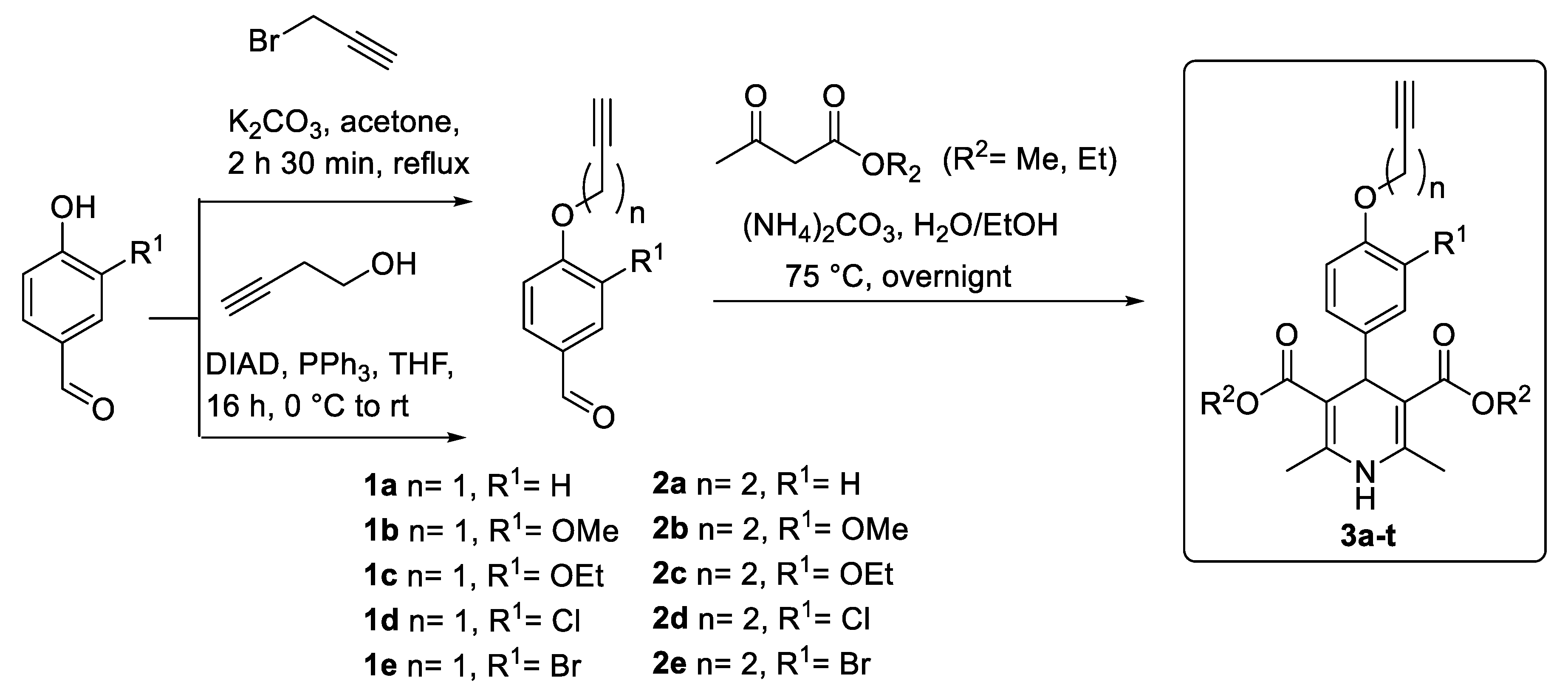
3.2. Synthesis of Propargylic Aldehydes 1a–e
A suspension of the corresponding 4-hydroxybenzaldehyde (1 equiv) and K2CO3 (1.3 equiv) in acetone (1.6 mmol/mL) was stirred at reflux for 30 min. The mixture was cooled to rt and propargyl bromide (1.6 equiv) was added dropwise. The resulting suspension was stirred at reflux for 2 h 30 min. After that time, the solvent was removed under pressure conditions. The residue was dissolved in water and extracted with ethyl acetate three times. Organic layers were joined and dried over Na2SO4. Activated carbon was added to the solution and the mixture is stirred over 15 min at 40 °C. The crude was finally filtered over Celite®, the filtrate was evaporated and the obtained residue was recrystallized from EtOAct/hexane (1:2 v/v) to afford the desired products in yields ranging from 34% to 98%.
4-(Prop-2-yn-1-yloxy)benzaldehyde (1a). The crude was prepared according to the general procedure starting from commercially available 4-hydroxybenzaldehyde (1 equiv, 8.19 mmol, 1 g), K2CO3 (1.3 equiv, 10.65 mmol, 1.47 g) and propargyl bromide (1.6 equiv, 13.10 mmol, 0.992 mL) in acetone (20 mL) to afford compound 1a (1.00 g, 77%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.91 (s, 1H), 7.93–7.80 (m, 2H), 7.14–7.07 (m, 2H), 4.78 (d, J = 2.4 Hz, 2H), 2.57 (t, J = 2.4 Hz, 1H).
3-Methoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (1b). The crude was prepared according to the general procedure starting from commercially available 4-hydroxy-3-methoxybenzaldehyde (1 equiv, 6.58 mmol, 1 g), K2CO3 (1.3 equiv, 8.55 mmol, 1.181 g) and propargyl bromide (1.6 equiv, 10.52 mmol, 0.800 mL) in acetone (16 mL) to afford compound 1b (1.16 g, 93%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.87 (s, 1H), 7.52–7.39 (m, 2H), 7.14 (d, J = 8.1 Hz, 1H), 4.86 (d, J = 2.3 Hz, 2H), 3.94 (s, 3H), 2.56 (t, J = 2.4 Hz, 1H).
3-Ethoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (1c). The crude was prepared according to the general procedure starting from commercially available 3-ethoxy-4-hydroxybenzaldehyde (1 equiv, 6.02 mmol, 1.00 g), K2CO3 (1.3 equiv, 7.82 mmol, 1.081 g) and propargyl bromide (1.6 equiv, 9.63 mmol, 0.730 mL) in acetone (15 mL) to afford compound 1c (428.32 mg, 35%). The crude was used without further purification.
3-Chloro-4-(prop-2-yn-1-yloxy)benzaldehyde (1d). The crude was prepared according to the general procedure starting from commercially available 3-chloro-4-hydroxybenzaldehyde (1 equiv, 6.39 mmol, 1.00 g), K2CO3 (1.3 equiv, 8.31 mmol, 1.148 g) and propargyl bromide (1.6 equiv, 10.22 mmol, 0.78 mL) in acetone (10 mL) to afford compound 1d (852.80 mg, 69%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.87 (s, 1H), 7.93 (d, J = 2.0 Hz, 1H), 7.79 (dd, J = 8.5, 2.0 Hz, 1H), 7.21 (d, J = 8.5 Hz, 1H), 4.88 (d, J = 2.4 Hz, 2H), 2.60 (t, J = 2.4 Hz, 1H).
3-Bromo-4-(prop-2-yn-1-yloxy)benzaldehyde (1e). The crude was prepared according to the general procedure starting from commercially available 3-bromo-4-hydroxybenzaldehyde (1 equiv, 4.98 mmol, 1.00 g), K2CO3 (1.3 equiv, 6.47 mmol, 893.81 mg) and propargyl bromide (1.6 equiv, 7.96 mmol, 0.60 mL) in acetone (12 mL) to afford compound 1e (1.163 g 98%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.86 (s, 1H), 8.09 (d, J = 1.0 Hz, 1H), 7.82 (dd, J = 8.5, 1.1 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 4.89–4.81 (m, 2H), 2.60 (t, J = 2.4 Hz, 1H).
3.3. Synthesis of Propargylic Aldehydes 2a–e
A solution of 4-hydroxybenzaldehyde (1 equiv), triphenylphosphine (2 equiv) and 3-butyn-1-ol (1.5 equiv) in THF (0.8 mmol/mL) was cooled to 0 °C. DIAD (1.5 equiv) is then added dropwise and the resulting mixture is stirred overnight at rt. The solvent was evaporated, the residue solubilized in ethyl acetate and washed 3 times with 1M NaOH solution and brine. The organic layers were dried over Na2SO4, filtered and evaporated under pressure conditions. The residue was triturated with ethyl ether and filtered. The filtrate was purified by flash chromatography with hexane/EtOAc (9:1 v/v) to afford the desired products in yields ranging from 45% to 97%.
4-(But-3-yn-1-yloxy)benzaldehyde (2a). The crude was prepared according to the general procedure starting from commercially available 4-hydroxybenzaldehyde (1 equiv, 16.38 mmol, 2.00 g), triphenylphosphine (2 equiv, 32.76 mmol, 8.59 g), DIAD (1.5 equiv, 24.57 mmol, 4.84 mL) and 3-butyn-1-ol (1.5 equiv, 27.57 mmol, 1.860 mL) in THF (120 mL) to afford compound 2a (1.30 g, 46%). The crude was used without further purification. 1H-NMR (400 MHz, CDCl3): δ 2.06 (br s, 1 H, CH), 2.73 (dt, J = 2.4, 7.1 Hz, 2 H, CH2), 4.18 (t, J = 7.1 Hz, 2 H, CH2O), 7.02 (d, J = 8.5 Hz, 2 H, Harom), 7.84 (d, J = 8.5 Hz, 2 H, Harom), 9.90 (s, 1 H, CHO).
4-(But-3-yn-1-yloxy)-3-methoxybenzaldehyde (2b). The crude was prepared according to the general procedure starting from commercially available 4-hydroxy-3-methoxybenzaldehyde (1 equiv, 6.57 mmol, 1.00 g), triphenylphosphine (2 equiv, 13.15 mmol, 3.45 g), DIAD (1.5 equiv, 9.86 mmol, 1.94 mL) and 3-butyn-1-ol (1.5 equiv, 9.86 mmol, 0.746 mL) in THF (60 mL) to afford compound 2b (674.9 mg, 50%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.85 (s, J = 4.2 Hz, 1H), 7.48–7.38 (m, 2H), 6.99 (dd, J = 8.1, 3.5 Hz, 1H), 4.28–4.18 (m, 2H), 3.97–3.89 (m, 3H), 2.77 (td, J = 7.3, 2.7 Hz, 2H), 2.09–2.00 (m, 1H).
4-(But-3-yn-1-yloxy)-3-ethoxybenzaldehyde (2c). The crude was prepared according to the general procedure starting from commercially available 3-ethoxy-4-hydroxybenzaldehyde (1 equiv, 6.02 mmol, 1.00 g), triphenylphosphine (2 equiv, 12.04 mmol, 3.16 g), DIAD (1.5 equiv, 9.03 mmol, 1.78 mL) and 3-butyn-1-ol (1.5 equiv, 9.03 mmol, 0.683 mL) in THF (60 mL) to afford compound 2c (1.423 g, 70%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.85 (s, 1H), 7.42 (dt, J = 5.5, 1.8 Hz, 2H), 6.99 (d, J = 8.0 Hz, 1H), 4.23 (t, J = 7.3 Hz, 2H), 4.15 (q, J = 7.0 Hz, 2H), 2.77 (td, J = 7.3, 2.7 Hz, 2H), 2.05 (t, J = 2.7 Hz, 1H), 1.47 (t, J = 7.0 Hz, 3H), 1.26 (d, J = 6.3 Hz, 3H).
