
Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair




Abstract
:1. Introduction
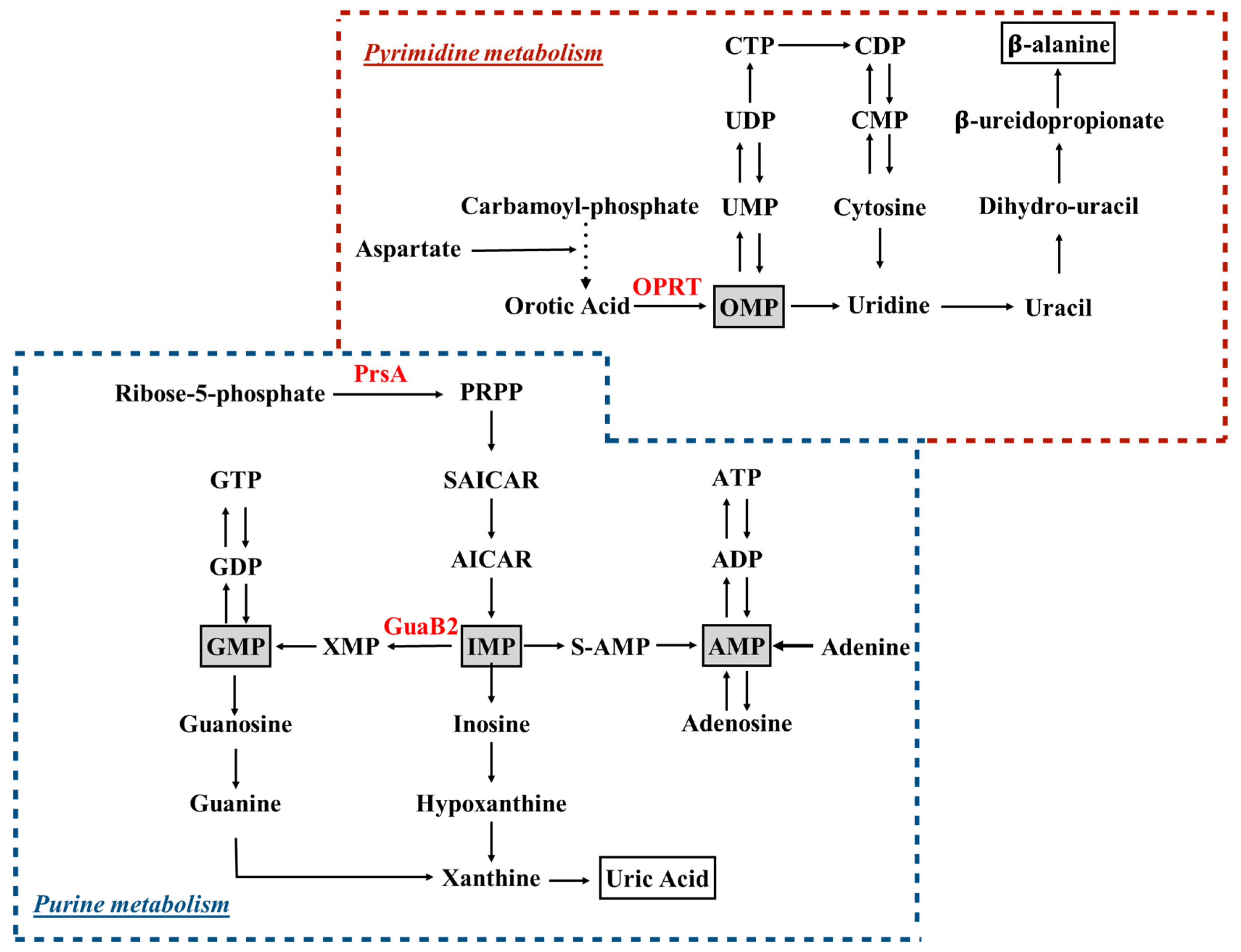
2. Targeting Purine and Pyrimidine Ribonucleotide Synthesis
2.1. Purine Biosynthesis
2.2. Pyrimidine Biosynthesis
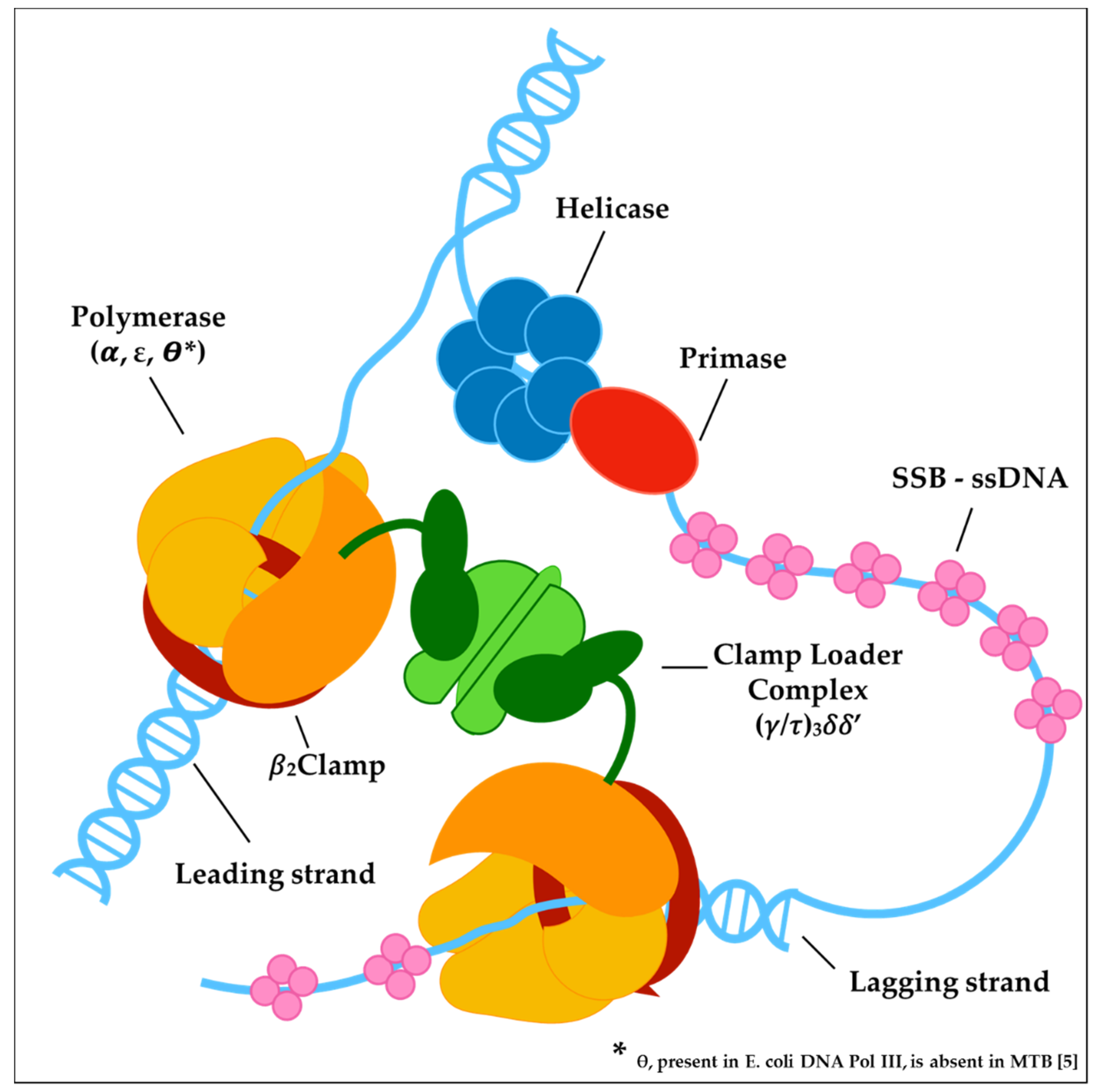
3. Targeting MTB DNA Replication
3.1. The Helicase-Primase Complex
3.2. The Core Complex and the Clamp Loader Complex
3.3. DNA Topology Control and Regulation
4. Targeting MTB DNA Repair
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Dheda, K.; Gumbo, T.; Gandhi, N.R.; Murray, M.; Theron, G.; Udwadia, Z.; Migliori, G.B.; Warren, R. Global control of tuberculosis: From extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2014, 4, 321–338. [Google Scholar] [CrossRef] [Green Version]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, 147–195. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Centis, R.; D’ambrosio, L.; Migliori, G.B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry III, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Maeurer, M.; Mwaba, P.; Chakaya, J.; Rustomjee, R.; Migliori, G.B.; Marais, B.; Schito, M.; Churchyard, G.; Swaminathan, S.; et al. Tuberculosis—Advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect. Dis. 2016, 16, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.T.; Riccardi, G. New tuberculosis drugs on the horizon. Curr. Opin. Microbiol. 2011, 14, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Global Alliance for TB Drug Development. Tuberculosis. Scientific blueprint for tuberculosis drug development. Tuberculosis (Edinb) 2001, 81, 1–52. [Google Scholar] [CrossRef]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhavar, P.S.; Vaja, M.D.; Dhameliya, T.M.; Chakraborti, A.K. Oxazolidinones as Anti-tubercular Agents: Discovery, Development and Future Perspectives. Curr. Med. Chem. 2015, 22, 4379–4397. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Pym, A.S.; Diacon, A.H.; Tang, S.J.; Conradie, F.; Danilovits, M.; Chuchottaworn, C.; Vasilyeva, I.; Andries, K.; Bakare, N.; De Marez, T.; et al. Bedaquiline in the treatment of multidrug- and extensively drug-resistant tuberculosis. Eur. Respir. J. 2016, 47, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Nuermberger, E.L.; Yoshimatsu, T.; Tyagi, S.; O’Brien, R.J.; Vernon, A.N.; Chaisson, R.E.; Bishai, W.R.; Grosset, J.H. Moxifloxacin-containing regimen greatly reduces time to culture conversion in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2004, 169, 421–426. [Google Scholar] [CrossRef]
- Reiche, M.A.; Warner, D.F.; Mizrahi, V. Targeting DNA Replication and Repair for the Development of Novel Therapeutics against Tuberculosis. Front. Mol. Biosci. 2017, 4, 75. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, G.; Doig, P. Bacterial DNA replication enzymes as targets for antibacterial drug discovery. Expert Opin. Drug Discov. 2012, 7, 327–339. [Google Scholar] [CrossRef]
