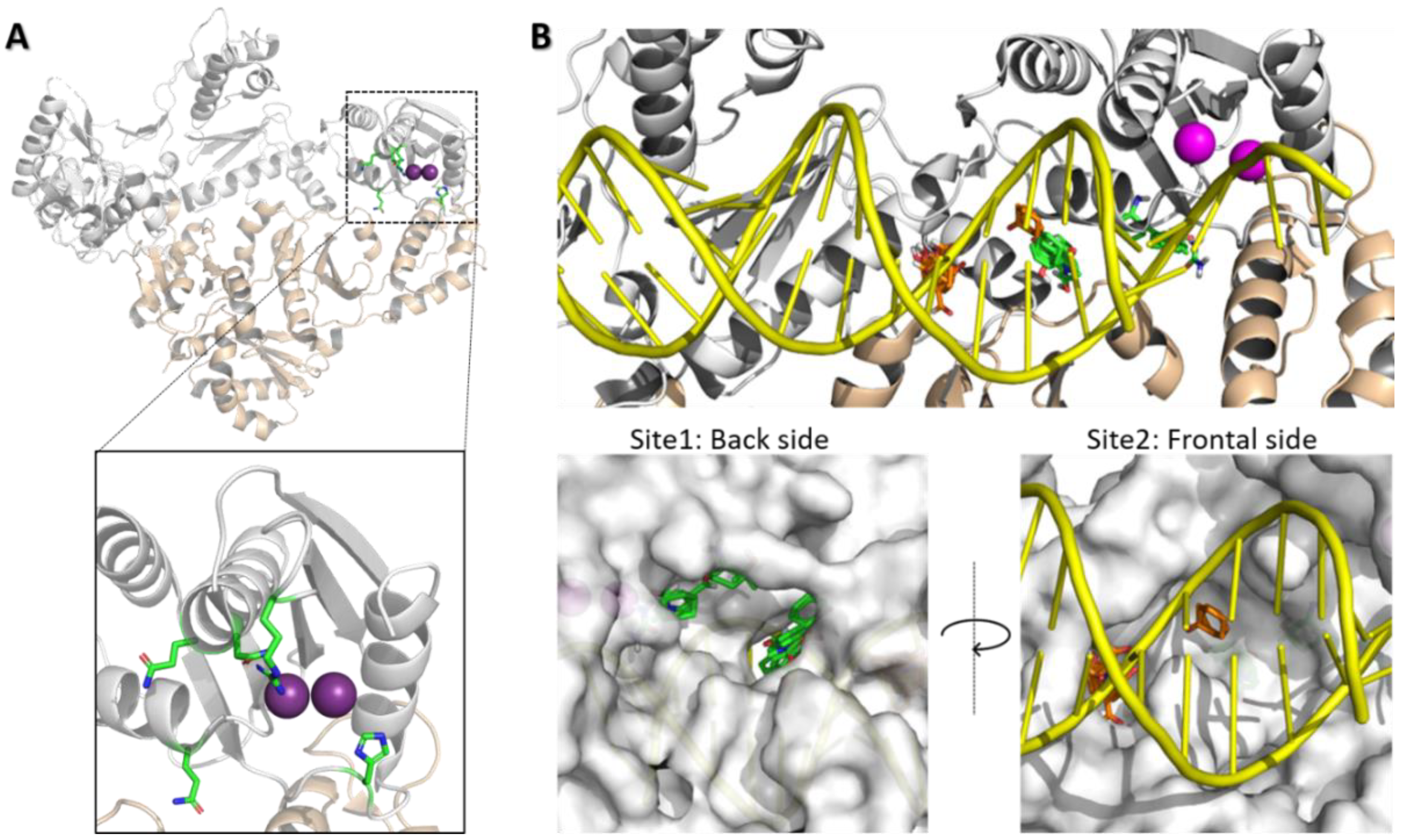
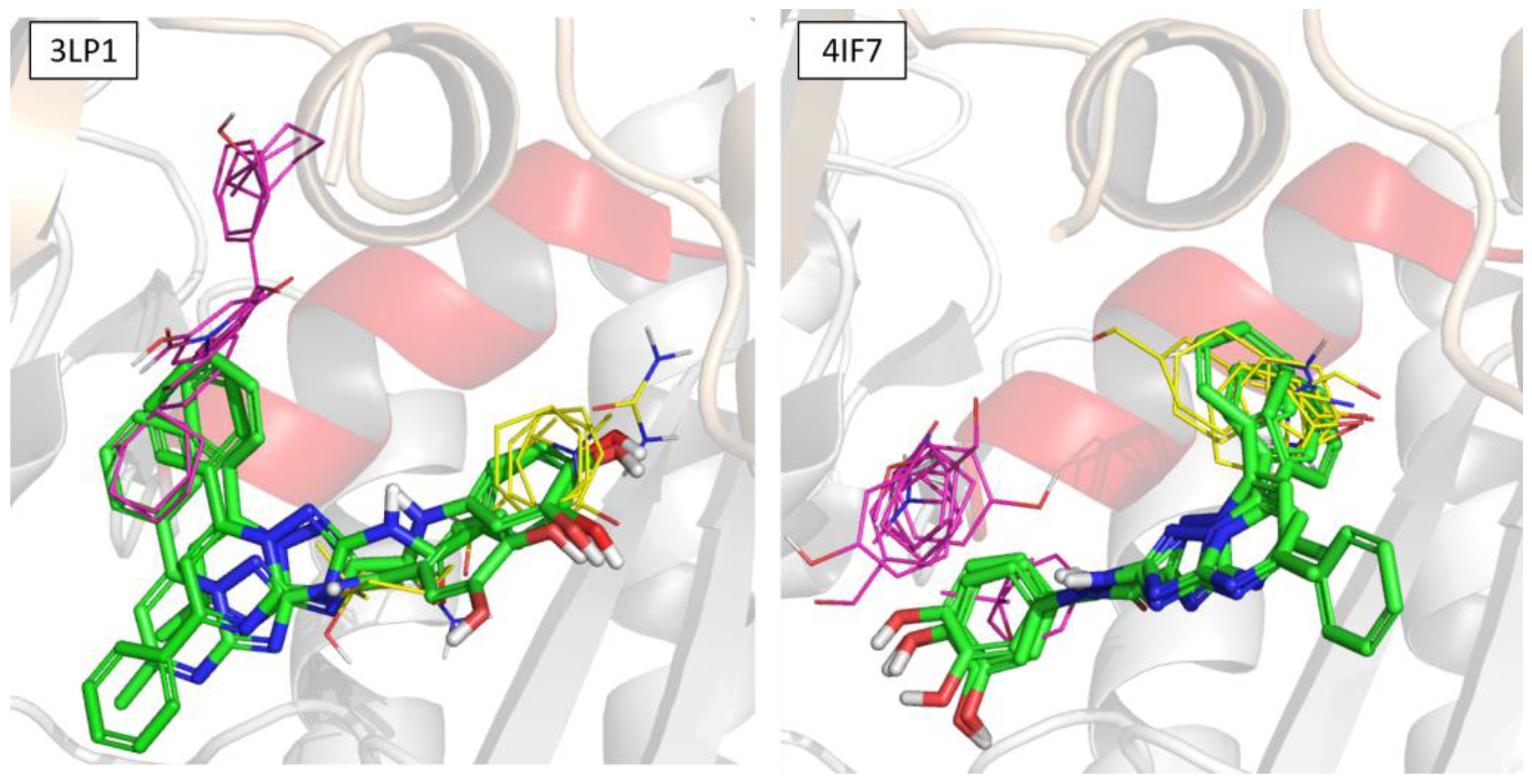
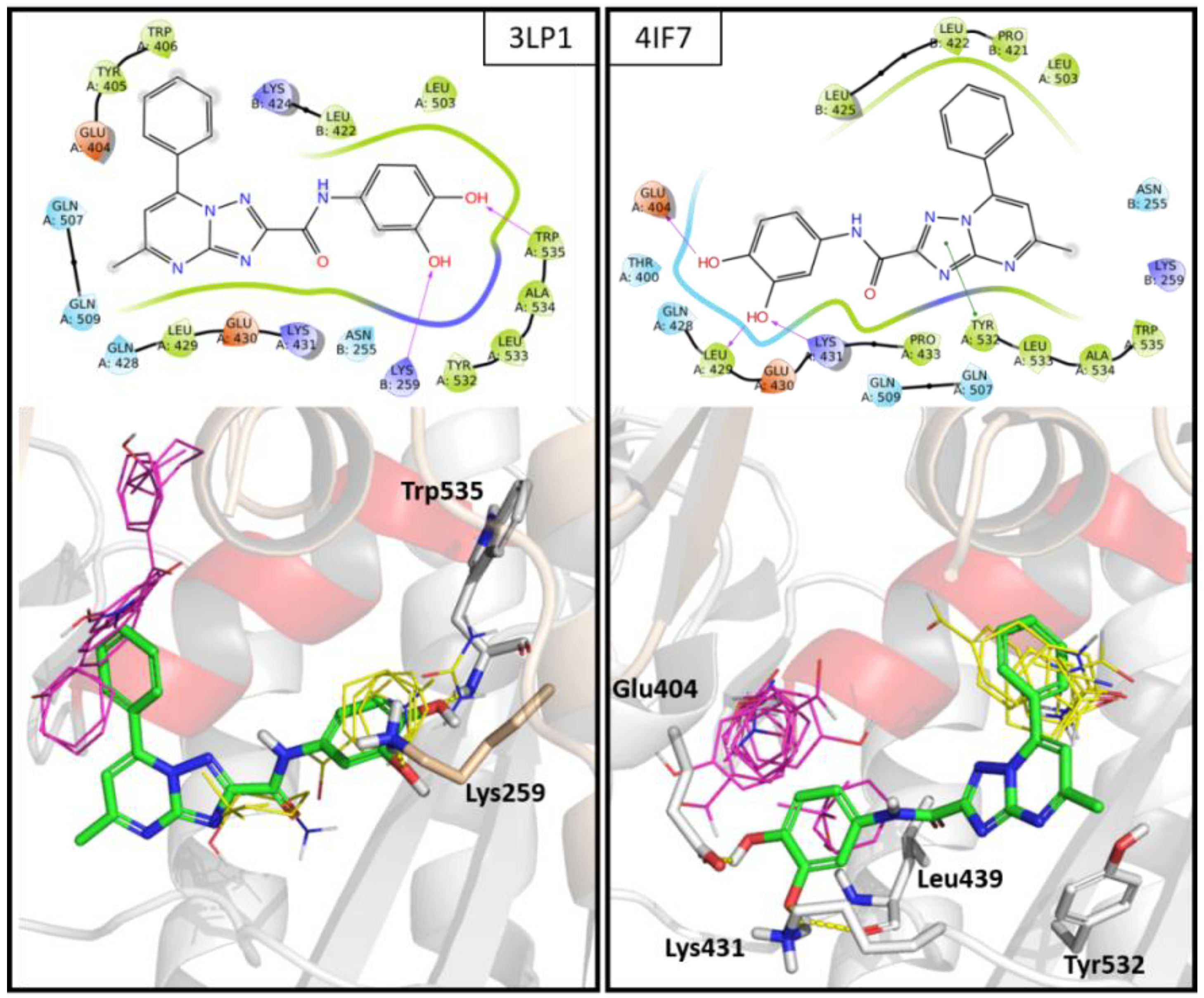
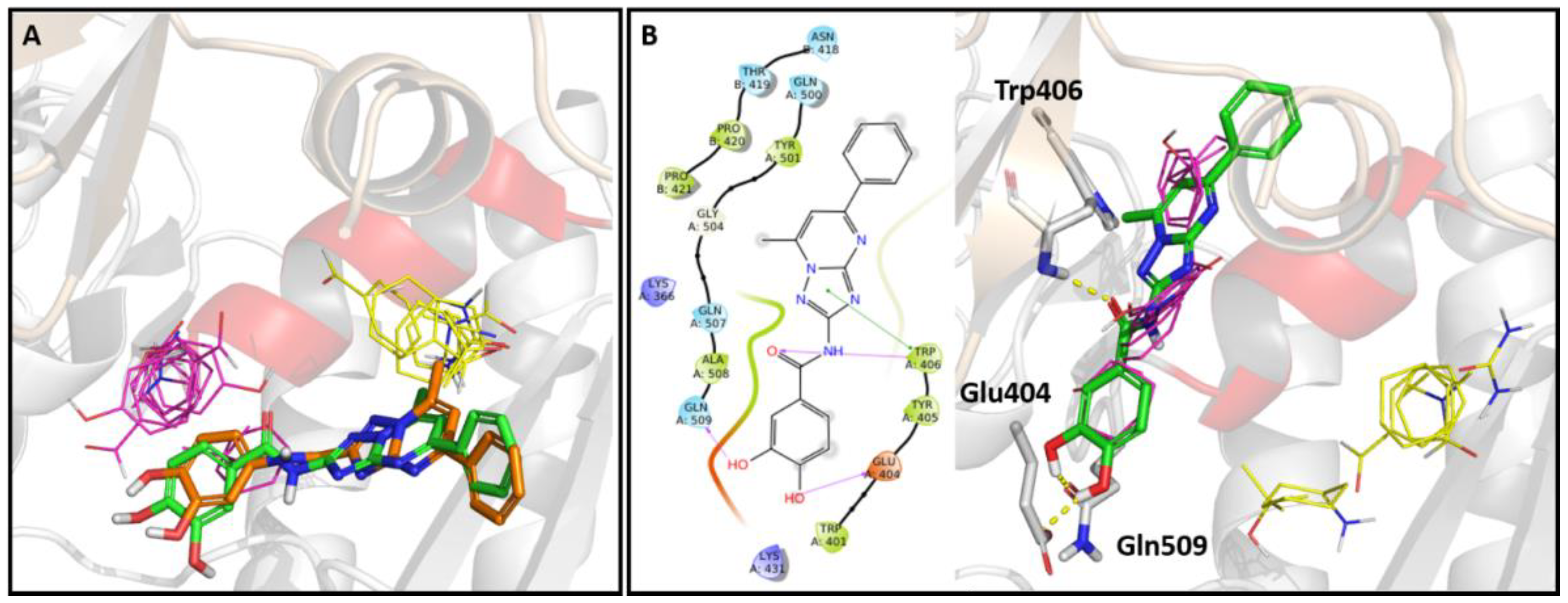
2.5. In Silico Studies
2.6. Site Directed Mutagenesis
2.7. Anti-HIV-1 Activity
3. Materials and Methods
3.1. Chemistry
3.1.1. General Synthesis Methods and Analysis
General Procedure for C-2 Amidation (Method A)
General Procedure for O-demethylation Reaction (Method B)
General Procedure for the Preparation of 5,7-(substitued)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (Method C)
General Procedure for C-2 Amidation (Method D)
3.2. Biological Evaluation
3.2.1. Expression and purification of recombinant HIV-1 RT
3.2.2. Site directed mutagenesis
3.2.3. HIV-1 DNA polymerase-independent RNase H activity determination
3.2.4. HIV-1 RNA-dependent DNA polymerase activity determination
3.2.5. In Vitro Antiviral Assays
3.3. Evaluation of MgCl2 Coordination
3.4. Evaluation of Fluorescence
3.5. In Silico Studies
3.5.1. Selection of Crystal Structure Subset
3.5.2. Binding Site Identification
3.5.3. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Taddei, T.H.; Lo Re, V.; Justice, A.C. HIV, aging, and viral coinfections: Taking the long view. Curr. HIV/AIDS Rep. 2016, 13, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The role of HIV-1 drug-resistant minority variants in treatment failure. J. Infect. Dis. 2017, 216, S847–S850. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Corona, A.; Spöring, I.; Jordan, M.; Buchholz, B.; Maccioni, E.; Di Santo, R.; Bodem, J.; Tramontano, E.; Wöhrl, B.M. Biochemical characterization of a multi-drug resistant HIV-1 subtype AG reverse transcriptase: Antagonism