Nucleophilic Substitution at Tetracoordinate Sulfur. Kinetics and Mechanism of the Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides: Counterintuitive Acceleration of Substitution at Sulfonyl Sulfur by ortho-Alkyl Groups and Its Origin


 and
and
Abstract
:
1. Introduction

2. Results and Discussion
2.1. Kinetics of Isotopic Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides and Their Reactivity
| 1 | (3-Me)-1 | (4-Me)-1 | (3,4-Me2)-1 | |
| 102 × k25 | 117.3 | 102.0 | 67.0 | 64.4 |
| (2-Et)-1 | (2-Me)-1 | (2-iPr)-1 | 1 | ||
| 102 × k25 | 248.3 | 128.1 | 124.0 | 117.3 | |
| (2,4,6-Me3)-1 | (2,4,6-Et3)-1 | (2,4,6-iPr3)-1 | 1 | ||
| 102 × k25 | 517.8 | 446.6 | 378.4 | 117.3 |
2.2. Structural Features of Arenesulfonyl Chlorides 1: DFT Calculations and X-Ray Diffraction Data
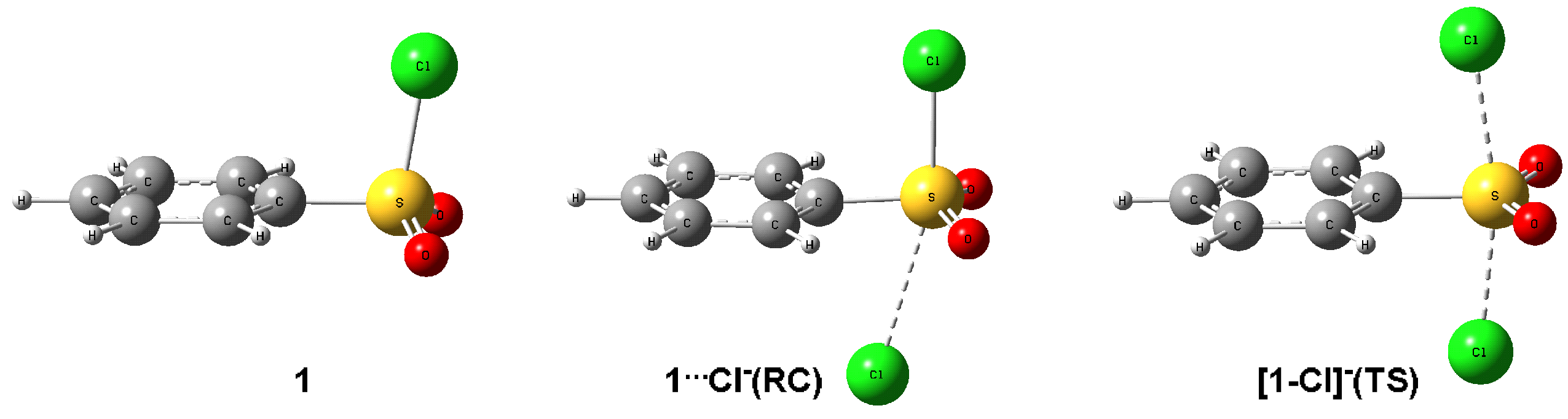
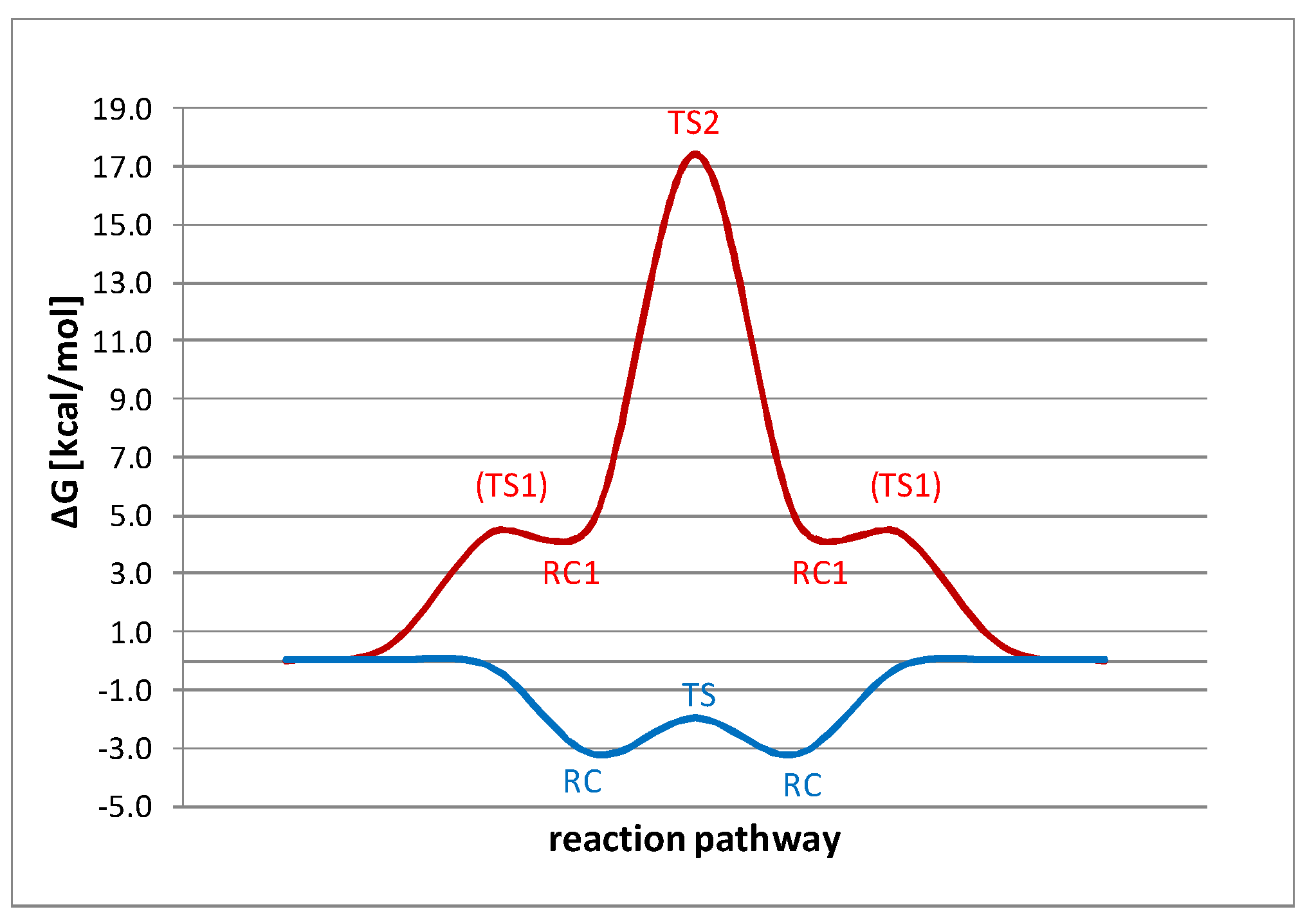
2.3. DFT Study of Identity Chloride Exchange Reaction in Arenesulfonyl Chlorides 1
- Rotation of sulfonyl group around C(1)-S bond within the observed range does not noticeably affect the energies of electron delocalization in terms of the NBO theory (Table S5, Supplementary Materials).
- Variation in steric repulsion between SO2Cl moiety and aromatic ring in 1 upon its rotation around C(1)-S bond is also insignificant.
- The NBO analysis does not provide any clear rationalization for the observed kinetic effect. The differences in delocalization pattern between simple benzenesulfonyl chlorides 1 and (4-Me)-1 and sterically hindered (2,6-Me2)-1, (2,4,6-Me3)-1, (2,6-iPr2)-1 are not significant.