- Robinson, A.; Causer, R.J.; Dixon, N.E. Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets. 2012, 13, 352–372. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef]
- Płocinska, R.; Korycka-Machala, M.; Plocinski, P.; Dziadek, J. Mycobacterial DNA Replication as a Target for Antituberculosis. Drug Discov. Curr. Top. Med. Chem. 2017, 17, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Donini, S.; Garavaglia, S.; Ferraris, D.M.; Miggiano, R.; Mori, S.; Shibayama, K.; Rizzi, M. Biochemical and structural investigations on phosphoribosylpyrophosphate synthetase from Mycobacterium smegmatis. PLoS ONE 2017, 12, e0175815. [Google Scholar] [CrossRef] [PubMed]
- Alderwick, L.J.; Lloyd, G.S.; Lloyd, A.J.; Lovering, A.L.; Eggeling, L.; Besra, G.S. Biochemical characterization of the Mycobacterium tuberculosis phosphoribosyl-1-pyrophosphate synthetase. Glycobiology 2011, 21, 410–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breda, A.; Martinelli, L.K.; Bizarro, C.V.; Rosado, L.A.; Borges, C.B.; Santos, D.S.; Basso, L.A. Wild-type phosphoribosylpyrophosphate synthase (PRS) from Mycobacterium tuberculosis: A bacterial class II PRS? PLoS ONE 2012, 7, e39245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Weerd, R.; Boot, M.; Maaskant, J.; Sparrius, M.; Verboom, T.; van Leeuwen, L.M.; Burggraaf, M.J.; Paauw, N.J.; Dainese, E.; Manganelli, R.; et al. Inorganic Phosphate Limitation Modulates Capsular Polysaccharide Composition in Mycobacteria. J. Biol. Chem. 2016, 291, 11787–11799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraris, D.M.; Spallek, R.; Oehlmann, W.; Singh, M.; Rizzi, M. Crystal structure of the Mycobacterium tuberculosis phosphate binding protein PstS3. Proteins 2014, 82, 2268–2274. [Google Scholar] [CrossRef] [Green Version]
- Warner, D.F.; Evans, J.C.; Mizrahi, V. Nucleotide Metabolism and DNA Replication. In Molecular Genetics of Mycobacteria, 2nd ed.; Hatfull, G.F., Jacobs, W.R., Jr., Eds.; American Society for Microbiology: Washington, WA, USA, 2014. [Google Scholar]
- Usha, V.; Gurcha, S.S.; Lovering, A.L.; Lloyd, A.J.; Papaemmanouil, A.; Reynolds, R.C.; Besra, G.S. Identification of novel diphenyl urea inhibitors of Mt-GuaB2 active against Mycobacterium tuberculosis. Microbiology 2011, 157, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [Green Version]
- Hedstrom, L. IMP dehydrogenase: Structure, mechanism, and inhibition. Chem. Rev. 2009, 109, 2903–2928. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.; Nair, V. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med. Res. Rev. 2008, 28, 219–232. [Google Scholar] [CrossRef]