4-(But-3-yn-1-yloxy)-3-chlorobenzaldehyde (2d). The crude was prepared according to the general procedure starting from commercially available 3-ethoxy-4-hydroxybenzaldehyde (1 equiv, 6.39 mmol, 1.00 g), triphenylphosphine (2 equiv, 12.78 mmol, 3.35 g), DIAD (1.5 equiv, 9.58 mmol, 1.89 mL) and 3-butyn-1-ol (1.5 equiv, 9.58 mmol, 0.725 mL) in THF (60 mL) to afford compound 2d (1.30 g, 97%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.84 (s, 1H), 7.90 (d, J = 2.0 Hz, 1H), 7.76 (dd, J = 8.5, 2.0 Hz, 1H), 7.04 (d, J = 8.5 Hz, 1H), 4.26 (t, J = 7.1 Hz, 2H), 2.78 (td, J = 7.1, 2.7 Hz, 2H), 2.06 (t, J = 2.7 Hz, 1H).
3-Bromo-4-(but-3-yn-1-yloxy)benzaldehyde (2e). The crude was prepared according to the general procedure starting from commercially available 3-bromo-4-hydroxybenzaldehyde (1 equiv, 4.97 mmol, 1.00 g), triphenylphosphine (2 equiv, 9.94 mmol, 2.61 g), DIAD (1.5 equiv, 7.46 mmol, 1.47 mL) and 3-butyn-1-ol (1.5 equiv, 7.46 mmol, 0.565 mL) in THF (60 mL) to afford compound 2e (631.20 mg, 50%). The crude was used without further purification. 1H-NMR (CDCl3) δ 9.85 (s, 1H), 8.09 (d, J = 2.0 Hz, 1H), 7.81 (dd, J = 8.5, 2.0 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 4.25 (t, J = 7.1 Hz, 2H), 2.80 (td, J = 7.1, 2.7 Hz, 2H), 2.07 (t, J = 2.7 Hz, 1H).
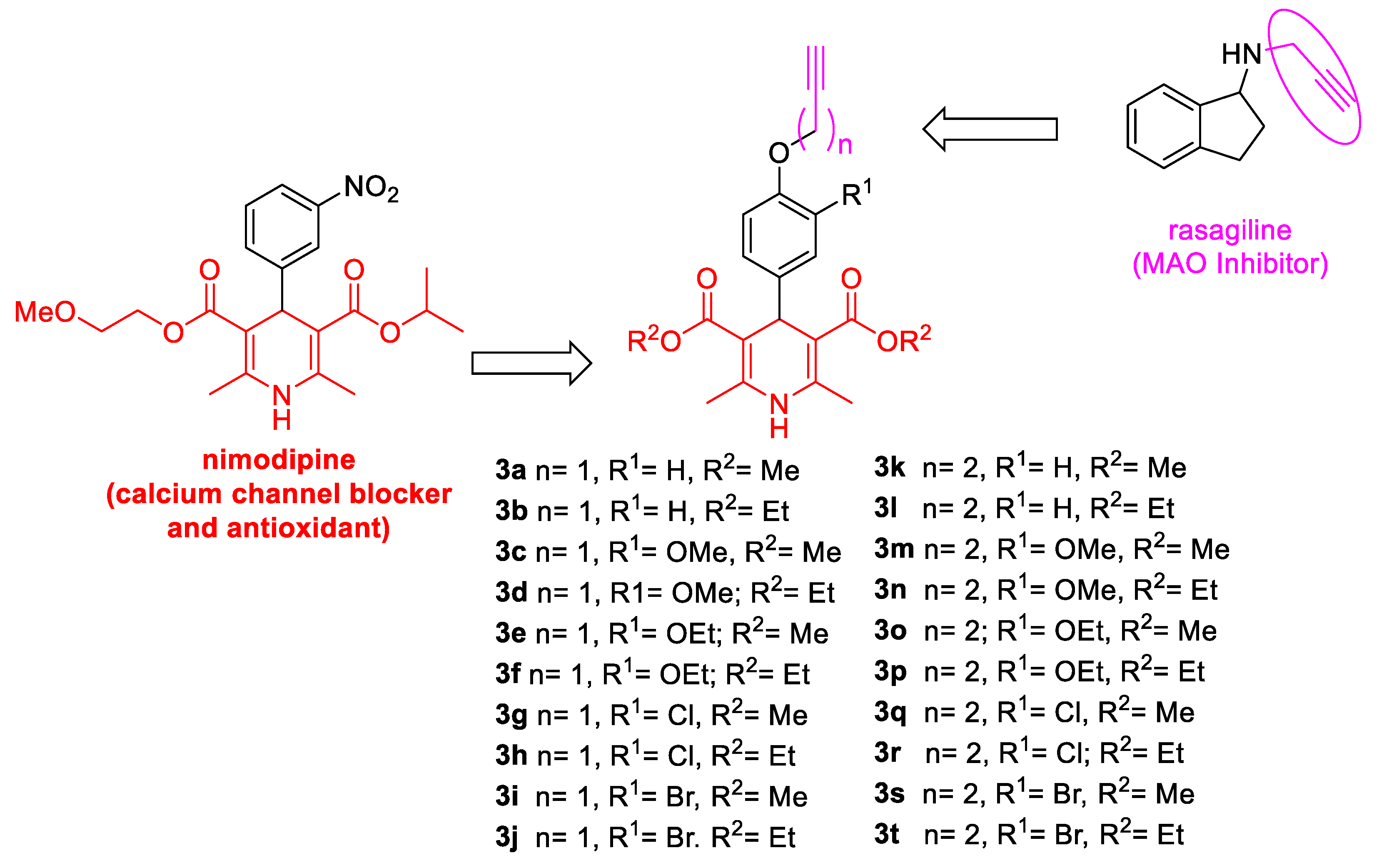
3.4. General procedure of compounds 3a–t
3-Substitued-4-alkynyloxy-benzaldehydes 1a–e and 2a–e (1 equiv) and the corresponding acetoacetate (3.5 equiv) were dissolved in a mixture of EtOH (4 mmol/mL) and the same volume of H2O. The resulting mixture is stirred and heated at 75 °C for 1 h. Next, ammonium carbonate (2.5 equiv) was added to the mixture, and the reaction stirred and heated at 75 °C overnight. The desired product precipitated once the crude reaction reached rt, or was triturated with diethyl ether. The solid was then filtered and washed with diethyl ether again to finally afford compounds 3a–t in yields ranging from 12% to 76%.
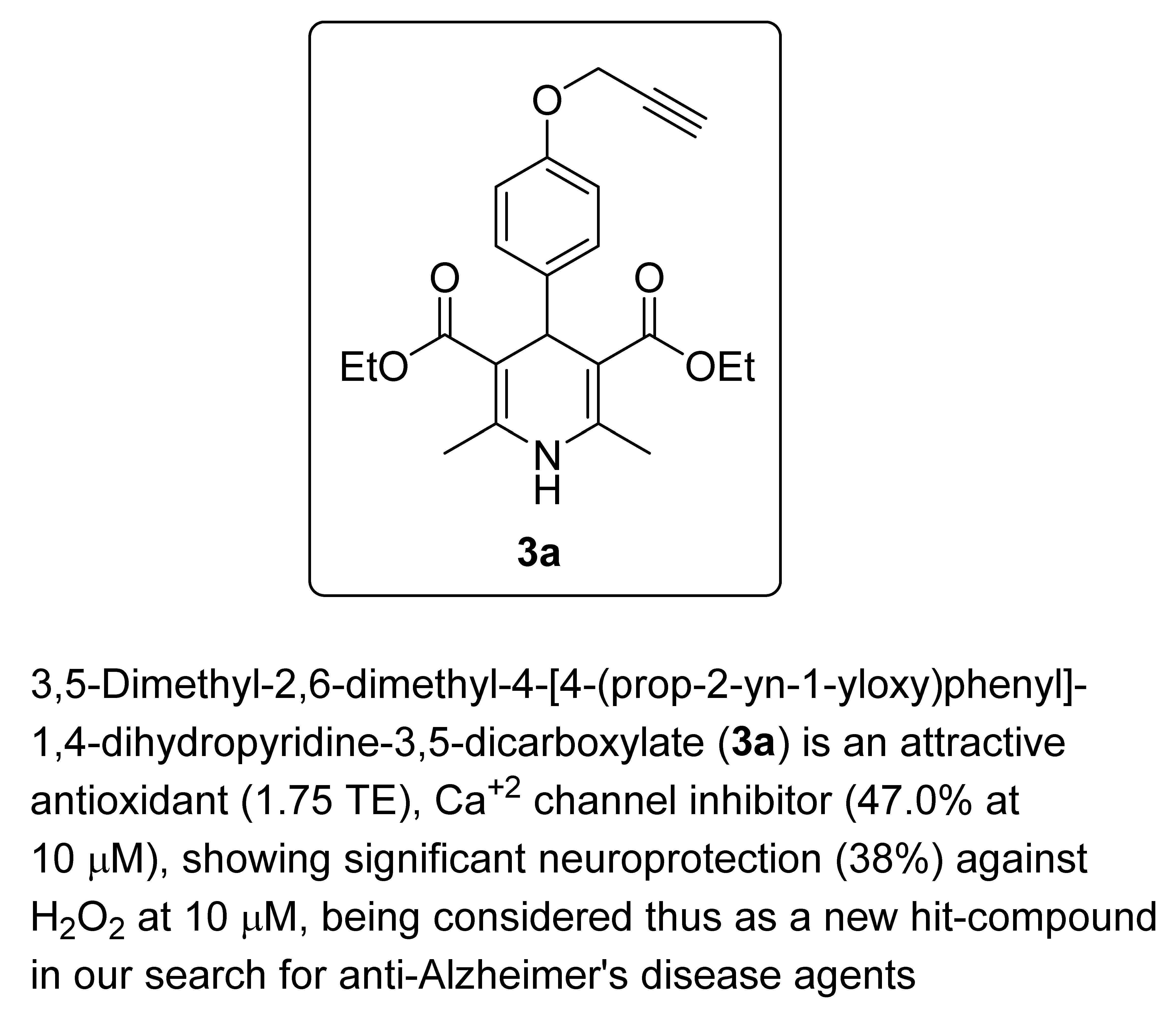
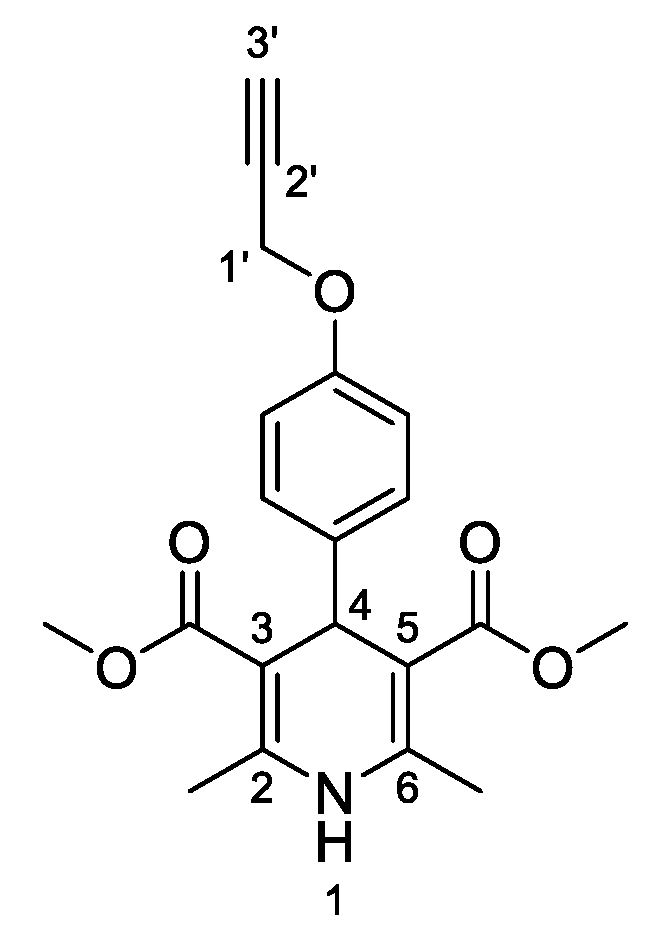
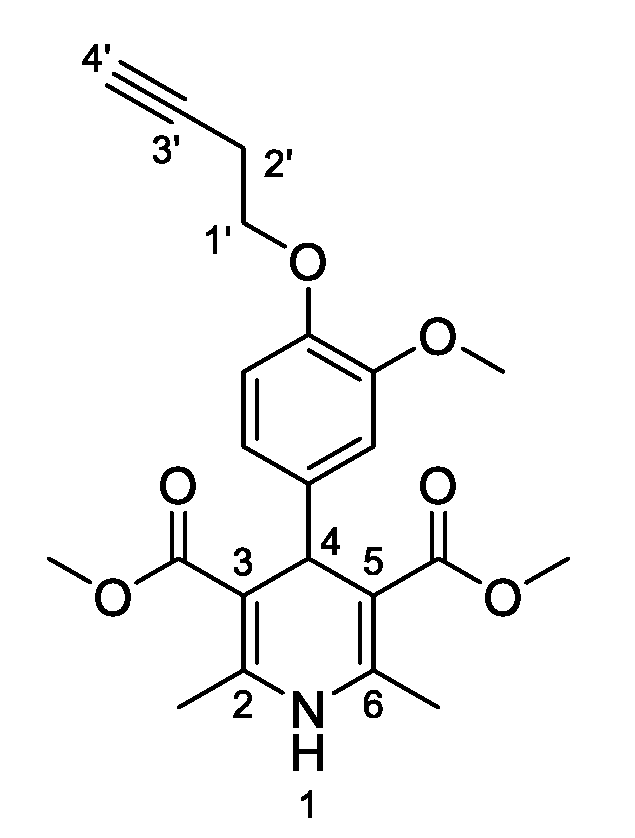
3,5-Dimethyl-2,6–dimethyl–4-[4-(prop–2–yn–1-yloxy)phenyl]-1,4-dihydropyridine-3,5-dicarboxylate (
3a, Figure 2). Following the general procedure starting from 4-(prop-2-yn-1-yloxy)benzaldehyde (
1a, 2.25 mmol, 360.02 mg), methyl acetoacetate (3.5 equiv, 7.88 mmol, 0.849 mL) and ammonium carbonate (2.5 equiv, 5.63 mmol, 540.51 mg) at 75 °C over 15 h, compound
3a (509.4 mg, 64%) was isolated as white powder:
1H-NMR (CDCl
3) δ 7.18 (d,
J = 8.7 Hz, 2H,
2HAr), 6.82 (d,
J = 8.7 Hz, 2H, 2
HAr), 5.67 (s, 1H, N
H), 4.95 (s, 1H,
H4), 4.62 (d,
J = 2.4 Hz, 2H,
2H1’), 3.64 (s, 6H,
2CO
2C
H3), 2.50 (t,
J = 2.4 Hz, 1H,
H3’), 2.32 (s, 6H, 2C
H3).