2.4. Concluding Remarks
3. Materials and Methods
3.1. Materials
3.2. Kinetic Methods
3.3. Kinetic Calculations
3.4. Computational Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mikołajczyk, M.; Drabowicz, J. Chiral Organosulfur Compounds. In Topic in Stereochemistry; Allinger, N.L., Eliel, E.I., Wilen, S.H., Eds.; John Wiley and Sons, Inc.: New York, NY, USA, 1982; Volume 13, pp. 333–468. [Google Scholar]
- Oae, S. Organic Sulfur Chemistry: Structure and Mechanism; Doi, J.T., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 119–200. [Google Scholar]
- Drabowicz, J.; Mikołajczyk, M.; Łyżwa, P. High-coordinated sulfur compounds. In The Chemistry of Sulphur-Containing Functional Groups; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 1993; pp. 799–956. [Google Scholar]
- Mikolajczyk, M.; Drabowicz, J.; Slebocka-Tilk, H. Nucleophilic substitution at sulfur. Kinetic evidence for inversion of configuration at sulfinyl sulfur in acid-catalyzed transesterification of sulfinates. J. Am. Chem. Soc. 1979, 101, 1302–1303. [Google Scholar] [CrossRef]
- Westheimer, F.H. Pseudo-rotation in the hydrolysis of phosphate esters. Acc. Chem. Res. 2002, 1, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, M.; Witczak, M. Stereochemistry of organophosphorus cyclic compounds. Part 5. Synthesis and geometry of some 4,5-disubstituted 1,3,2-dioxaphospholan-2-thiones and stereochemistry of nucleophilic displacement reactions at phosphorus. J. Chem. Soc. Perkin Trans. 1977, 1, 2213–2222. [Google Scholar] [CrossRef]
- Oae, S.; Yokoyama, M.; Kise, M.; Furukawa, N. Oxygen exchange reaction of sulfoxides with dimethyl sulfofide. Tetrahedron Lett. 1968, 9, 4131–4134. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J. Asymmetric synthesis of amidothiosulphites. Substitution at sulphur with retention of configuration. J. Chem. Soc. Chem. Commun. 1974, 775–776. [Google Scholar] [CrossRef]
- Bujnicki, B.; Drabowicz, J.; Mikolajczyk, M. Acid catalyzed alcoholysis of sulfinamides: Unusual stereochemistry, kinetics and a question of mechanism involving sulfurane intermediates and their pseudorotation. Molecules 2015, 20, 2949–2972. [Google Scholar] [CrossRef] [Green Version]
- Sabol, M.A.; Andersen, K.K. Nucleophilic substitution at tetracoordinate hexavalent sulfur. Reaction of (-) -menthyl phenylmethanesulfonate with p-tolylmagnesium bromide. J. Am. Chem. Soc. 1969, 91, 3603–3605. [Google Scholar] [CrossRef]
- Jonsson, E.U.; Johnson, C.R. Chemistry of sulfoxides and related compounds. XXXIII. Stereochemistry of substitution at tetracoordinate hexavalent sulfur. Nucleophilic reactions at sulfur in sulfonimidoyl compounds. J. Am. Chem. Soc. 1971, 93, 5308–5309. [Google Scholar] [CrossRef]
- Gordon, I.M.; Maskill, H.; Ruasse, M.-F. Sulphonyl transfer reactions. Chem. Soc. Rev. 1989, 18, 123–151. [Google Scholar] [CrossRef]