- Chen, L.; Wilson, D.J.; Xu, Y.; Aldrich, C.C.; Felczak, K.; Sham, Y.Y.; Pankiewicz, K.W. Triazole-linked inhibitors of inosine monophosphate dehydrogenase from human and Mycobacterium tuberculosis. J. Med. Chem. 2010, 53, 4768–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ioerger, T.R.; O’Malley, T.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Mohaideen, N.; Murphy, K.C.; Boshoff, H.I.; Mizrahi, V.; Rubin, E.J.; et al. Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS ONE 2013, 8, e75245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Pacitto, A.; Bayliss, T.; Cleghorn, L.A.T.; Wang, Z.; Hartman, T.; Arora, K.; Ioerger, T.R.; Sacchettini, J.; Rizzi, M.; et al. Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect. Dis. 2017, 3, 18–33. [Google Scholar] [CrossRef]
- Singh, V.; Donini, S.; Pacitto, A.; Sala, C.; Hartkoorn, R.C.; Dhar, N.; Keri, G.; Ascher, D.B.; Mondésert, G.; Vocat, A.; et al. The Inosine Monophosphate Dehydrogenase, GuaB2, Is a Vulnerable New Bactericidal Drug Target for Tuberculosis. ACS Infect. Dis. 2017, 3, 5–17. [Google Scholar] [CrossRef]
- Sahu, N.U.; Singh, V.; Ferraris, D.M.; Rizzi, M.; Kharkar, P.S. Hit discovery of Mycobacterium tuberculosis inosine 5′-monophosphate dehydrogenase, GuaB2, inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1714–1718. [Google Scholar] [CrossRef]
- Chacko, S.; Boshoff, H.I.M.; Singh, V.; Ferraris, D.M.; Gollapalli, D.R.; Zhang, M.; Lawson, A.P.; Pepi, M.J.; Joachimiak, A.; Rizzi, M.; et al. Expanding Benzoxazole-Based Inosine 5′-Monophosphate Dehydrogenase (IMPDH) Inhibitor Structure-Activity As Potential Antituberculosis Agents. J. Med. Chem. 2018, 61, 4739–4756. [Google Scholar] [CrossRef]
- Makowska-Grzyska, M.; Kim, Y.; Gorla, S.K.; Wei, Y.; Mandapati, K.; Zhang, M.; Maltseva, N.; Modi, G.; Boshoff, H.I.; Gu, M.; et al. Mycobacterium tuberculosis IMPDH in Complexes with Substrates, Products and Antitubercular Compounds. PLoS ONE 2015, 10, e0138976. [Google Scholar] [CrossRef] [Green Version]
- Umejiego, N.N.; Gollapalli, D.; Sharling, L.; Volftsun, A.; Lu, J.; Benjamin, N.N.; Stroupe, A.H.; Riera, T.V.; Striepen, B.; Hedstrom, L. Targeting a prokaryotic protein in a eukaryotic pathogen: Identification of lead compounds against cryptosporidiosis. Chem. Biol. 2008, 15, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Maurya, S.K.; Gollapalli, D.R.; Kirubakaran, S.; Zhang, M.; Johnson, C.R.; Benjamin, N.N.; Hedstrom, L.; Cuny, G.D. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2009, 52, 4623–4630. [Google Scholar] [CrossRef] [Green Version]
- Kirubakaran, S.; Gorla, S.K.; Sharling, L.; Zhang, M.; Liu, X.; Ray, S.S.; MacPherson, I.S.; Striepen, B.; Hedstrom, L.; Cuny, G.D. Structure-activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg. Med. Chem. Lett. 2012, 22, 1985–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharling, L.; Liu, X.; Gollapalli, D.R.; Maurya, S.K.; Hedstrom, L.; Striepen, B. A screening pipeline for antiparasitic agents targeting cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl. Trop. Dis. 2010, 4, e794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Breda, A.; Rosado, L.A.; Lorenzini, D.M.; Basso, L.A.; Santos, D.S. Molecular, kinetic and thermodynamic characterization of Mycobacterium tuberculosis orotate phosphoribosyltransferase. Mol. Biosyst. 