13C-NMR (CDCl
3) δ 168.20, 156.19, 144.14, 140.94, 128.98, 128.79, 114.45, 114.37, 104.17, 79.03, 75.42, 55.91, 51.13, 38.58, 19.74. Anal. Calcd. for C
20H
21NO
5: C, 67.59; H, 5.96; N, 3.94. Found: C, 67.33; H, 6.04; N, 3.89.
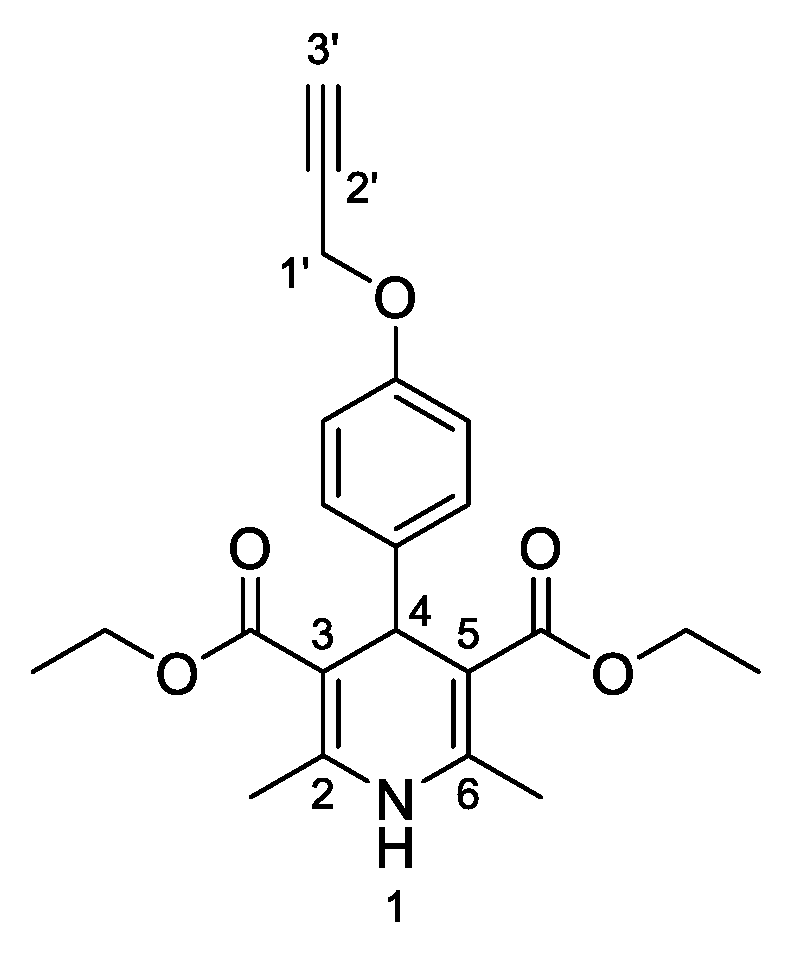
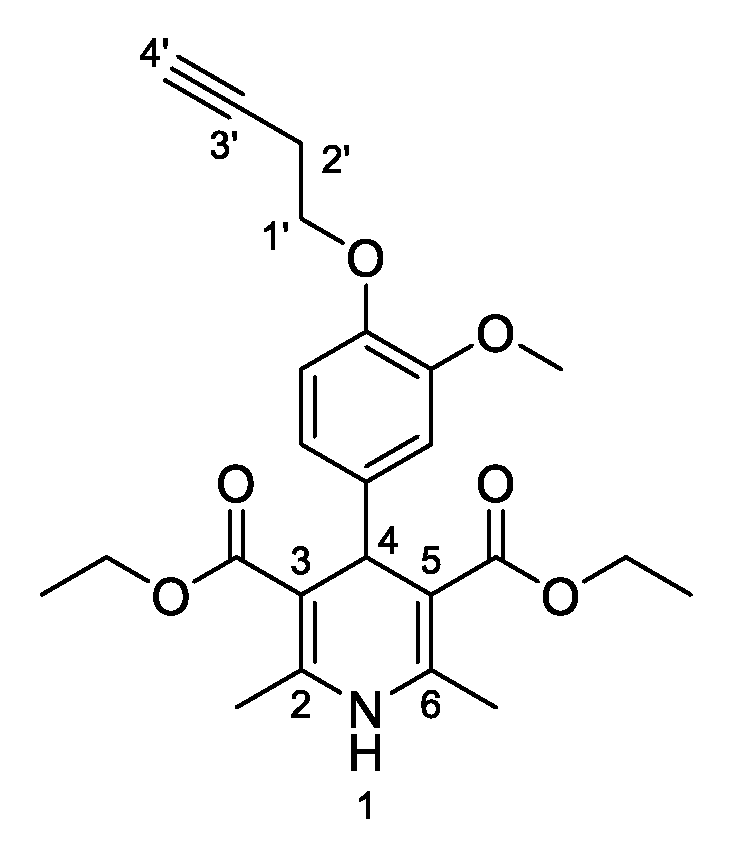
3,5-Diethyl-2,6-dimethyl-4-[4-(prop-2-yn-1-yloxy)phenyl]-1,4-dihydropyridine-3,5-dicarboxylate (
3b,
Figure 3). Following the general procedure starting from 4-(prop-2-yn-1-yloxy)benzaldehyde (
1a, 1 equiv, 2.25 mmol, 360.02 mg), ethyl acetoacetate (3.5 equiv, 7.88 mmol, 1.00 mL) and ammonium carbonate (2.5 equiv, 5.63 mmol, 540.51 mg) at 75 °C over 15 h, the solvent was reduced under pressure conditions and the residue was triturated with diethyl ether. The resulting solid was stirred in diethyl ether overnight and filtered to afford compound
3b (564.10 mg, 47%) as white powder:
1H-NMR (CDCl
3) δ 7.23 – 7.12 (m, 2H,
2HAr), 6.86 – 6.75 (m, 2H,
2HAr), 5.58 (bs, 1H, N
H), 4.94 (s, 1H,
H4), 4.63 (d,
J = 2.4 Hz, 2H,
2H1’), 4.17 – 3.99 (m, 4H, 2OC
H2CH
3), 2.49 (t,
J = 2.4 Hz, 1H,
H3’), 2.32 (s, 6H, 2C
H3), 1.22 (t,
J = 7.1 Hz, 6H,
2CO
2CH
2C
H3).
13C-NMR (101 MHz, CDCl
3) δ 167.79, 156.12, 143.72, 141.36, 129.17, 114.29, 104.49, 79.04, 75.38, 59.85, 56.00, 55.94, 38.94, 19.76, 14.41. HRMS ESI-TOF [M]
+ m/z Calcd. for C
22H
25NO
5: 383,1726. Found: 383,1733.
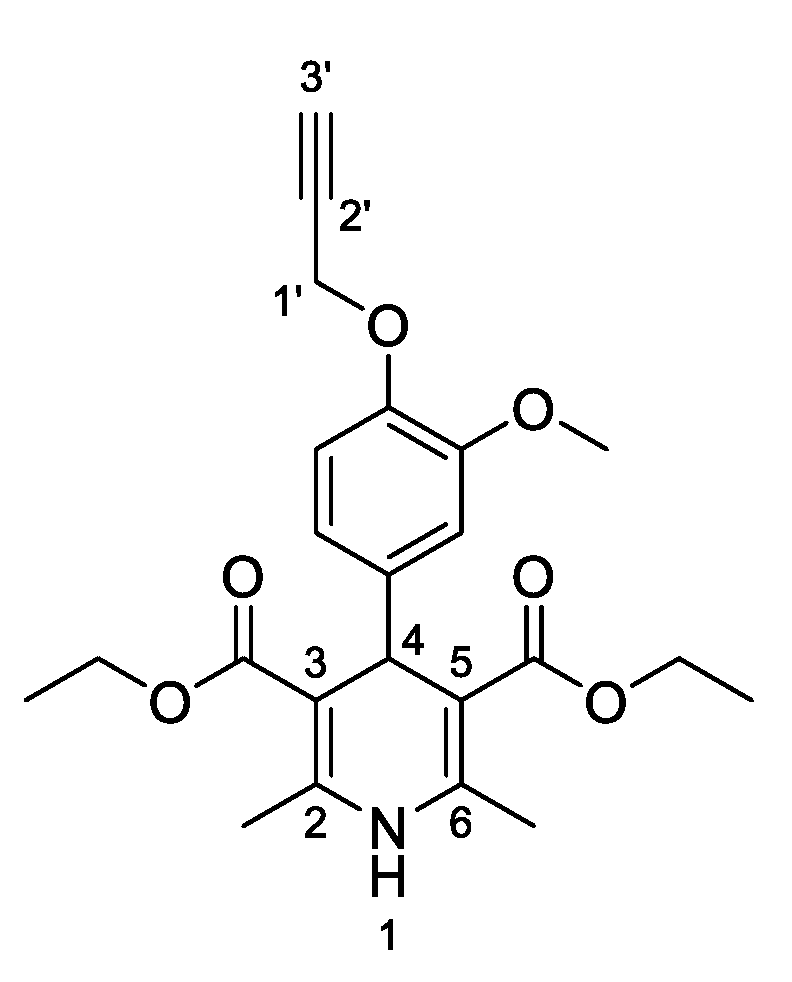
3,5-Dimethyl-4- [3-methoxy-4-(prop-2-yn-1-yloxy) phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5- dicarboxylate (
3c,
Figure 4). Following the general procedure starting from 3-methoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (
1b, 1 equiv, 1.84 mmol, 350.00 mg), methyl acetoacetate (3.5 equiv, 6.44 mmol, 0.695 mL) and ammonium carbonate (2.5 equiv, 4.60 mmol, 442.01 mg) at 75 °C over 15 h, compound
3c (324.10 mg, 46%) precipitated as beige crystals:
1H-NMR (CDCl
3) δ 6.88 (dd,
J = 5.0, 3.1 Hz, 2H,
2HAr), 6.75 (dd,
J = 8.3, 1.9 Hz, 1H,
HAr), 5.70 (d,
J = 32.2 Hz, 1H, N
H), 4.97 (s, 1H,
H4), 4.69 (d,
J = 2.3 Hz, 2H,
2H1’), 3.82 (s, 3H, OC
H3), 3.66 (s, 6H, 2CO
2C
H3), 2.48 (t,
J = 2.3 Hz, 1H,
H3’), 2.33 (s, 6H, 2C
H3).