- Iazykov, M.; Rublova, L.; Canle L, M.; Santaballa, J.A. Solvent network at the transition state in the solvolysis of hindered sulfonyl compounds. J. Phys. Org. Chem. 2017, 30, e3588. [Google Scholar] [CrossRef]
- Iazykov, M.; Canle, L.M.; Santaballa, J.A.; Rublova, L. Isotope Effects in the Solvolysis of Sterically Hindered Arenesulfonyl Chlorides. Int. J. Chem. Kinet. 2015, 47, 744–750. [Google Scholar] [CrossRef]
- D’Rozario, P.; Smyth, R.L.; Williams, A. Evidence for a single transition state in the intermolecular transfer of a sulfonyl group between oxyanion donor and acceptors. J. Am. Chem. Soc. 1984, 106, 5027–5028. [Google Scholar] [CrossRef]
- Baxter, N.J.; Rigoreau, L.J.M.; Laws, A.P.; Page, M.I. Reactivity and Mechanism in the Hydrolysis of β-Sultams. J. Am. Chem. Soc. 2000, 122, 3375–3385. [Google Scholar] [CrossRef]
- Perkins, C.W.; Martin, J.C. Isolable species from nucleophilic attack at sulfinyl and sulfonyl sulfur: A sulfuran dioxide (10-S-5) salt and a sulfuran oxide (10-S-4) salt. J. Am. Chem. Soc. 1983, 105, 1377–1379. [Google Scholar] [CrossRef]
- Hall, H.K. Kinetics of Reactions of Acyl Chlorides. II. Mechanisms of Hydrolysis of Sulfonyl Chlorides. J. Am. Chem. Soc. 1956, 78, 1450–1454. [Google Scholar] [CrossRef]
- Hall, H.K.; Lueck, C.H. The Ionization Mechanism for the Hydrolysis of Acyl Chlorides. J. Org. Chem. 1963, 28, 2818–2825. [Google Scholar] [CrossRef]
- Tommila, E.; Hirsjärvi, P.; Haug, C.M.; Stene, J.; Sörensen, N.A. The Alcoholysis of Some Aromatic Sulphonyl Chlorides. Acta Chem. Scand. 1951, 5, 659–664. [Google Scholar] [CrossRef] [Green Version]
- Tonnet, M.L.; Hambly, A.N. Solvolysis of sulphonyl halides. VII. Hydrolysis of some aromatic sulphonyl chlorides in aqueous dioxan. Aust. J. Chem. 1971, 24, 703–712. [Google Scholar] [CrossRef]
- Vizgert, R.V. Mechanisms of hydrolysis of aromatic sulfonyl chlorides XII. J. Gen. Chem. USSR 1962, 32, 619–624. [Google Scholar]
- Vizgert, R.V.; Savchuk, E.K. Influence of polarity of medium on hydrolysis of aromatic sulfochlorides. J. Gen. Chem. USSR 1964, 34, 3437–3443. [Google Scholar]
- Vizgert, R.V. Mechanisms of hydrolysis of aromatic sulfonyl chlorides, alkyl and aryl sulfonates. Russ. Chem. Rev. 1963, 32, 1–20. [Google Scholar] [CrossRef]
- Rogne, O. Kinetics of the neutral and alkaline hydrolysis of aromatic sulphonyl chlorides in water. J. Chem. Soc. B 1968, 1294–1296. [Google Scholar] [CrossRef]
- Senatore, L.; Sagramora, L.; Ciuffarin, E. Neutral and alkaline hydrolysis of 2,4,6-trimethylbenzenesulphonyl chloride. J. Chem. Soc. Perkin Trans. 1974, 2, 722–723. [Google Scholar] [CrossRef]