of AZT discrimination and excision pathways and sensitivity to RNase H inhibitors. Nucleic Acids Res. 2016, 44, 2310–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, T. Understanding and managing the adverse effects of antiretroviral therapy. Antivir. Res. 2010, 85, 201–209. [Google Scholar] [CrossRef]
- Stein, J.; Storcksdieck Genannt Bonsmann, M.; Streeck, H. Barriers to HIV cure. HLA 2016, 88, 155–163. [Google Scholar] [CrossRef]
- Badowski, M.E.; Pérez, S.E.; Biagi, M.; Littler, J.A. New antiretroviral treatment for HIV. Infect. Dis. Ther. 2016, 5, 329–352. [Google Scholar] [CrossRef] [Green Version]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Abbondanzieri, E.A.; Rausch, J.W.; Le Grice, S.F.; Zhuang, X. Slide into action: Dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science 2008, 322, 1092–1097. [Google Scholar] [CrossRef]
- Tisdale, M.; Schulze, T.; Larder, B.A.; Moelling, K. Mutations within the RNase H domain of human immunodeficiency virus type 1 reverse transcriptase abolish virus infectivity. J. Gen. Virol. 1991, 72, 59–66. [Google Scholar] [CrossRef]
- Volkmann, S.; Wöhrl, B.M.; Tisdale, M.; Moelling, K. Enzymatic analysis of two HIV-1 reverse transcriptase mutants with mutations in carboxyl-terminal amino acid residues conserved among retroviral ribonucleases H. J. Biol. Chem. 1993, 268, 2674–2683. [Google Scholar]
- Tanese, N.; Goff, S.P. Domain structure of the Moloney murine leukemia virus reverse transcriptase: Mutational analysis and separate expression of the DNA polymerase and RNase H activities. Proc. Natl. Acad. Sci. USA 1988, 85, 1777–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, O.; Cromme, F.V.; Grüninger-Leitch, F.; Le