13C-NMR (CDCl
3) δ 168.19, 149.09, 145.43, 144.19, 141.75, 119.52, 114.06, 112.02, 104.00, 79.10, 75.63, 56.87, 55.95, 51.15, 38.94, 19.75. HRMS ESI-TOF [M]
+ m/z Calcd. for C
21H
23NO
6: 385,1513. Found: 385,1525.
3,5-Diethyl-4-[3-methoxy-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3d,
Figure 5). Following the general procedure starting from 3-methoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (
1b, 1 equiv, 1.84 mmol, 350.00 mg), ethyl acetoacetate (3.5 equiv, 6.44 mmol, 0.822 mL) and ammonium carbonate (2.5 equiv, 4.60 mmol, 442.01 mg) at 75 °C over 15 h, compound
3d (581.40 mg, 76%) precipitated as a yellow powder:
1H-NMR (CDCl
3) δ 6.89 (dd,
J = 6.3, 5.3 Hz, 2H,
2HAr), 6.78 (dd,
J = 8.3, 2.0 Hz, 1H,
HAr), 5.61 (s, 1H, N
H), 4.95 (s, 1H,
H4), 4.69 (d, J = 2.4 Hz, 1H,
2H1’), 4.18 – 4.04 (m, 4H, 2CO
2C
H2CH
3), 3.83 (s, 3H, OC
H3), 2.47 (t,
J = 2.4 Hz, 1H,
H3’), 2.33 (s, 6H, 2C
H3), 1.23 (t,
J = 7.1 Hz, 6H,
2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 167.79, 148.96, 145.35, 143.79, 142.23, 119.91, 114.07, 112.37, 104.34, 79.11, 75.59, 59.87, 56.92, 55.92, 39.28, 19.78, 14.48. HRMS ESI-TOF [M]
+ m/z Calcd. for C
23H
27NO
6: 413,1829. Found: 413,1838.
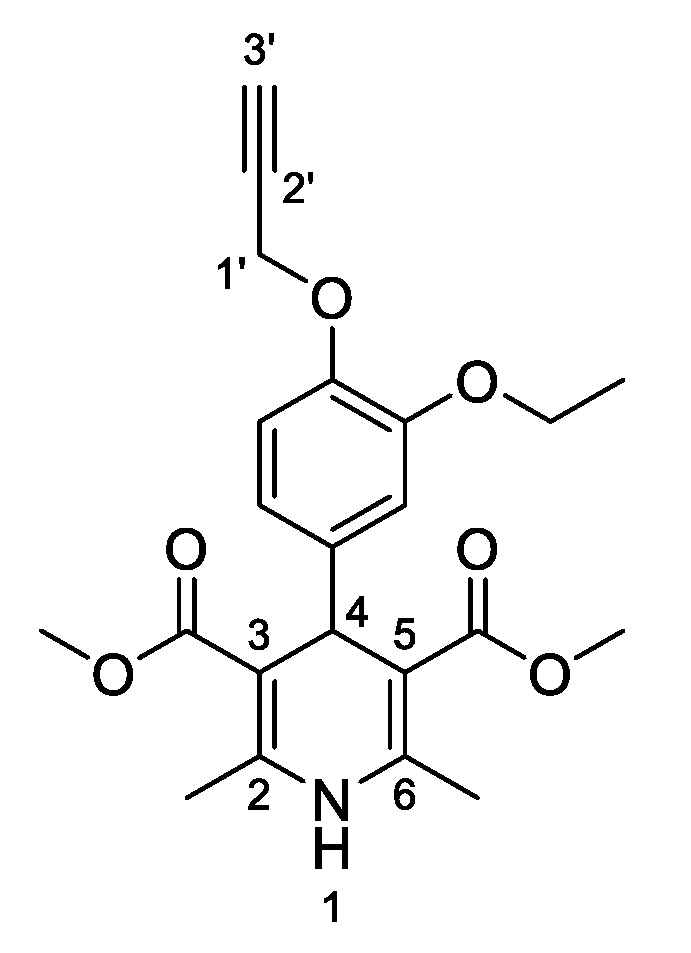
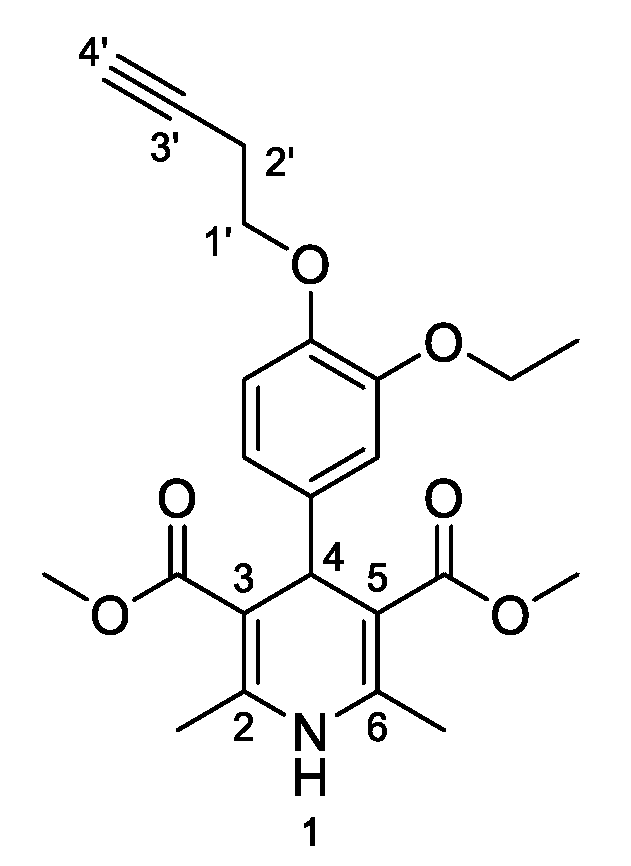
3,5-Dimethyl-4-[3-ethoxy-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3e,
Figure 6). Following the general procedure starting from 3-ethoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (
1c, 1 equiv, 1.22 mmol, 191.30 mg), methyl acetoacetate (3.5 equiv, 4.30 mmol, 0.462 mL) and ammonium carbonate (2.5 equiv, 3.06 mmol, 294.04 mg) at 75° C over 15 h, a precipitated was isolated that was filtered and washed in diethyl ether:pentane (1:2 v/v) to afford compound
3a (192.70 mg, 40%) as a white powder:
1H-NMR (CDCl
3) δ 6.88 (dd,
J = 7.7, 5.1 Hz, 2H, 2
HAr), 6.75 (dd,
J = 8.3, 1.9 Hz, 1H,
HAr), 5.61 (s, 1H, N
H), 4.95 (s, 1H,
H4), 4.69 (d, J = 2.2 Hz, 1H, 2
H1’), 4.06 (q,
J = 7.0 Hz, 2H, OC
H2CH
3), 3.65 (s, 6H, 2CO
2C
H3), 2.47 (t,
J = 2.3 Hz, 1H,
H3’), 2.33 (s, 6H, 2C
H3), 1.42 (t,
J = 7.0 Hz, 3H, OCH
2C
H3).
13C-NMR (CDCl
3) δ 168.21, 148.45, 145.76, 144.17, 141.89, 119.62, 114.91, 113.68, 104.02, 79.32, 75.50, 64.44, 57.10, 51.15, 38.90, 19.74, 14.96. HRMS ESI-TOF [M]
+ m/z Calcd. for C
22H
25NO
6: 399,1673. Found: 399,1682.