- Kevill, D.N.; Park, B.C.; Park, K.H.; D’Souza, M.J.; Yaakoubd, L.; Mlynarski, S.L.; Kyong, J.B. Rate and product studies in the solvolyses of N,N-dimethylsulfamoyl and 2-propanesulfonyl chlorides. Org. Biomol. Chem. 2006, 4, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Vizgert, R.V.; Rubleva, L.I.; Maksimenko, N.N. Reactivity of sterically hindered aromatic sulfonic acid derivatives. I. Effect of substituents on rate and mechanism of hydrolysis of several substituted benzenesulfonyl chlorides. Zh. Org. Khim. 1989, 25, 810–814. [Google Scholar]
- Bunnett, J.F.; Bassett, J.Y. Kinetics of Reactions of Piperidine with Nitrophenyl Esters of p-Toluenesulfonic and Mesitylenesulfonic Acids. The Miserable Effect of ortho-Methyl Groups. J. Org. Chem. 1962, 27, 2345–2348. [Google Scholar] [CrossRef]
- Darwish, D.; Noreyko, J. The Solvolysis of Arenesulfinates, Sulfur–Oxygen Versus Carbon–Oxygen Bond Fission. Can. J. Chem. 1965, 43, 1366–1374. [Google Scholar] [CrossRef]
- Lee, I.; Kim, C.K.; Li, H.G.; Sohn, C.K.; Kim, C.K.; Lee, H.W.; Lee, B.-S. Acyl-Transfer Mechanisms Involving Various Acyl Functional Groups: >XY with X = C, S, P and Y = O, S. J. Am. Chem. Soc. 2000, 122, 11162–11172. [Google Scholar] [CrossRef]
- Sung, D.D.; Kim, T.J.; Lee, I. Theoretical studies of the nucleophilic substitution of halides and amine at a sulfonyl center. J. Phys. Chem. A 2009, 113, 7073–7079. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Gajl, M.; Reimschüssel, W. Chlorine isotopic exchange at sulphur in sulphonyl chlorides. Accelerating effect of the ortho-alkyl groups. Tetrahedron Lett. 1975, 16, 1325–1326. [Google Scholar] [CrossRef]
- Swain, C.G.; Langsdorf, W.P. Concerted Displacement Reactions. VI. m- and p-Substituent Effects as Evidence for a Unity of Mechanism in Organic Halide Reactions1. J. Am. Chem. Soc. 1951, 73, 2813–2819. [Google Scholar] [CrossRef]
- Ciuffarin, E.; Senatore, L. A hammett study of the alkaline hydrolysis of benzenesulphonyl fluorides. Tetrahedron Lett. 1974, 15, 1635–1636. [Google Scholar] [CrossRef]
- Brown, H.C.; McDonald, G.J. Secondary Isotope Effects in the Reactions of Methyl-d3-pyridines with Alkyl Iodides. Evidence for a Smaller Steric Requirement of the Methyl-d3 over the Methyl-d0 Group1,2. J. Am. Chem. Soc. 1966, 88, 2514–2519. [Google Scholar] [CrossRef]
- Lohrmann, R.; Khorana, H.G. Studies on Polynucleotides. LII.1 The Use of 2,4,6-Triisopropylbenzenesulfonyl Chloride for the Synthesis of Internucleotide Bonds2. J. Am. Chem. Soc. 1966, 88, 829–833. [Google Scholar] [CrossRef]
- Zhdanov, R.I.; Zhenodarova, S.M. Chemical Methods of Oligonucleotide Synthesis. Synthesis 1975, 1975, 222–245. [Google Scholar] [CrossRef]
- Reese, C.B.; Pei-Zhuo, Z. Phosphotriester approach to the synthesis of oligonucleotides: A reappraisal. J. Chem. Soc. Perkin Trans. 1993, 1, 2291–2301. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Cantone, A.; Maccarone, E.; Tomaselli, G.A.; Tripolone, M. Nucleophilic substitution at sulphonyl sulphur. Part 2. Hydrolysis and alcoholysis of aromatic sulphonyl chlorides. J. Chem. Soc. Perkin Trans. 1981, 2, 438–441. [Google Scholar] [CrossRef]