Grice, S.F. Point mutations in conserved amino acid residues within the C-terminal domain of HIV-1 reverse transcriptase specifically repress RNase H function. FEBS Lett. 1989, 257, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Corona, A.; Masaoka, T.; Tocco, G.; Tramontano, E.; Le Grice, S.F. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Future Med. Chem. 2013, 5, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Song, W.; De Clercq, E.; Zhan, P.; Liu, X. Recent progress in the research of small molecule HIV-1 RNase H inhibitors. Curr. Med. Chem. 2014, 21, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, P.; Menendez-Arias, L.; Liu, X.; Zhan, P. Update on recent developments in small molecular HIV-1 RNase H inhibitors (2013-2016): Opportunities and challenges. Curr. Med. Chem. 2018, 25, 1682–1702. [Google Scholar] [CrossRef]
- Tramontano, E.; Corona, A.; Menéndez-Arias, L.; Ribonuclease, H. An unexploited target for antiviral intervention against HIV and hepatitis B virus. Antivir. Res. 2019, 171, 104613. [Google Scholar] [CrossRef]
- Himmel, D.M.; Sarafianos, S.G.; Dharmasena, S.; Hossain, M.M.; McCoy-Simandle, K.; Ilina, T.; Clark, A.D., Jr.; Knight, J.L.; Julias, J.G.; Clark, P.K.; et al. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 2006, 1, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Christen, M.T.; Menon, L.; Myshakina, N.S.; Ahn, J.; Parniak, M.A.; Ishima, R. Structural basis of the allosteric inhibitor interaction on the HIV-1 reverse transcriptase RNase H domain. Chem. Biol. Drug Des. 2012, 80, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Distinto, S.; Esposito, F.; Kirchmair, J.; Cardia, M.C.; Gaspari, M.; Maccioni, E.; Alcaro, S.; Markt, P.; Wolber, G.; Zinzula, L.; et al. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur. J. Med. Chem. 2012, 50, 216–229. [Google Scholar] [CrossRef]