2012, 8, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Donini, S.; Ferraris, D.M.; Miggiano, R.; Massarotti, A.; Rizzi, M. Structural investigations on orotate phosphoribosyltransferase from Mycobacterium tuberculosis, a key enzyme of the de novo pyrimidine biosynthesis. Sci. Rep. 2017, 7, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breda, A.; Machado, P.; Rosado, L.A.; Souto, A.A.; Santos, D.S.; Basso, L.A. Pyrimidin-2(1H)-ones based inhibitors of Mycobacterium tuberculosis orotate phosphoribosyltransferase. Eur. J. Med. Chem. 2012, 54, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef] [Green Version]
- Wolucka, B.A. Biosynthesis of D-arabinose in mycobacteria - a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 2008, 275, 2691–2711. [Google Scholar] [CrossRef]
- Lucarelli, A.P.; Buroni, S.; Pasca, M.R.; Rizzi, M.; Cavagnino, A.; Valentini, G.; Riccardi, G.; Chiarelli, L.R. Mycobacterium tuberculosis phosphoribosylpyrophosphate synthetase: Biochemical features of a crucial enzyme for mycobacterial cell wall biosynthesis. PLoS ONE 2010, 5, e15494. [Google Scholar] [CrossRef] [Green Version]
- Fivian-Hughes, A.S.; Houghton, J.; Davis, E.O. Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology 2012, 158, 308–318. [Google Scholar] [CrossRef]
- Luciani, R.; Saxena, P.; Surade, S.; Santucci, M.; Venturelli, A.; Borsari, C.; Marverti, G.; Ponterini, G.; Ferrari, S.; Blundell, T.L.; et al. Virtual Screening and X-ray Crystallography Identify Non-Substrate Analog Inhibitors of Flavin-Dependent Thymidylate Synthase. J. Med. Chem. 2016, 59, 9269–9275. [Google Scholar] [CrossRef] [PubMed]
- Hirmondo, R.; Lopata, A.; Suranyi, E.V.; Vertessy, B.G.; Toth, J. Differential control of dNTP biosynthesis and genome integrity maintenance by the dUTPase superfamily enzymes. Sci. Rep. 2017, 7, 6043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ditse, Z.; Lamers, M.H.; Warner, D.F. DNA Replication in Mycobacterium tuberculosis. In Tuberculosis and the Tubercle Bacillus, 2nd ed.; Jacobs, W.R., Jr., McShane, H., Mizrahi, V., Orme, I.M., Eds.; American Society for Microbiology: Washington, WA, USA, 2018. [Google Scholar]
- McHenry, C.S. DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 2011, 80, 403–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brézellec, P.; Vallet-Gely, I.; Possoz, C.; Quevillon-Cheruel, S.; Ferat, J.L. DciA is an ancestral replicative helicase operator essential for bacterial replication initiation. Nat. Commun. 2016, 7, 13271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, K.M.; Huang, D.L.; Hooppaw, A.J.; Logsdon, M.M.; Richardson, K.; Lee, H.J.; Kimmey, J.M.; Aldridge, B.B.; Stallings, C.L. Rv0004 is a new essential member of the mycobacterial DNA replication machinery. PLoS Genet. 2017, 13, e1007115. [Google Scholar] [CrossRef] [PubMed]