- Denehy, E.; White, J.M.; Williams, S.J. Electronic Structure of the Sulfonyl and Phosphonyl Groups: A Computational and Crystallographic Study. Inorg. Chem. 2007, 46, 8871–8886. [Google Scholar] [CrossRef]
- Tillmann, J.; Bolte, M. 4-Methylbenzenesulfonyl chloride. CSD Commun. CCDC 889573 Exp. Cryst. Struct. Determinat 2012. [Google Scholar] [CrossRef]
- Laba, V.I.; Sviridova, A.V.; Nesterov, V.N. 2,4,6-tri(isopropyl)benzenesulfonyl chloride. CCDC 685972 Exp. Cryst. Struct. Determinat. 2011. [Google Scholar] [CrossRef]
- Laba, V.I.; Sviridova, A.V.; Nesterov, V.N. Structural study of 2,4,6-triisopropylbenzenesulfonyl chloride. Crystallogr. Rep. 2009, 54, 44–47. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Martin-Diaconescu, V.; Kennepohl, P. Effects of Hyperconjugation on the Electronic Structure and Photoreactivity of Organic Sulfonyl Chlorides. Inorg. Chem. 2009, 48, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Weyl, T.; Houben, H.J. Houben-Weyl Methoden der Organischen Chemie, 4 ed.; Georg Thieme Verlag: Stuttgart, Germany, 1955; Volume 9, pp. 561–597. [Google Scholar]
- Uhlenbroek, J.H. Preparation of diaryl sulphides. Recl. Trav. Chim. Pays-Bas 1961, 80, 1057–1065. [Google Scholar] [CrossRef]
- Bamford, C.H.; Tripper, C.F.H. The Practice of Kinetics; Bamford, C.H., Tripper, C.F.H., Eds.; Elsevier: Amsterdam, The Netherlands, 1969; Volume 1, pp. 343–407. [Google Scholar]
- Svehla, G. Comprehensive Analytical Chemistry; Svehla, G., Ed.; Elsevier: Amsterdam, The Netherlands, 1981; Volume 11. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Coote, M.L. A universal approach for continuum solvent pK a calculations: Are we there yet? Theor. Chem. Acc. 2010, 125, 3–21. [Google Scholar] [CrossRef]
- Ho, J.; Ertem, M.Z. Calculating Free Energy Changes in Continuum Solvation Models. J. Phys. Chem. B 2016, 120, 1319–1329. [Google Scholar] [CrossRef]
- Besora, M.; Vidossich, P.; Lledós, A.; Ujaque, G.; Maseras, F. Calculation of Reaction Free Energies in Solution: A Comparison of Current Approaches. J. Phys. Chem. A 2018, 122, 1392–1399. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | 102·k0 [1·mol−1·s−1] | E [kJ·mol−1] | Log A [1·mol−1·s−1] | Temp. Range | Number of Runs |
|---|---|---|---|---|---|
| (4-Me2N)-1 | 0.78 ± 0.02 | 59.8 ± 2.0 | 9.35 ± 0.35 | 8.3 ÷ 35.0 | 8 |
| (4-MeO)-1 | 2.80 ± 0.08 | 66.8 ± 4.9 | 11.18 ± 0.94 | −6.2 ÷ 10.9 | 6 |
| (4-Me)-1 | 6.04 ± 0.08 | 62.6 ± 1.3 | 10.77 ± 1.82 | −23.9 ÷ 0.0 | 5 |
| (3-Me)-1 | 11.09 ± 0.17 | 60.7 ± 2.7 | 10.62 ± 0.54 | −20.1 ÷ 0.0 | 7 |
| 1 | 16.07 ± 0.46 | 57.1 ± 5.1 | 10.06 ± 0.98 | −27.0 ÷ 0.0 | 8 |
| (4-F)-1 | 14.41 ± 0.27 | 64.5 ± 2.7 | 11.48 ± 0.54 | −22.0 ÷ 0.0 | 5 |
| (4-Cl)-1 | 29.40 ± 0.33 | 64.3 ± 1.8 | 11.79 ± 0.35 | −22.0 ÷ 0.0 | 10 |
| (4-Br)-1 | 39.34 ± 0.80 | 65.2 ± 2.9 | 12.06 ± 0.78 | −20.6 ÷ 0.0 | 5 |