- Distinto, S.; Maccioni, E.; Meleddu, R.; Corona, A.; Alcaro, S.; Tramontano, E. Molecular aspects of the RT/drug interactions. Perspective of dual inhibitors. Curr. Pharm. Des. 2013, 19, 1850–1859. [Google Scholar] [CrossRef]
- Corona, A.; Meleddu, R.; Esposito, F.; Distinto, S.; Bianco, G.; Masaoka, T.; Maccioni, E.; Menéndez-Arias, L.; Alcaro, S.; Le Grice, S.F.; et al. Ribonuclease H/DNA polymerase HIV-1 reverse transcriptase dual inhibitor: Mechanistic studies on the allosteric mode of isatin-based compound RMNC6. PLoS ONE 2016, 11, e0147225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendeler, M.; Lee, H.F.; Bermingham, A.; Miller, J.T.; Chetov, O.; Bona, M.K.; Baichoo, N.S.; Ehteshami, M.; Beutler, J.; O’Keefe, B.R.; et al. Vinylogous ureas as a novel class of inhibitors of reverse transcriptase-associated ribonuclease H activity. ACS Chem. Biol. 2008, 3, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Wendeler, M.; Rausch, J.W.; Beilhartz, G.; Gotte, M.; O’Keefe, B.R.; Bermingham, A.; Beutler, J.A.; Liu, S.; Zhuang, X.; et al. Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 2010, 54, 3913–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.; Miller, J.T.; Johnson, B.C.; Hughes, S.H.; Le Grice, S.F. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J. Biol. Chem. 2012, 287, 4066–4075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaoka, T.; Chung, S.; Caboni, P.; Rausch, J.W.; Wilson, J.A.; Taskent-Sezgin, H.; Beutler, J.A.; Tocco, G.; Le Grice, S.F. Exploiting drug-resistant enzymes as tools to identify thienopyrimidinone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 2013, 56, 5436–5445. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Nannetti, G.; Goracci, L.; Sancineto, L.; Muratore, G.; Sabatini, S.; Manfroni, G.; Mercorelli, B.; Cecchetti, V.; Facchini, M.; et al. Structural investigation of cycloheptathiophene-3-carboxamide derivatives targeting influenza virus polymerase assembly. J. Med. Chem. 2013, 56, 10118–10131. [Google Scholar] [CrossRef]
- Corona, A.; Desantis, J.; Massari, S.; Distinto, S.; Masaoka, T.; Sabatini, S.; Esposito, F.; Manfroni, G.; Maccioni, E.; Cecchetti, V.; et al. Studies on cycloheptathiophene-3-carboxamide derivatives as allosteric HIV-1 ribonuclease H inhibitors. ChemMedChem 2016, 11, 1709–1720. [Google Scholar] [CrossRef]
- Massari, S.; Corona, A.; Distinto, S.; Desantis, J.; Caredda, A.; Sabatini, S.; Manfroni, G.; Felicetti, T.; Cecchetti, V.; Pannecouque, C.; et al. From cycloheptathiophene-3-carboxamide to oxazinone-based derivatives as allosteric HIV-1 ribonuclease H inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 55–74. [Google Scholar] [CrossRef] [Green Version]
- Sonar, V.P.; Corona, A.; Distinto, S.; Maccioni, E.; Meleddu, R.; Fois, B.; Floris, C.; Malpure, N.V.; Alcaro, S.; Tramontano, E.; et al. Natural product-inspired esters and amides of ferulic and caffeic acid as dual inhibitors of HIV-1 reverse transcriptase. Eur. J. Med. Chem. 2017, 130, 248–260. [Google Scholar] [CrossRef]