- Frank, J. Time-resolved cryo-electron microscopy: Recent progress. J. Struct. Biol. 2017, 200, 303–306. [Google Scholar] [CrossRef]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Bernstein, D.A.; Satyshur, K.A.; Keck, J.L. Small-molecule tools for dissecting the roles of SSB/protein interactions in genome maintenance. Proc. Natl. Acad. Sci. USA 2010, 107, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.C.; Smith, K.C. Mechanism of sbcB-suppression of the recBC-deficiency in postreplication repair in UV-irradiated Escherichia coli K-12. Mol. Gen. Genet. 1985, 201, 186–191. [Google Scholar] [CrossRef]
- Marceau, A.H.; Bernstein, D.A.; Walsh, B.W.; Shapiro, W.; Simmons, L.A.; Keck, J.L. Protein interactions in genome maintenance as novel antibacterial targets. PLoS ONE 2013, 8, e58765. [Google Scholar] [CrossRef] [Green Version]
- Hegde, V.R.; Pu, H.; Patel, M.; Black, T.; Soriano, A.; Zhao, W.; Gullo, V.P.; Chan, T.-M. Two new bacterial DNA primase inhibitors from the plant Polygonum cuspidatum. Bioorg. Med. Chem. Lett. 2004, 14, 2275–2277. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Mierzwa, R.; Xu, L.; He, L.; Terracciano, J.; Patel, M.; Gullo, V.; Black, T.; Zhao, W.; Chan, T.-M.; et al. Isolation and structure elucidation of Sch 642305, a novel bacterial DNA primase inhibitor produced by Penicillium verrucosum. J. Nat. Prod. 2003, 66, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Griep, M.A.; Blood, S.; Larson, M.A.; Koepsell, S.A.; Hinrichs, S.H. Myricetin inhibits Escherichia coli DnaB helicase but not primase. Bioorg. Med. Chem. 2007, 15, 7203–7208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Huang, C.Y. Characterization of flavonol inhibition of DnaB helicase: Real-time monitoring, structural modeling, and proposed mechanism. J. Biomed. Biotechnol. 2012, 2012, 735368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.S.; Lennon, C.W.; Li, Z.; Miller, M.R.; Banavali, N.K.; Li, H.; Belfort, M. Mycobacterial DnaB helicase intein as oxidative stress sensor. Nat. Commun. 2018, 9, 4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C.; et al. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA gyrase inhibitors: Progress and synthesis of potent compounds as antibacterial agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.J.; Singh, O.M.; Smith, C.V.; Skarzynski, T.; Maxwell, A.; Wonacott, A.J.; Wigley, D.B. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996, 15, 1412–1420. [Google Scholar] [CrossRef]