| (4-I)-1 | 40.56 ± 1.19 | 65.2 ± 3.0 | 12.08 ± 0.59 | −20.0 ÷ 0.0 | 7 |
| (3-CF3)-1 | 120.89 ± 4.80 | 65.2 ± 2.9 | 12.52 ± 1.30 | −23.3 ÷ 0.0 | 8 |
| (2-Me)-1 | 12.98 ± 0.46 | 59.5 ± 5.6 | 10.52 ± 1.09 | −20.4 ÷ 1.0 | 5 |
| (2-Et)-1 | 22.02 ± 0.47 | 64.7 ± 3.4 | 11.71 ± 0.69 | −28.5 ÷ 0.0 | 6 |
| (2-iPr)-1 | 13.63 ± 0.68 | 57.4 ± 6.0 | 10.13 ± 1.19 | −24.5 ÷ 0.0 | 6 |
| (2,4-Me2)-1 | 11.32 ± 0.30 | 62.8 ± 2.6 | 11.05 ± 0.33 | −20.0 ÷ 0.0 | 6 |
| (2,5-Me2)-1 | 20.26 ± 0.28 | 61.3 ± 1.2 | 11.02 ± 0.24 | −20.0 ÷ 0.0 | 6 |
| (3,4-Me2)-1 | 6.19 ± 0.05 | 63.5 ± 3.3 | 10.92 ± 0.62 | −6.4 ÷ 14.2 | 5 |
| (2,4,6-Me3)-1 | 74.88 ± 3.19 | 52.9 ± 6.9 | 9.96 ± 1.36 | −20.0 ÷ 0.0 | 7 |
| (2,4,6-Et3)-1 | 45.27 ± 1.05 | 61.4 ± 5.2 | 11.39 ± 1.02 | −20.0 ÷ 0.0 | 7 |
| (2,4,6-iPr3)-1 | 43.22 ± 1.65 | 54.4 ± 3.4 | 10.10 ± 0.67 | −26.7 ÷ 0.0 | 6 |
| (2,3,4,5,6-Me5)-1 | 19.83 ± 0.39 | 67.6 ± 2.7 | 11.90 ± 0.53 | −16.2 ÷ 3.8 | 5 |
| (2-CD3)-1 | 15.08 ± 0.55 | 61.0 ± 5.3 | 10.89 ± 1.05 | −15.1 ÷ 1.0 | 6 |
| (4-CD3)-1 | 6.43 ± 0.21 | 62.5 ± 3.4 | 10.79 ± 0.65 | −20.2 ÷ 1.0 | 5 |
 | 102·k25 [1·mol−1·s−1] | |||
|---|---|---|---|---|
| (4-Me2N)-1 | 7.8 ± 0.1 | 57.3 | −74.3 | 79.5 |
| (4-MeO)-1 | 31.3 ± 2.4 | 64.3 | −39.3 | 76.0 |
| (4-Me)-1 | 67.0 ± 2.1 | 60.1 | −47.0 | 74.1 |
| (3-Me)-1 | 102.0 ± 6.3 | 58.2 | −50.0 | 73.1 |
| 1 | 117.3 ± 2.3 | 54.6 | −60.8 | 72.8 |
| (4-F)-1 | 156.9 ± 0.4 | 62.0 | −33.4 | 72.0 |
| (4-Cl)-1 | 348.9 ± 2.9 | 61.8 | −27.5 | 70.0 |
| (4-Br)-1 | 448.5 ± 99.0 | 62.7 | −22.3 | 69.3 |
| (4-I)-1 | 467.3 ± 34.0 | 62.8 | −22.0 | 69.4 |
| (3-CF3)-1 | 1295.0 ± 209.8 | 62.8 | −13.5 | 66.8 |
| (2-Me)-1 | 128.1 ± 15.4 | 57.0 | −56.7 | 72.5 |
| (2-Et)-1 | 248.3 ± 21.5 | 62.2 | −29.1 | 70.9 |
| (2-iPr)-1 | 124.0 ± 17.7 | 54.9 | −59.3 | 72.6 |
| (2,4-Me2)-1 | 116.7 ± 8.1 | 60.3 | −41.7 | 72.8 |
| (2,5-Me2)-1 | 194.0 ± 6.5 | 58.9 | −42.4 | 71.5 |
| (3,4-Me2)-1 | 64.4 ± 2.6 | 61.0 | −44.3 | 74.2 |
| (2,4,6-Me3)-1 | 517.8 ± 84.6 | 50.4 | −62.6 | 67.1 |
| (2,4,6-Et3)-1 | 446.6 ± 52.2 | 61.4 | −35.2 | 71.9 |
| (2,4,6-iPr3)-1 | 378.4 ± 27.8 | 52.0 | −60.0 | 69.9 |
| (2,3,4,5,6-Me5)-1 | 119.1 ± 7.3 | 65.1 | −25.5 | 72.7 |
| (2-CD3)-1 | 157.6 ± 18.9 | 58.5 | −45.2 | 72.0 |
| (4-CD3)-1 | 71.3 ± 5.1 | 60.0 | −46.8 | 74.0 |
| S-Cl1 [Å] | S-Cl2 [Å] | <θ((ring)-S-Cl) [°] | |
|---|---|---|---|
| 1 | 2.137 (2.129) | - | 90 |
| 1···Cl- (RC) | 2.269 (2.262) | 3.219 (3.075) | 90 |
| 1-Cl- (TS) | 2.564 (2.514) | 2.564 (2.514) | 90 |
| (4-Me)-1 | 2.140 (2.132) | - | 90 |
| (4-Me)-1···Cl- (RC) | 2.262 (2.253) | 3.324 | 90 |
| [(4-Me)-1-Cl]- (TS) | 2.568 (2.514) | 2.574 (2.524) | 90 |
| (2,6-Me2)-1 | 2.152 (2.143) | - | 80.9 |
| (2,6-Me2)-1···Cl- (RC) | 2.285 (2.272) | 3.252 (3.127) | 75.2 |
| [(2,6-Me2)-1-Cl]- (TS) | 2.591 (2.533) | 2.591 (2.533) | 79.8 |
| (2,4,6-Me3)-1 | 2.156 (2.147) | - | 80.8 |