- Massari, S.; Nannetti, G.; Desantis, J.; Muratore, G.; Sabatini, S.; Manfroni, G.; Mercorelli, B.; Cecchetti, V.; Palù, G.; Cruciani, G.; et al. A broad anti-influenza hybrid small molecule that potently disrupts the interaction of polymerase acidic protein-basic protein 1 (PA-PB1) subunits. J. Med. Chem. 2015, 58, 3830–3842. [Google Scholar] [CrossRef]
- Massari, S.; Desantis, J.; Nannetti, G.; Sabatini, S.; Tortorella, S.; Goracci, L.; Cecchetti, V.; Loregian, A.; Tabarrini, O. Efficient and regioselective one-step synthesis of 7-aryl-5-methyl- and 5-aryl-7-methyl-2-amino-[1,2,4]triazolo[1,5-a]pyrimidine derivatives. Org. Biomol. Chem. 2017, 15, 7944–7955. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberger, A. Kondensationen mit hydrazin-N,N′-dicarbonsäure-diamidin, III. s-triazolo[4.3-a]pyrimidine und s-triazolo[1.5-a]pyrimidine. Chem. Ber. 1966, 99, 2237–2245. [Google Scholar] [CrossRef]
- Desenko, S.M.; Kolos, N.N.; Tueny, M.; Orlov, V.D. Cyclocondensation of chalcones with di- and triamino-1,2,4-triazoles. Khim. Geterotsikl. Soedin. 1990, 7, 938–941. [Google Scholar] [CrossRef]
- Chernyshev, V.M.; Chernysheva, A.V.; Taranushich, V.A. Synthesis of esters and amides of 5-Amino-1,2,4-triazole-3-carboxylic and 5-amino-1,2,4-triazol-3-ylacetic acids. Russ. J. Appl. Chem. 2006, 79, 783–786. [Google Scholar] [CrossRef]
- Tramontano, E.; Esposito, F.; Badas, R.; Di Santo, R.; Costi, R.; La Colla, P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antivir. Res. 2005, 65, 117–124. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Gerick, F.; Thornton, J.M. The structural basis of allosteric regulation in proteins. FEBS Lett. 2009, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- FTMap. Available online: http://ftmap.bu.edu (accessed on 2 December 2019).