- Chopra, S.; Matsuyama, K.; Tran, T.; Malerich, J.P.; Wan, B.; Franzblau, S.G.; Lun, S.; Guo, H.; Maiga, M.C.; Bishai, W.R.; et al. Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012, 67, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Inhibition of Mycobacterium tuberculosis topoisomerase I by m-AMSA, a eukaryotic type II topoisomerase poison. Biochem. Biophys. Res. Commun. 2014, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Godbole, A.A.; Ahmed, W.; Bhat, R.S.; Bradley, E.K.; Ekins, S.; Nagaraja, V. Targeting Mycobacterium tuberculosis topoisomerase I by small-molecule inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1549–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Martins, A.; Bongiorno, P.; Glickman, M.; Shuman, S. Biochemical and genetic analysis of the four DNA ligases of mycobacteria. J. Biol. Chem. 2004, 279, 20594–20606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.K.; Dube, D.; Tewari, N.; Dwivedi, N.; Tripathi, R.P.; Ramachandran, R. Mycobacterium tuberculosis NAD+-dependent DNA ligase is selectively inhibited by glycosylamines compared with human DNA ligase I. Nucleic Acids Res. 2005, 33, 7090–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.K.; Dube, D.; Kukshal, V.; Jha, A.K.; Hajela, K.; Ramachandran, R. NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis: Novel structure-function relationship and identification of a specific inhibitor. Proteins 2007, 69, 97–111. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Tripathi, R.P.; Ramachandran, R. NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis. Crystal structure of the adenylation domain and identification of novel inhibitors. J. Biol. Chem. 2005, 280, 30273–30281. [Google Scholar]
- Gorna, A.E.; Bowater, R.P.; Dziadek, J. DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: Varying activities at different stages of infection. Clin. Sci. (Lond.) 2010, 119, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Durbach, S.I.; Springer, B.; Machowski, E.E.; North, R.J.; Papavinasasundaram, K.G.; Colston, M.J.; Böttger, E.C.; Mizrahi, V. DNA alkylation damage as a sensor of nitrosative stress in Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 997–1000. [Google Scholar] [CrossRef] [Green Version]
- Drabløs, F.; Feyzi, E.; Aas, P.A.; Vaagbø, C.B.; Kavli, B.; Bratlie, M.S.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Mizrahi, V.; Andersen, S.J. DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol. Microbiol. 1998, 29, 1331–1339. [Google Scholar] [CrossRef]
- Castañeda-García, A.; Prieto, A.I.; Rodríguez-Beltrán, J.; Alonso, N.; Cantillon, D.; Costas, C.; Pérez-Lago, L.; Zegeye, E.D.; Herranz, M.; Plociński, P.; et al. A non-canonical mismatch repair pathway in prokaryotes. Nat. Commun. 2017, 8, 14246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truglio, J.J.; Croteau, D.L.; Van Houten, B.; Kisker, C. Prokaryotic nucleotide excision repair: The UvrABC system. Chem. Rev. 2006, 106, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Khanduja, J.S.; Bortoluzzi, A.; Houghton, J.; Sander, P.; Güthlein, C.; Davis, E.O.; Springer, B.; Böttger, E.C.; Relini, A.; et al. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 2011, 39, 7316–7328. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Kumar, M.B.; Muniyappa, K. Mycobacterium tuberculosis UvrB Is a Robust DNA-Stimulated ATPase That Also Possesses Structure-Specific ATP-Dependent DNA Helicase Activity. Biochemistry 2016, 55, 5865–5883. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Rizzi, M.; Rossi, F.; Miggiano, R. Mycobacterium tuberculosis UvrB forms dimers in solution and interacts with UvrA in the absence of ligands. Proteins 2018, 86, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraris, D.M.; Miggiano, R.; Rossi, F.; Rizzi, M. Mycobacterium tuberculosis Molecular Determinants of Infection, Survival Strategies, and Vulnerable Targets. Pathogens 2018, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazloum, N.; Stegman, M.A.; Croteau, D.L.; Houten, B.V.; Kwon, N.S.; Ling, Y.; Dickinson, C.; Venugopal, A.; Towheed, M.A.; Nathan, C. Identification of a chemical that inhibits the mycobacterial UvrABC complex in nucleotide excision repair. Biochemistry 2011, 50, 1329–1335. [Google Scholar] [CrossRef]
- Yang, M.; Aamodt, R.M.; Dalhus, B.; Balasingham, S.; Helle, I.; Andersen, P.; Tønjum, T.; Alseth, I.; Rognes, T.; Bjørås, M. The ada operon of Mycobacterium tuberculosis encodes two DNA methyltransferases for inducible repair of DNA alkylation damage. DNA Repair 2011, 10, 595–602. [Google Scholar] [CrossRef]
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Miggiano, R.; Casazza, V.; Garavaglia, S.; Ciaramella, M.; Perugino, G.; Rizzi, M.; Rossi, F. Biochemical and structural studies of the Mycobacterium tuberculosis O6-methylguanine methyltransferase and mutated variants. J. Bacteriol. 2013, 195, 2728–2736. [Google Scholar] [CrossRef] [Green Version]
- Miggiano, R.; Perugino, G.; Ciaramella, M.; Serpe, M.; Rejman, D.; Páv, O.; Pohl, R.; Garavaglia, S.; Lahiri, S.; Rizzi, M.; et al. Crystal structure of Mycobacterium tuberculosis O6-methylguanine-DNA methyltransferase protein clusters assembled on to damaged DNA. Biochem. J. 2016, 473, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Miggiano, R.; Valenti, A.; Rossi, F.; Rizzi, M.; Perugino, G.; Ciaramella, M. Every OGT Is Illuminated … by Fluorescent and Synchrotron Lights. Int. J. Mol. Sci. 2017, 18, 2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perugino, G.; Miggiano, R.; Serpe, M.; Vettone, A.; Valenti, A.; Lahiri, S.; Rossi, F.; Rossi, M.; Rizzi, M.; Ciaramella, M. Structure-function relationships governing activity and stability of a DNA alkylation damage repair thermostable protein. Nucleic Acids Res. 2015, 43, 8801–8816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrone, C.; Miggiano, R.; Serpe, M.; Massarotti, A.; Valenti, A.; del Monaco, G.; Rossi, M.; Rossi, F.; Rizzi, M.; Perugino, G.; et al. Interdomain interactions rearrangements control the reaction steps of a thermostable DNA alkyltransferase. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Inoue, T.; Nishioka, M.; Fujiwara, S.; Takagi, M.; Imanakac, T.; Kai, Y. Hyperthermostable protein structure maintained by intra and inter-helix ion-pairs in archaeal O6-methylguanine-DNA methyltransferase. J. Mol. Biol. 1999, 292, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pelton, J.G.; Wemmer, D.E. Structural studies of MJ1529, an O6-methylguanine-DNA methyltransferase. Magn. Reson. Chem. 2006, 44, S71–S82. [Google Scholar] [CrossRef] [PubMed]
- Wibley, J.E.; Pegg, A.E.; Moody, P.C. Crystal structure of the human O6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000, 28, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Target Protein | In Vitro Essentiality [30,45] | Inhibitor Molecules/Classes |
|---|---|---|
| GuaB2 | Essential | diphenyl urea derivatives: DPU-2, DPU-3 [29] |
| triazole-linked mycophenolic adenine [33] | ||
| indazole sulfonamides [36] | ||
| VCC234718 5-(4-cyclohexanecarbonylpiperazine-1-sulfonyl)isoquinoline [37] | ||
| 5-amidophthalide derivative [38] | ||
| Benzoxazole derivatives [39] | ||
| OPRT | Essential | Hydroxy-2-oxo-1,2-dihydropyridine-4-carboxylic acid and its derivative 3-Benzylidene-2,6-dioxo-1,2,3,6-tetrahydropyridine-4-carboxylic acid [48] |
| SSB (E.coli) | Essential | Small-molecule inhibitors 1 [61] |
| DnaG (E.coli) | Essential | Phenolic monosaccharides 1 [64] |
| Bicyclic macrolide 1 [65] | ||
| DnaB (E.coli, K. Pneumoniae) | Essential | Flavonols 1 [66,67] |
| DnaN/β | Essential | Griselimycins [69] |
| GyrA | Essential | Quinolones [71] |
| GyrB | Essential | Novobiocin and coumarin derivatives [72,73] |
| TopA | Essential | m-AMSA [74] |
| Norclomipramine and Imipramin [75] | ||
| Hydroxycamptothecin derivatives [18] | ||
| LigA | Essential | Pyridochromanone [76] |
| Bis-xylofuranosylated diamines [77,79] | ||
| N-substituted tetracyclic indoles [78] | ||
| UvrABC complex | Essential | 2-(5-amino-1,3,4-thiadiazol-2-ylbenzo[f]chromen-3-one) (ATBC) [90] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miggiano, R.; Morrone, C.; Rossi, F.; Rizzi, M. Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair. Molecules 2020, 25, 1205. https://doi.org/10.3390/molecules25051205
Miggiano R, Morrone C, Rossi F, Rizzi M. Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair. Molecules. 2020; 25(5):1205. https://doi.org/10.3390/molecules25051205
Chicago/Turabian StyleMiggiano, Riccardo, Castrese Morrone, Franca Rossi, and Menico Rizzi. 2020. "Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair" Molecules 25, no. 5: 1205. https://doi.org/10.3390/molecules25051205
APA StyleMiggiano, R., Morrone, C., Rossi, F., & Rizzi, M. (2020). Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair. Molecules, 25(5), 1205. https://doi.org/10.3390/molecules25051205