| (2,4,6-Me3)-1···Cl- (RC) | 2.284 (2.267) | 3.319 (3.213) | 74.5 |
| [(2,4,6-Me3)-1-Cl]- (TS) | 2.600 (2.539) | 2.603 (2.542) | 78.6 |
| (2,6-iPr2)-1 | 2.153 (2.142) | - | 81.9 |
| (2,6-iPr2)-1···Cl- (RC) | 2.274 (2.254) | 3.325 (3.260) | 66.6 |
| [(2,6-iPr2)-1-Cl]- (TS) | 2.588 (2.532) | 2.588 (2.532) | 75.1 |
| (2,4,6-iPr3)-1 | 2.156 | - | 83.4 |
| [(2,4,6-iPr3)-1-Cl]- (TS) | 2.594 | 2.594 | 75.1 |
| F5-1 | 2.115 (2.107) | - | 78.8 |
| F5-1···Cl- (RC) | 2.222 | 3.256 | 85.2 |
| [F5-1-Cl]- (TS) | 2.523 (2.475) | 2.523 (2.475) | 90 |
| ΔHcompl [kcal/mol] | ΔG‡ [kcal/mol] | kn/k1 (calc) | kn/k1 (exp)a | |
|---|---|---|---|---|
| 1 | −9.9 (−9.7) | 17.7 (17.4) | 1 (1) | 1 |
| (4-Me)-1 | −8.8 (−8.7) | 18.2 (17.5) | 0.39 (0.97) | 0.5 |
| (2,6-Me2)-1 | −12.6 (−12.5) | 16.6 (16.8) | 6.23 (2.89) | N/A |
| (2,4,6-Me3)-1 | −11.7 (−11.5) | 16.5 (16.7) | 7.17 (3.64) | 4.6 |
| (2,6-iPr2)-1 | −14.5 (−14.4) | 17.3 (16.8) | 1.81 (3.17) | N/A |
| (2,4,6-iPr3)-1 | - | (17.1) | (1.82) | 2.8 |
| F5-1 | −20.0 (−20.5) | 16.2 (16.2) | 12.4 (7.70) | N/A |
| Reaction | ΔH | ΔG |
|---|---|---|
| 5a | 5.2 (6.5) | 8.1 (9.7) |
| 5b | 0.2 (4.2) | 3.4 (7.6) |
| 5c | –1.0 | 0.4 |
| 5d | 0.3 | 1.8 |
| 5e | 6.4 | 9.9 |
| 5f | 2.5 | 7.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikołajczyk, M.; Gajl, M.; Błaszczyk, J.; Cypryk, M.; Gostyński, B. Nucleophilic Substitution at Tetracoordinate Sulfur. Kinetics and Mechanism of the Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides: Counterintuitive Acceleration of Substitution at Sulfonyl Sulfur by ortho-Alkyl Groups and Its Origin. Molecules 2020, 25, 1428. https://doi.org/10.3390/molecules25061428
Mikołajczyk M, Gajl M, Błaszczyk J, Cypryk M, Gostyński B. Nucleophilic Substitution at Tetracoordinate Sulfur. Kinetics and Mechanism of the Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides: Counterintuitive Acceleration of Substitution at Sulfonyl Sulfur by ortho-Alkyl Groups and Its Origin. Molecules. 2020; 25(6):1428. https://doi.org/10.3390/molecules25061428
Chicago/Turabian StyleMikołajczyk, Marian, Monika Gajl, Jarosław Błaszczyk, Marek Cypryk, and Bartłomiej Gostyński. 2020. "Nucleophilic Substitution at Tetracoordinate Sulfur. Kinetics and Mechanism of the Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides: Counterintuitive Acceleration of Substitution at Sulfonyl Sulfur by ortho-Alkyl Groups and Its Origin" Molecules 25, no. 6: 1428. https://doi.org/10.3390/molecules25061428
APA StyleMikołajczyk, M., Gajl, M., Błaszczyk, J., Cypryk, M., & Gostyński, B. (2020). Nucleophilic Substitution at Tetracoordinate Sulfur. Kinetics and Mechanism of the Chloride-Chloride Exchange Reaction in Arenesulfonyl Chlorides: Counterintuitive Acceleration of Substitution at Sulfonyl Sulfur by ortho-Alkyl Groups and Its Origin. Molecules, 25(6), 1428. https://doi.org/10.3390/molecules25061428