- Bauman, J.D.; Patel, D.; Dharia, C.; Fromer, M.W.; Ahmed, S.; Frenkel, Y.; Vijayan, R.S.K.; Eck, J.T.; Ho, W.C.; Das, K.; et al. Detecting allosteric sites of HIV-1 reverse transcriptase by X-ray crystallographic fragment screening. J. Med. Chem. 2013, 56, 2738–2746. [Google Scholar] [CrossRef] [Green Version]
- Felts, A.K.; Labarge, K.; Bauman, J.D.; Patel, D.V.; Himmel, D.M.; Arnold, E.; Parniak, M.A.; Levy, R.M. Identification of alternative binding sites for inhibitors of HIV-1 ribonuclease H through comparative analysis of virtual enrichment studies. J. Chem. Inf. Model. 2011, 51, 1986–1998. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2019-4: QikProp; Schrödinger, LLC: New York, NY, USA, 2019.
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15-ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Majid, T.N.; Hopkins, C.; Pedgrift, B.L.; Collar, N.; Wirtz-Brugger, F.; Merrill, J. Pyrazoloisoquinoline derivatives as kinase inhibitors, and their preparation, pharmaceutical compositions, and use in the treatment of diseases involving increased NIK activity. PCT Int. Appl. 2005012301, 10 February 2005. [Google Scholar]
- Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Gagliardi, M.G.; Ambrosio, F.A.; Ortuso, F.; et al. Novel natural non-nucleoside inhibitors of HIV-1 reverse transcriptase identified by shape- and structure-based virtual screening techniques. Eur. J. Med. Chem. 2019, 161, 1–10. [Google Scholar] [CrossRef]
- Xu, L.; Grandi, N.; Del Vecchio, C.; Mandas, D.; Corona, A.; Piano, D.; Esposito, F.; Parolin, C.; Tramontano, E. From the traditional Chinese medicine plant Schisandra chinensis new scaffolds effective on HIV-1 reverse transcriptase resistant to non-nucleoside inhibitors. J. Microbiol. 2015, 53, 288–293. [Google Scholar] [CrossRef]
- Pala, N.; Esposito, F.; Rogolino, D.; Carcelli, M.; Sanna, V.; Palomba, M.; Naesens, L.; Corona, A.; Grandi, N.; Tramontano, E.; et al. Inhibitory effect of 2,3,5,6-tetrafluoro-4-[4-(aryl)-1H-1,2,3-triazol-1-yl]benzenesulfonamide derivatives on HIV reverse transcriptase associated RNase H activities. Int. J. Mol. Sci. 2016, 17, E1371. [Google Scholar] [CrossRef] [Green Version]
- Pannecouque, C.; Daelemans, D.; De Clercq, E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: Revisited 20 years later. Nat. Protoc. 2008, 3, 427–434. [Google Scholar] [CrossRef]
- Tabarrini, O.; Massari, S.; Daelemans, D.; Meschini, F.; Manfroni, G.; Bottega, L.; Gatto, B.; Palumbo, M.; Pannecouque, C.; Cecchetti, V. Studies of anti-HIV transcription inhibitor quinolones: Identification of potent N1-vinyl derivatives. ChemMedChem 2010, 5, 1880–1892. [Google Scholar] [CrossRef]
- Esposito, F.; Sanna, C.; Del Vecchio, C.; Cannas, V.; Venditti, A.; Corona, A.; Bianco, A.; Serrilli, A.M.; Guarcini, L.; Parolin, C.; et al. Hypericum hircinum L. components as new single-molecule inhibitors of both HIV-1 reverse transcriptase-associated DNA polymerase and ribonuclease H activities. Pathog. Dis. 2013, 68, 116–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger Release 2019-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2019.
- Schrödinger Release 2019-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2019.
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2019.