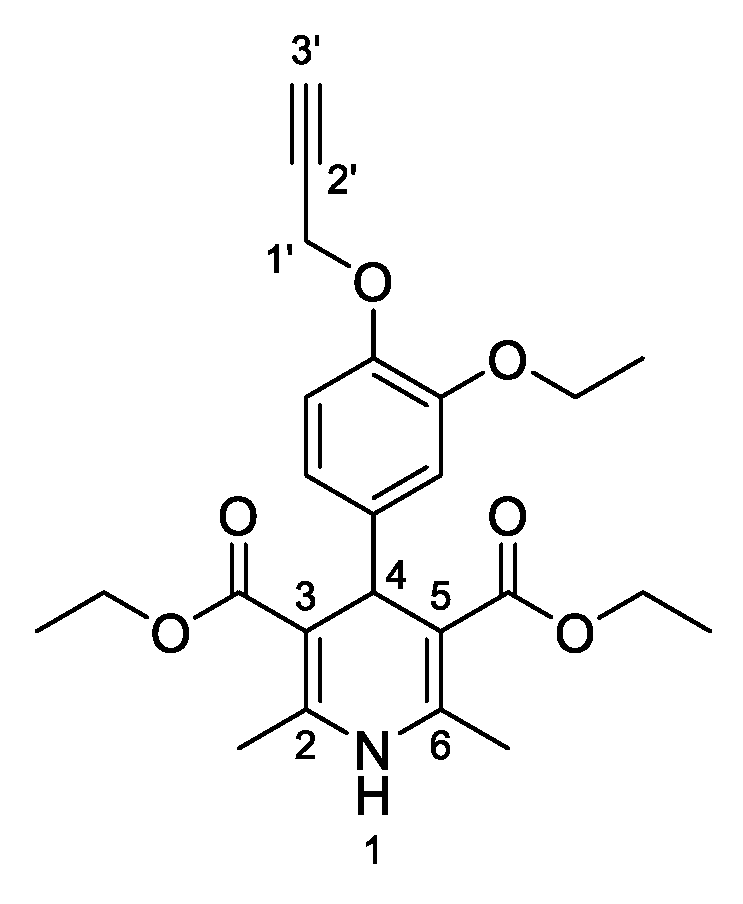
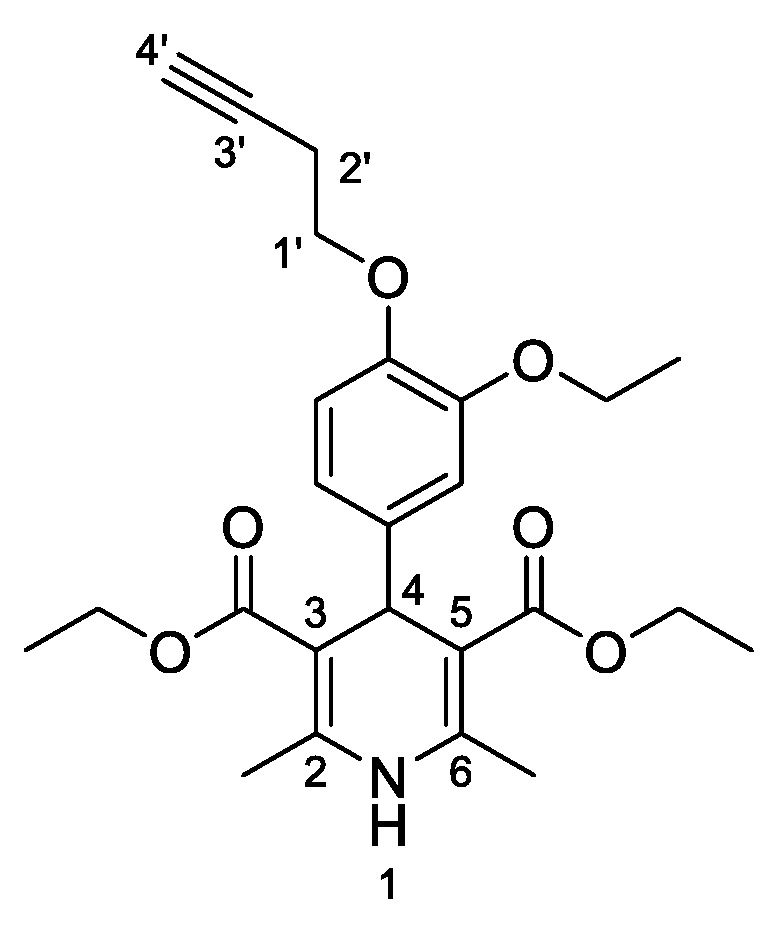
3,5-Diethyl-4-[3-ethoxy-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3f,
Figure 7). Following the general procedure starting from 3-ethoxy-4-(prop-2-yn-1-yloxy)benzaldehyde (
1c, 1 equiv, 1.01 mmol, 205.00 mg), ethyl acetoacetate (3.5 equiv, 3.54 mmol, 0.452 mL) and ammonium carbonate (2.5 equiv, 2.53 mmol, 242.63 mg) at 75 °C over 15 h, the solvent was removed under pressure conditions and the residue was triturated with diethyl ether. The resulting solid was stirred in diethyl ether overnight and filtered to afford compound
3f (272.90 mg, 52%) as abrown powder:
1H-NMR (CDCl
3) δ 6.89 (dd,
J = 5.0, 3.2 Hz, 2H, 2
HAr), 6.77 (dd,
J = 8.3, 1.8 Hz, 1H,
HAr), 5.54 (s, 1H, N
H), 4.94 (s, 1H,
H4), 4.69 (d,
J = 2.3 Hz, 2H, 2
H1’), 4.16 – 3.98 (m, 6H, OC
H2CH
3 and 2CO
2C
H2CH
3), 2.46 (t,
J = 2.3 Hz, 1H,
H3’), 2.33 (s, 6H, 2C
H3), 1.42 (t,
J = 7.0 Hz, 3H, OCH
2C
H3), 1.23 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2CH3).
13C-NMR (CDCl
3) δ 167.80, 148.45, 145.71, 143.72, 142.29, 120.02, 114.93, 114.01, 104.38, 79.37, 75.45, 64.46, 59.88, 57.18, 39.22, 19.81, 15.04, 14.48. HRMS ESI-TOF [M]
+ m/z Calcd. for C
24H
29NO
6: 427,1977. Found: 427,1995.
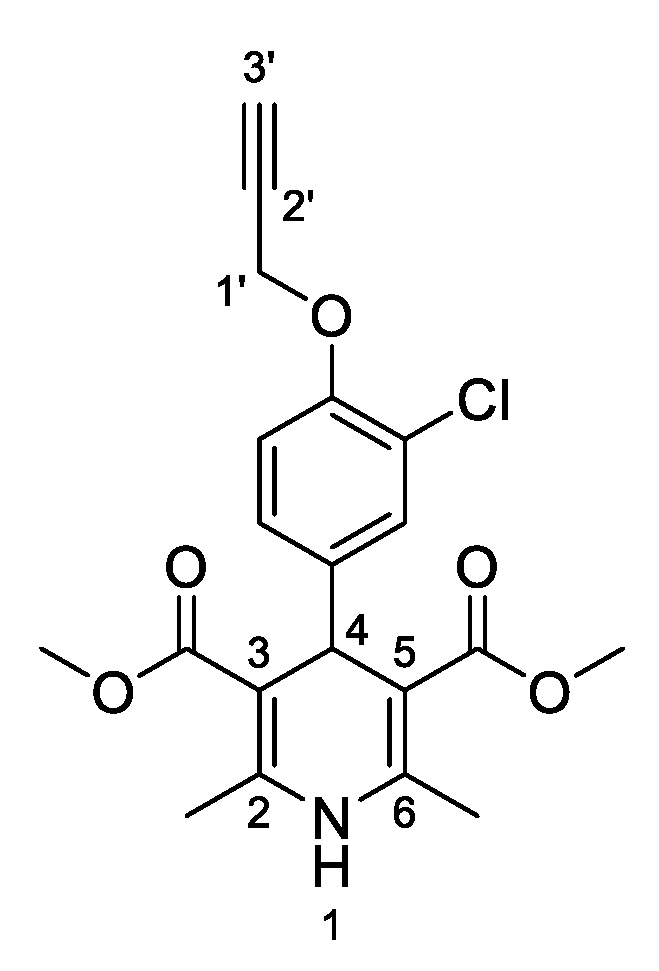
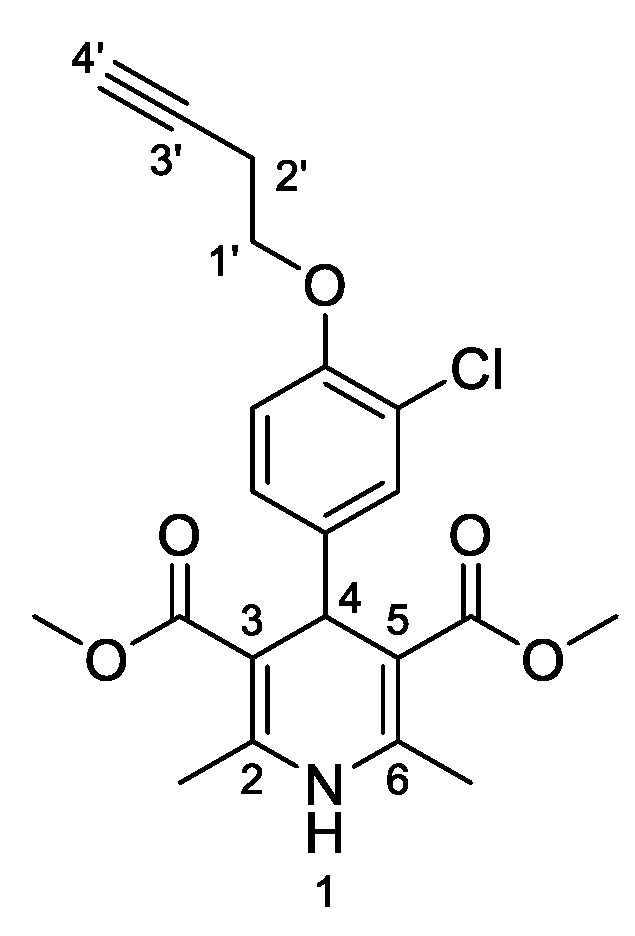
3,5-Dimethyl-4-[3-chloro-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3g,
Figure 8). Following the general procedure starting from 3-chloro-4-(prop-2-yn-1-yloxy)benzaldehyde (
1d, 1 equiv, 1.54 mmol, 300.00 mg), methyl acetoacetate (3.5 equiv, 5.39 mmol, 0.578 mL) and ammonium carbonate (2.5 equiv, 3.85 mmol, 369.95 mg) at 75 °C over 15 h, the observed precipitate was filtered and washed in diethyl ether to afford compound
3g (405.71 mg, 68%) as a white solid:
1H-NMR (400 MHz, CDCl
3) δ 7.23 (d,
J = 2.1 Hz, 1H,
HAr), 7.13 (dd,
J = 8.5, 2.1 Hz, 1H,
HAr), 6.94 (d,
J = 8.5 Hz, 1H,
HAr), 5.64 (s, 1H, N
H), 4.94 (s, 1H,
H4), 4.71 (d,
J = 2.3 Hz, 2H, 2
H1’), 3.66 (s, 6H, 2CO
2C
H3), 2.52 (t,
J = 2.3 Hz, 1H,
H3’), 2.34 (s, 6H, 2C
H3).
13C-NMR (101 MHz, CDCl
3) δ 167.96, 151.68, 144.38, 142.15, 129.74, 126.98, 122.80, 114.02, 103.76, 78.46, 76.09, 57.02, 51.22, 38.67, 19.83. HRMS ESI-TOF [M]
+ m/z Calcd. for C
20H
20ClNO
5: 389,1024. Found: 389,1030.
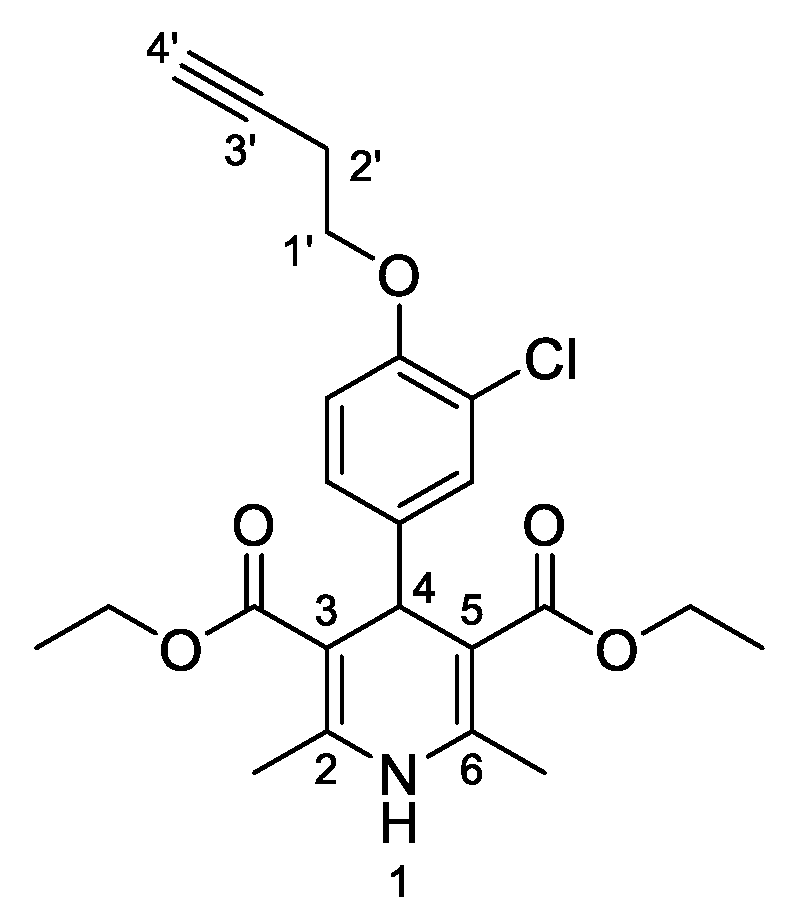
3,5-Diethyl-4-[3-chloro-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3h,
Figure 9). Following the general procedure starting from 3-chloro-4-(prop-2-yn-1-yloxy)benzaldehyde (
1d, 1 equiv, 1.54 mmol, 300.00 mg), ethyl acetoacetate (3.5 equiv, 5.39 mmol, 0.688 mL) and ammonium carbonate (2.5 equiv, 3.85 mmol, 369.95 mg) at 75 °C over 15 h, the precipitate was filtered and washed with diethyl ether to afford compound
3h (416.9 mg, 68%) as a white solid:
1H-NMR (CDCl
3) δ 7.29 (s, 1H,
HAr), 7.16 (dd,
J = 8.5, 2.2 Hz, 1H,
HAr), 6.95 (d,
J = 8.5 Hz, 1H,
HAr), 5.61 (s, 1H, N
H), 4.94 (s, 1H,
H4), 4.74 (d,
J = 2.4 Hz, 2H, 2
H1’), 4.21 – 4.03 (m, 4H, 2CO
2C
H2CH
3), 2.54 (t,
J = 2.4 Hz, 1H,
H3’), 2.36 (s, 6H, 2C
H3), 1.25 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 296.64, 280.65, 273.12, 271.69, 259.32, 256.41, 251.67, 243.07, 233.13, 207.55, 205.15, 189.07, 186.15, 168.17, 148.91, 143.51. Anal. Calcd. for C
22H
24ClNO
5: C, 63.23; H, 5.79; N, 3.35. Found: C, 62.93; H, 5.88; N, 3.39.
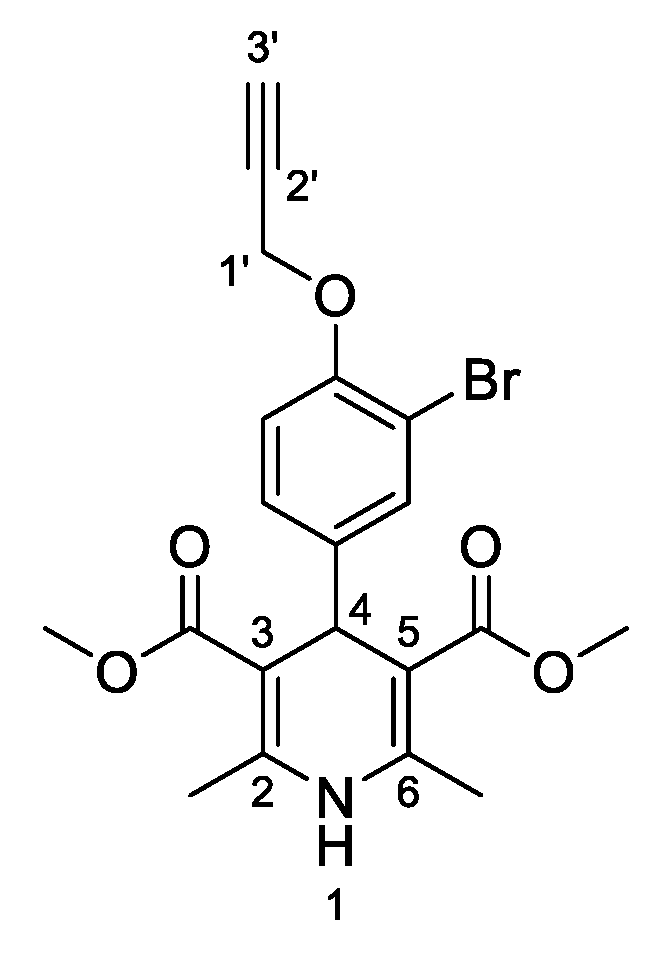
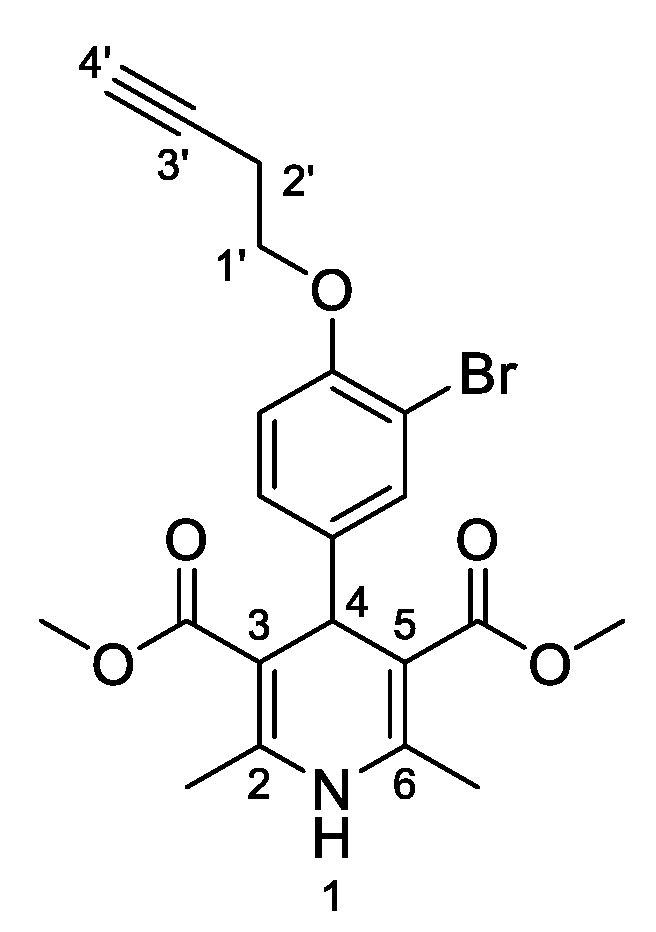
3,5-Dimethyl-4-[3-bromo-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3i,
Figure 10). Following the general procedure starting from 3-bromo-4-(prop-2-yn-1-yloxy)benzaldehyde (
1e, 1 equiv, 1.05 mmol, 250.00 mg), methyl acetoacetate (3.5 equiv, 3.66 mmol, 0.395 mL) and ammonium carbonate (2.5 equiv, 2.63 mmol, 252.24 mg) at 75 °C over 15 h, the precipitate was filtered and washed with diethyl diethyl ether:pentane (1:1 v/v) to afford compound
3i (266.20 mg, 58%) as a white solid:
1H-NMR (CDCl
3) δ 7.40 (d,
J = 2.2 Hz, 1H,
HAr), 7.17 (dd,
J = 8.5, 2.2 Hz, 1H,
HAr), 6.92 (d,
J = 8.5 Hz, 1H,
HAr), 5.63 (s, 1H, N
H), 4.94 (s, 1H,
H4), 4.71 (d,
J = 2.4 Hz, 2H, 2
H1’), 3.66 (s,
J = 6.2 Hz, 6H, 2CO
2C
H3), 2.52 (t,
J = 2.4 Hz, 1H,
H3’), 2.34 (s, 6H, 2C
H3).
13C-NMR (CDCl
3) δ 167.95, 152.59, 144.38, 142.54, 132.78, 127.78, 113.81, 112.07, 103.77, 78.45, 76.10, 57.07, 51.22, 38.62, 19.84. HRMS ESI-TOF [M]
+ m/z Calcd. for C
20H
20BrNO
5: 433,0516. Found: 433,0525.
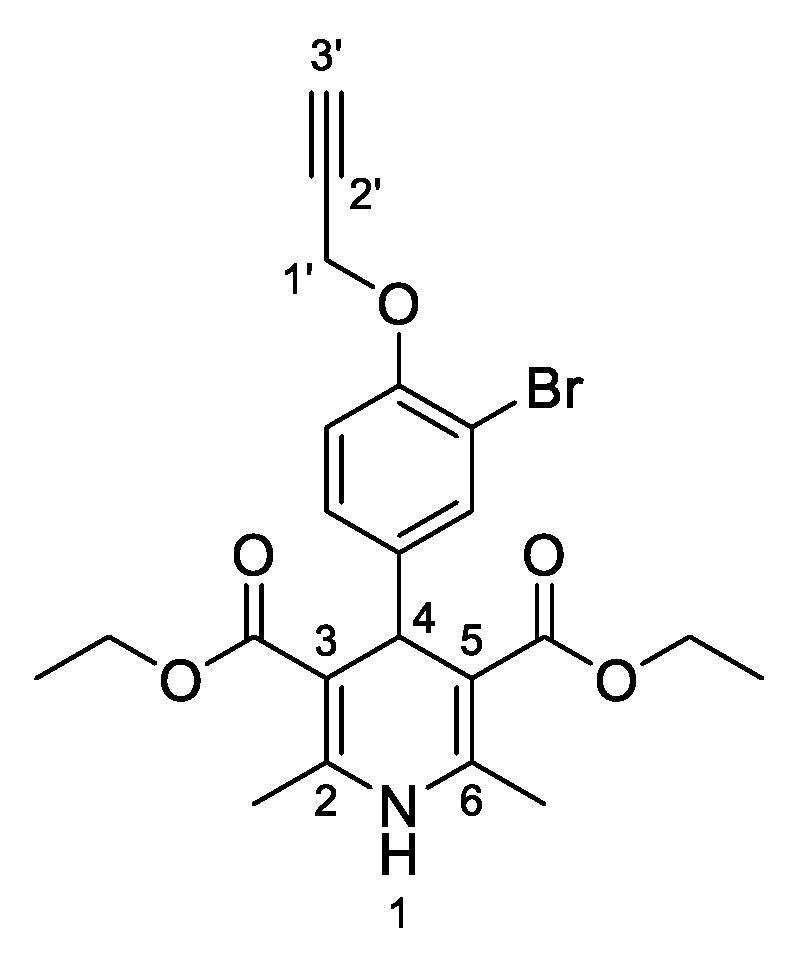
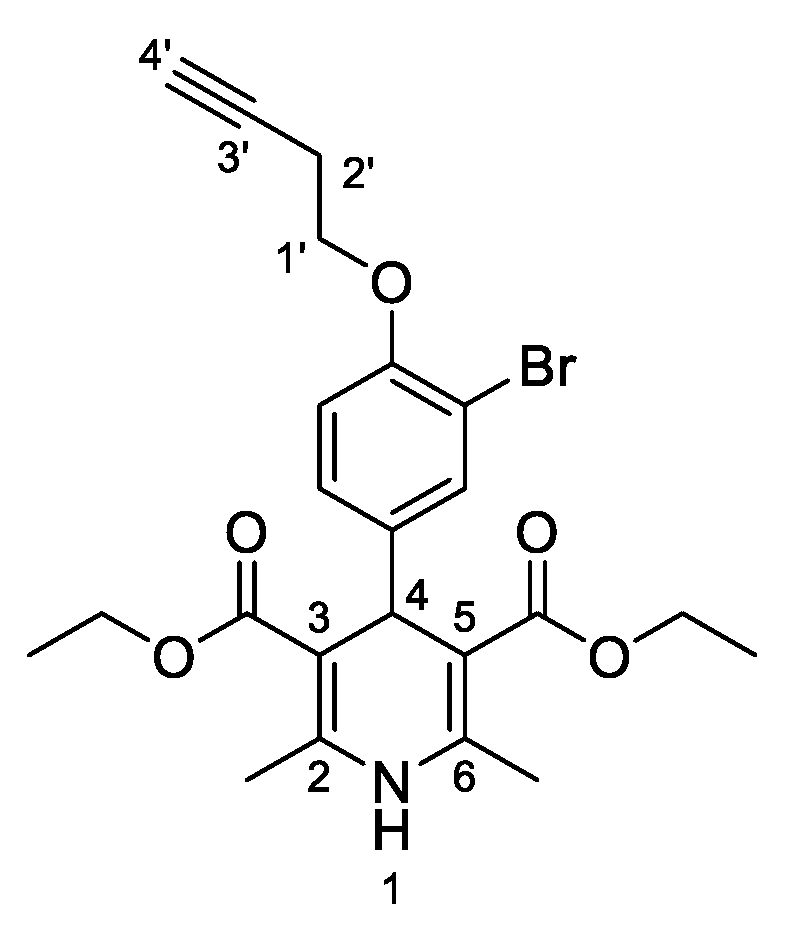
3,5-Diethyl-4-[3-bromo-4-(prop-2-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3j,
Figure 11). Following the general procedure starting from 3-bromo-4-(prop-2-yn-1-yloxy)benzaldehyde (
1e, 1 equiv, 1.05 mmol, 205.00 mg), ethyl acetoacetate (3.5 equiv, 3.66 mmol, 0.467 mL) and ammonium carbonate (2.5 equiv, 2.63 mmol, 252.24 mg) at 70 °C over 15 h, the solvent was removed under pressure conditions and the residue was triturated with diethyl ether. The resulting solid was stirred in diethyl ether:pentane (1:1 v/v) overnight and filtered to afford compound
3j (202.71 mg, 42%) as white powder:
1H-NMR (CDCl
3) δ 7.43 (d,
J = 2.1 Hz, 1H,
HAr), 7.18 (dd,
J = 8.5, 2.1 Hz, 1H,
HAr), 6.91 (d,
J = 8.5 Hz, 1H,
HAr), 5.56 (s, 1H, N
H), 4.91 (s, 1H,
H4), 4.71 (d,
J = 2.4 Hz, 2H, 2
H1’), 4.22 – 4.01 (m, 4H, 2CO
2C
H2CH
3), 2.51 (t,
J = 2.4 Hz, 1H,
H3’), 2.34 (s, 6H, 2C
H3), 1.23 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 167.51, 152.46, 143.99, 142.98, 133.29, 128.09, 113.76, 111.78, 104.06, 78.44, 76.06, 59.97, 57.10, 39.03, 19.83, 14.42. HRMS ESI-TOF [M]
+ m/z Calcd. for C
22H
24BrNO
5: 461,0829. Found: 461,0838.