- Vernekar, S.K.V.; Tang, J.; Wu, B.; Huber, A.D.; Casey, M.C.; Myshakira, N.; Wilson, D.; Kankanala, J.; Kirby, K.A.; Parniak, M.A.; et al. Double-winged 3-hydroxypyrimidine-2,4-diones: Potent and selective inhibition against HIV-1 RNase H with significant antiviral activity. J. Med. Chem. 2017, 60, 5045–5056. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, S.; Artese, A.; Ceccherini-Silberstein, F.; Chiarella, V.; Dimonte, S.; Ortuso, F.; Perno, C.F. Computational analysis of Human Immunodeficiency Virus (HIV) Type-1 reverse transcriptase crystallographic models based on significant conserved residues found in Highly Active Antiretroviral Therapy (HAART)-treated patients. Curr. Med. Chem. 2010, 17, 290–308. [Google Scholar] [CrossRef] [Green Version]
- Edwards, T.C.; Ponzar, N.L.; Tavis, J.E. Shedding light on RNaseH: A promising target for hepatitis B virus (HBV). Expert Opin. Ther. Targets 2019, 23, 559–563. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | HIV-1 RNaseH IC50, µMa | HIV-1 RDDP IC50, µMb | |
|---|---|---|---|---|
 | 11a |  | 17.7 ± 2.7 | >100 (74%)c |
| 11b |  | 13.1 ± 2.2 | >100 (74%) | |
 | 12b | 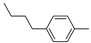 | 12.3 ± 1.5 | >100 (59%) |
| 12c |  | >100 (75%) | >100 (59%) | |
| 12d | 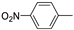 | >100 (77%) | >100 (100%) | |
| 12e | 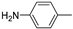 | >100 (91%) | >100 (62%) | |
| 12f | 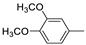 | >100 (100%) | >100 (63%) | |
| 12g | 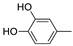 | 0.8 ± 0.2 | >100 (73%) | |
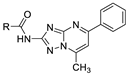 | 13c |  | >100 (90%) | >100 (62%) |
| 13d | 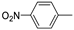 | 60.1 ± 3.3 | >100 (72%) | |
| 13e | 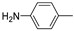 | >100 (98%) | >100 (81%) | |
| 13f | 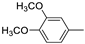 | >100 (100%) | >100 | |
| 13g | 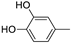 | 3.5 ± 1.5 | >100 | |
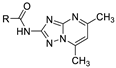 | 14b | 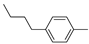 | >100(100%) | |
| 14c | 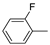 | >100 (100%) | >100 (80%) | |
| 14d | 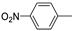 | >100 (75%) | >100 (86%) | |
| 14e | 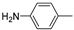 | >100 (91%) | >100 (70%) | |
| 14f | 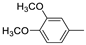 | >100 (93%) | >100 (90%) | |
| 14g |  | 6.23 ± 0.01 | >100 (78%) | |
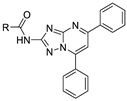 | 15g |  | 1.86 ± 0.54 | 20.5 ± 4.1 |
 | 17h | 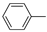 | 1.13 ± 0.33 | >100 (60%) |
 | 16hh | - | 6.74 ± 0.55 | >100 (55%) |
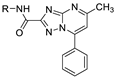 | 18g | 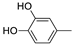 | 0.41 ± 0.005 | >100 (67%) |
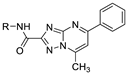 | 19g |  | 43.1 ± 2.1 | N.T.d |
| 5 (NSC727447) | - | 4.0 ± 0.5 | N.T. | |
| RDS1643 | - | 9.0 ± 2.0 | N.T. | |
| efavirenz | - | - | 0.023 ± 0.0016 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desantis, J.; Massari, S.; Corona, A.; Astolfi, A.; Sabatini, S.; Manfroni, G.; Palazzotti, D.; Cecchetti, V.; Pannecouque, C.; Tramontano, E.; et al. 1,2,4-Triazolo[1,5-a]pyrimidines as a Novel Class of Inhibitors of the HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Molecules 2020, 25, 1183. https://doi.org/10.3390/molecules25051183
Desantis J, Massari S, Corona A, Astolfi A, Sabatini S, Manfroni G, Palazzotti D, Cecchetti V, Pannecouque C, Tramontano E, et al. 1,2,4-Triazolo[1,5-a]pyrimidines as a Novel Class of Inhibitors of the HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Molecules. 2020; 25(5):1183. https://doi.org/10.3390/molecules25051183
Chicago/Turabian StyleDesantis, Jenny, Serena Massari, Angela Corona, Andrea Astolfi, Stefano Sabatini, Giuseppe Manfroni, Deborah Palazzotti, Violetta Cecchetti, Christophe Pannecouque, Enzo Tramontano, and et al. 2020. "1,2,4-Triazolo[1,5-a]pyrimidines as a Novel Class of Inhibitors of the HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity" Molecules 25, no. 5: 1183. https://doi.org/10.3390/molecules25051183
APA StyleDesantis, J., Massari, S., Corona, A., Astolfi, A., Sabatini, S., Manfroni, G., Palazzotti, D., Cecchetti, V., Pannecouque, C., Tramontano, E., & Tabarrini, O. (2020). 1,2,4-Triazolo[1,5-a]pyrimidines as a Novel Class of Inhibitors of the HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity. Molecules, 25(5), 1183. https://doi.org/10.3390/molecules25051183