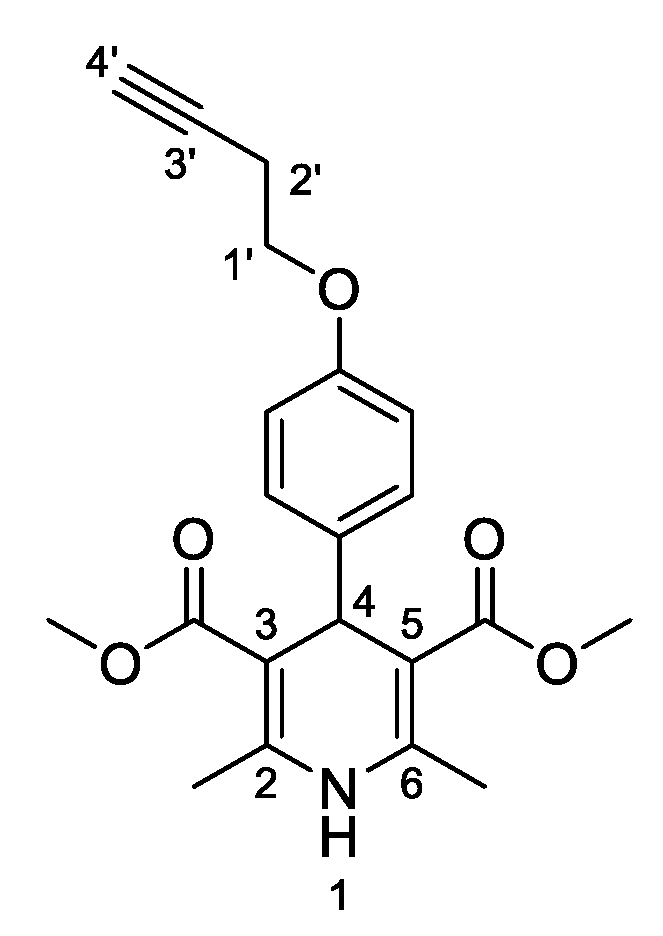
3,5-Dimethyl-4-[4-(but-3-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3k,
Figure 12). Following the general procedure starting from 4-(but-3-yn-1-yloxy)benzaldehyde (
2a) (1 equiv, 1.43 mmol, 250.00 mg), methyl acetoacetate (3.5 equiv, 5.02 mmol, 0.640 mL) and ammonium carbonate (2.5 equiv, 3.58 mmol, 343.52 mg) at 75 °C over 15 h, the solvent was removed under pressure conditions and the resulting residue was purified by flash column chromatography using hexane/EtOAc (7/·3) + 1% Et
3N, to afford compound
3k (144.2 mg, 27%) as white crystals:
1H-NMR (CDCl
3) δ 7.21–7.10 (m, 2H, 2
HAr), 6.79–6.72 (m, 2H, 2
HAr), 5.61 (s, 1H, N
H), 4.94 (s, 1H,
H4), 4.04 (t,
J = 7.0 Hz, 2H, 2
H1’), 3.64 (s, 6H, 2CO
2C
H3), 2.64 (td,
J = 7.0, 2.7 Hz, 2H, 2
H2’), 2.33 (s, 6H, 2C
H3), 2.01 (t,
J = 2.7 Hz, 1H,
H4’).
13C-NMR (CDCl
3) δ 168.20, 156.93, 144.03, 140.51, 129.01, 128.82, 114.28, 114.19, 104.27, 80.72, 69.90, 66.01, 51.13, 38.60, 19.77, 19.70. HRMS ESI-TOF [M]
+ m/z Calcd. for C
21H
23NO
5: 397,1879. Found: 397,1889.
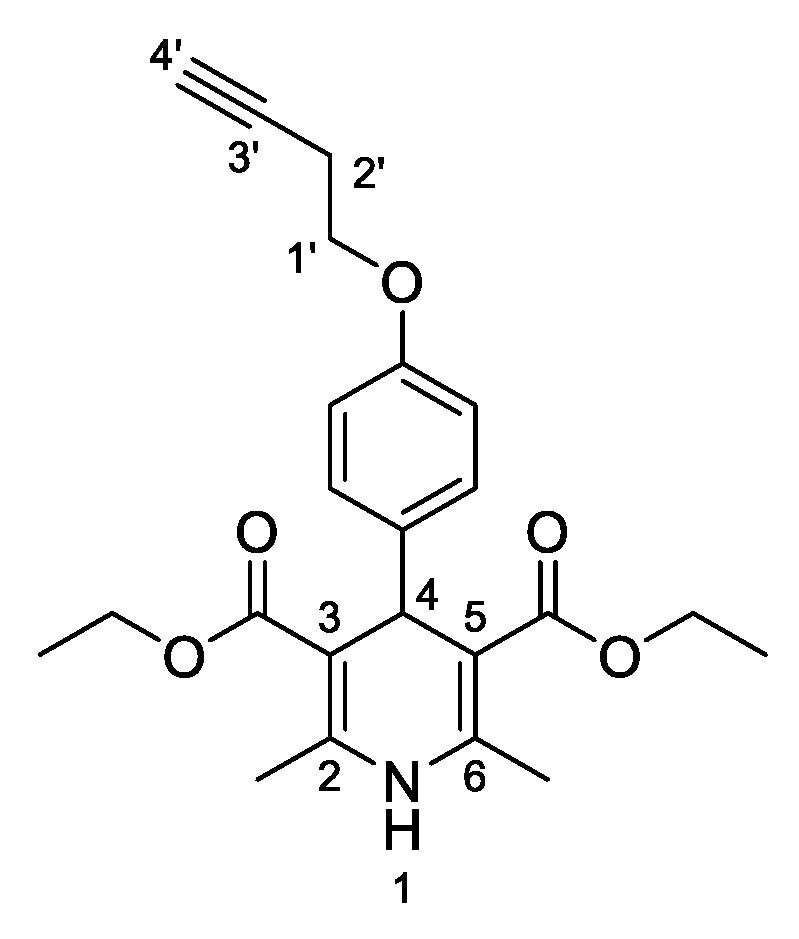
3,5-Diethyl-4-[4-(but-3-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3l,
Figure 13). Following the general procedure starting from 4-(but-3-yn-1-yloxy)benzaldehyde (
2a, 1 equiv, 1.43 mmol, 250.00 mg), ethyl acetoacetate (3.5 equiv, 5.02 mmol, 0.540 mL) and ammonium carbonate (2.5 equiv, 3.58 mmol, 343.52 mg) at 75 °C over 15 h, the solvent was removed under pressure conditions and the residue was triturated with diethyl ether. The resulting residue was recrystallized in DCM/diethyl ether (1:1 v/v) to afford compound
3l (70.10 mg, 12%) as a white solid:
1H-NMR (CDCl
3) δ 7.23–7.11 (m, 2H, 2
HAr), 6.78–6.72 (m, 2H, 2
HAr), 5.57 (s, 1H, N
H), 4.92 (s, 1H,
H4), 4.15–3.99 (m, 6H, 2CO
2C
H2CH
3 and 2
H1’), 2.64 (td,
J = 7.0, 2.7 Hz, 2H, 2
H2’), 2.32 (s, 6H, 2C
H3), 2.01 (t,
J = 2.7 Hz, 1H,
H4’), 1.22 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 167.79, 156.86, 143.65, 140.92, 129.18, 114.11, 104.54, 80.73, 69.90, 66.03, 59.84, 38.93, 19.77, 19.70, 14.42. HRMS ESI-TOF [M]
+ m/z Calcd. for C
23H
27NO
5: 369,1570. Found: 369,1576.
3,5-Dimethyl-4-[4-(but-3-yn-1-yloxy)-3-methoxyphenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3m,
Figure 14). Following the general procedure starting from 4-(but-3-yn-1-yloxy)-3-methoxybenzaldehyde (
2b, 1 equiv, 1.47 mmol, 300.00 mg), methyl acetoacetate (3.5 equiv, 5.14 mmol, 0.594 mL) and ammonium carbonate (2.5 equiv, 3.68 mmol, 353.13 mg) at 75 °C over 15 h, the precipitate was filtered and washed in diethyl ether:pentane (1:1 v/v) to afford compound
3m (420.5 mg, 72%) as a white solid:
1H-NMR (CDCl
3) δ 6.87 (s, 1H,
HAr), 6.74 (d,
J = 0.8 Hz, 2H, 2
HAr), 5.63 (s, 1H, N
H), 4.95 (s, 1H,
H4), 4.10 (t,
J = 7.4 Hz, 2H, 2
H1’), 3.82 (s, 3H, OC
H3), 3.66 (s, 6H, 2CO
2C
H3), 2.68 (td,
J = 7.4, 2.7 Hz, 2H, 2
H2’), 2.34 (s, 6H, 2C
H3), 2.01 (t,
J = 2.7 Hz, 1H,
H4’).
13C-NMR (CDCl
3) δ 168.19, 149.13, 146.29, 144.09, 141.35, 119.75, 113.79, 112.39, 104.09, 80.54, 70.03, 67.23, 56.14, 51.14, 38.94, 19.78, 19.61. Anal. Calcd. for C
22H
25NO
5: C, 66.15; H, 6.31; N, 3.51. Found: C, 66.23; H, 6.36; N, 3.59.
3,5-Diethyl-4-[4-(but-3-yn-1-yloxy)-3-methoxyphenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3n,
Figure 15). Following the general procedure starting from 4-(but-3-yn-1-yloxy)-3-methoxybenzaldehyde (
2b, 1 equiv, 1.47 mmol, 300.00 mg), ethyl acetoacetate (3.5 equiv, 5.14 mmol, 0.656 mL) and ammonium carbonate (2.5 equiv, 3.68 mmol, 353.13 mg) at 75 °C over 15 h, the solvent was removed under pressure conditions and the residue was triturated with diethyl ether. The resulting solid was stirred in diethyl ether:pentane (1:1 v/v) overnight and filtered to afford compound
3n (520.92 mg, 83%) as a light yellowish solid:
1H-NMR (CDCl
3) δ 6.88 (d,
J = 1.7 Hz, 1H,
HAr), 6.79–6.70 (m, 2H, 2
HAr), 5.55 (s, 1H, N
H), 4.94 (s, 1H,
H4), 4.10 (pd,
J = 8.2, 3.7 Hz, 6H, 2CO
2C
H2CH
3 and 2
H1’), 3.82 (s, 3H, OC
H3), 2.68 (td,
J = 7.5, 2.7 Hz, 2H, 2
H2’), 2.33 (s, 6H, 2C
H3), 2.01 (t,
J = 2.7 Hz, 1H,
H4’), 1.23 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 167.78, 148.99, 146.21, 143.70, 141.81, 120.14, 113.78, 112.73, 104.42, 80.56, 70.01, 67.27, 59.88, 56.12, 39.26, 19.81, 19.62, 14.49. HRMS ESI-TOF [M]
+ m/z Calcd. for C
24H
29NO
6: 427,1985. Found: 427,1995.
3,5-Dimethyl-4-[4-(but-3-yn-1-yloxy)-3-ethoxyphenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3o,
Figure 16). Following the general procedure starting from 4-(but-3-yn-1-yloxy)-3-ethoxybenzaldehyde (
2c, 1 equiv, 1.60 mmol, 350.00 mg), methyl acetoacetate (3.5 equiv, 5.61 mmol, 0.679 mL) and ammonium carbonate (2.5 equiv, 4.00 mmol, 384.36 mg) at 75 °C over 15 h, the precipitate was filtered and washed in diethyl ether:pentane (1:1 v/v) to afford compound
3o (288.40 mg, 44%) as a white solid:
1H-NMR (CDCl
3) δ 6.89 (s, 1H,
HAr), 6.77 (s, 2H, 2
HAr), 5.67 (s, 1H, N
H), 4.95 (s, 1H,
H4), 4.09 (dt,
J = 2.3, 7.1 Hz, 4H, OC
H2CH
3 and 2
H1’), 3.67 (s, 6H, 2CO
2C
H3), 2.69 (td,
J = 7.4, 2.6 Hz, 2H, 2
H2’), 2.35 (s, 6H, 2C
H3), 2.02 (t,
J = 2.6 Hz, 1H,
H4’), 1.42 (t,
J = 7.0 Hz, 3H, OCH
2C
H3).
13C-NMR (CDCl
3) δ 168.20, 148.54, 146.73, 144.08, 141.58, 120.21, 120.01, 114.84, 114.43, 104.10, 80.72, 69.88, 67.61, 64.76, 51.13, 38.91, 19.75, 19.68, 15.03. HRMS ESI-TOF [M]
+ m/z Calcd. for C
23H
27NO
6: 413,1831. Found: 413,1838.
3,5-Diethyl-4-[4-(but-3-yn-1-yloxy)-3-ethoxyphenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3p,
Figure 17). Following the general procedure starting from 4-(but-3-yn-1-yloxy)-3-ethoxybenzaldehyde (
2c, 1 equiv, 1.60 mmol, 350.00 mg), ethyl acetoacetate (3.5 equiv, 5.61 mmol, 0.716 mL) and ammonium carbonate (2.5 equiv, 4.00 mmol, 384.36 mg) at 75 °C over 15 h, the precipitate was filtered and washed in diethyl ether:pentane (1:1 v/v) to afford compound
3p (340.31 mg, 48%) as a white solid: H-NMR (CDCl
3) δ 6.88 (d,
J = 1.3 Hz, 1H,
HAr), 6.81–6.70 (m, 2H, 2
HAr), 5.52 (s, 1H, N
H), 4.93 (s, 1H,
H4), 4.17 – 3.99 (m, 8H, OC
H2CH
3 and 2
H1’ and 2CO
2C
H2CH
3), 2.67 (td,
J = 7.4, 2.7 Hz, 2H, 2
H2’), 2.33 (s, 6H, 2C
H3), 2.00 (t,
J = 2.7 Hz, 1H,
H4’), 1.40 (t,
J = 7.0 Hz, 3H, OCH
2C
H3), 1.23 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 167.79, 148.52, 146.68, 143.64, 141.97, 120.41, 114.81, 114.76, 104.44, 80.71, 69.86, 67.66, 64.80, 59.86, 39.20, 19.81, 19.70, 15.10, 14.48. Anal. Calcd. for C
25H
31NO
6: C, 68.01; H, 7.08; N, 3.17. Found: C, 67.87; H, 7.15; N, 3.25. HRMS ESI-TOF [M]
+ m/z Calcd. for C
25H
31NO
6: 441,2137. Found: 441,2151.
3,5-Dimethyl-4-[4-(but-3-yn-1-yloxy)-3-chlorophenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3q,
Figure 18). Following the general procedure starting from 4-(but-3-yn-1-yloxy)-3-chlorobenzaldehyde (
2d, 1 equiv, 1.44 mmol, 300.00 mg), methyl acetoacetate (3.5 equiv, 5.03 mmol, 0.543 mL) and ammonium carbonate (2.5 equiv, 3.60 mmol, 345.92 mg) at 78 °C over 15 h, the solvent was removed under pressure conditions and the resulting residue was purified by flash column chromatography using hexane/EtOAc (6/4) + 1% Et
3N, to afford compound
3q (76.05 mg, 13%) as a white solid:
1H-NMR (CDCl
3) δ 7.22 (d,
J = 2.2 Hz, 1H,
HAr), 7.11 (dd,
J = 8.5, 2.2 Hz, 1H,
HAr), 6.78 (d,
J = 8.5 Hz, 1H,
HAr), 5.64 (s, 1H, N
H), 4.92 (s, 1H,
H4), 4.10 (t,
J = 7.2 Hz, 2H, 2
H1’), 3.65 (s,
J = 4.3 Hz, 6H, 2CO
2C
H3), 2.70 (td,
J = 7.2, 2.7 Hz, 2H, 2
H2’), 2.34 (s, 6H, 2C
H3), 2.02 (t,
J = 2.7 Hz, 1H,
H4’).
13C-NMR (CDCl
3) δ 167.84, 152.29, 144.19, 141.61, 129.55, 126.95, 122.72, 113.59, 103.67, 80.11, 70.01, 67.27, 51.06, 38.55, 19.68, 19.48. HRMS ESI-TOF [M]
+ m/z Calcd. for C
21H
22ClNO
5: 403,1180. Found: 403,1187.
3,5-Diethyl-4-[4-(but-3-yn-1-yloxy)-3-chlorophenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3r,
Figure 19). Following the general procedure starting from 4-(but-3-yn-1-yloxy)-3-chlorobenzaldehyde (
2d) (1 equiv, 1.44 mmol, 300.00 mg), ethyl acetoacetate (3.5 equiv, 5.03 mmol, 0.643 mL) and ammonium carbonate (2.5 equiv, 3.60 mmol, 345.92 mg) at 79 °C over 15 h, the solvent was removed under pressure conditions and the resulting residue was purified by flash column chromatography using hexane/EtOAc (6/4) + 1% Et
3N, to afford compound
3r (80.23 mg, 13%) as a white solid:
1H-NMR (CDCl
3) δ 7.25 (d,
J = 2.2 Hz, 1H,
HAr), 7.12 (dd,
J = 8.4, 2.2 Hz, 1H,
HAr), 6.78 (d,
J = 8.5 Hz, 1H,
HAr), 5.57 (s, 1H, N
H), 4.90 (s, 1H,
H4), 4.20–4.01 (m, 6H, 2
H1’ and 2CO
2CH2CH
3), 2.70 (td,
J = 7.3, 2.7 Hz, 2H, 2
H2’), 2.33 (s, 6H, 2CH
3), 2.02 (t,
J = 2.7 Hz, 1H,
H4’), 1.23 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2CH3).
13C-NMR (CDCl
3) δ 167.55, 152.33, 143.95, 142.16, 130.16, 127.41, 122.61, 113.64, 104.08, 80.26, 70.14, 67.44, 59.95, 39.06, 22.09, 19.81, 19.62, 14.41. HRMS ESI-TOF [M]
+ m/z Calcd. for C
23H
26ClNO
5: 431,1491. Found: 431,1500.
3,5-Dimethyl-4-[3-bromo-4-(but-3-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3s,
Figure 20). Following the general procedure starting from 3-bromo-4-(but-3-yn-1-yloxy)benzaldehyde (
2e, 1 equiv, 0.77 mmol, 195.00 mg), methyl acetoacetate (3.5 equiv, 2.69 mmol, 0.291 mL) and ammonium carbonate (2.5 equiv, 1.93 mmol, 189.97 mg) at 75 °C over 15 h, the precipitated was filtered and washed in diethyl ether:pentane (1:3 v/v) to afford compound
3s (142.40 mg, 23%) as a white solid:
1H-NMR (CDCl
3) δ 7.38 (d,
J = 1.8 Hz, 1H,
HAr), 7.16 (d,
J = 8.4 Hz, 1H,
HAr), 6.76 (d,
J = 8.4 Hz, 1H,
HAr), 5.62 (s, 1H, N
H), 4.92 (s, 1H,
H4), 4.10 (t,
J = 7.2 Hz, 2H, 2
H1’), 3.65 (s, 6H, 2CO
2C
H3), 2.70 (td,
J = 7.1, 2.5 Hz, 2H, 2
H2’), 2.34 (s, 6H, 2CH
3), 2.03 (d,
J = 2.4 Hz, 1H,
H4’).
13C-NMR (CDCl
3) δ 167.95, 153.32, 144.30, 142.15, 132.73, 127.89, 113.49, 103.82, 80.27, 70.15, 67.46, 51.20, 38.65, 19.83, 19.62. HRMS ESI-TOF [M]
+ m/z Calcd. for C
21H
22BrNO
5: 447,0666. Found: 447,0681.
3,5-Diethyl-4-[3-bromo-4-(but-3-yn-1-yloxy)phenyl]-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (
3t,
Figure 21). Following the general procedure starting from 3-bromo-4-(but-3-yn-1-yloxy)benzaldehyde (
2e, 1 equiv, 0.77 mmol, 195.00 mg), ethyl acetoacetate (3.5 equiv, 1.69 mmol, 0.343 mL) and ammonium carbonate (2.5 equiv, 1.93 mmol, 184.97 mg) at 85 °C over 15 h, the solvent was removed under pressure conditions and the residue was triturated with diethyl ether. The resulting solid was stirred in diethyl ether:pentane (1:1 v/v) overnight and filtered to afford compound
3t (280.92 mg, 49%) as a white solid:
1H-NMR (CDCl
3) δ 7.42 (d,
J = 2.1 Hz, 1H,
HAr), 7.16 (dd,
J = 8.4, 2.2 Hz, 1H,
HAr), 6.88 – 6.66 (m, 1H,
HAr), 5.55 (s, 1H, N
H), 4.90 (s, 1H,
H4), 4.31 – 4.02 (m, 6H, 2
H1’ and 2CO
2C
H2CH
3), 2.70 (td,
J = 7.3, 2.7 Hz, 2H, 2
H2’), 2.33 (s, 6H, 2CH
3), 2.03 (t,
J = 2.7 Hz, 1H,
H4’), 1.23 (t,
J = 7.1 Hz, 6H, 2CO
2CH
2C
H3).
13C-NMR (CDCl
3) δ 167.56, 153.21, 144.02, 142.58, 133.20, 128.18, 113.42, 111.93, 104.03, 80.27, 70.14, 67.48, 59.95, 39.01, 19.77, 19.61, 14.41. HRMS ESI-TOF [M]
+ m/z Calcd. for C
23H
26BrNO
5: 475,0983. Found: 475,0994.


 ,
, 























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}