Abstract
In diagnostic microbiology, culture media are widely used for detection of pathogenic bacteria. Such media employ various ingredients to optimize detection of specific pathogens such as chromogenic enzyme substrates and selective inhibitors to reduce the presence of commensal bacteria. Despite this, it is rarely possible to inhibit the growth of all commensal bacteria, and thus pathogens can be overgrown and remain undetected. One approach to attempt to remedy this is the use of “suicide substrates” that can target specific bacterial enzymes and selectively inhibit unwanted bacterial species. With the purpose of identifying novel selective inhibitors, six novel phosphonopeptide derivatives based on d/l-fosfalin and β-chloro-l-alanine were synthesized and tested on 19 different strains of clinically relevant bacteria. Several compounds show potential as useful selective agents that could be exploited in the recovery of several bacterial pathogens including Salmonella, Pseudomonas aeruginosa, and Listeria.
1. Introduction
Antimicrobial resistance (AMR) is rapidly becoming a global crisis. According to the Centers for Disease Control and Prevention (CDC), around 2.8 million people are infected with antibiotic-resistant bacteria and approximately 35,000 deaths occur each year in the USA due to these infections [1,2]. Moreover, the O’Neill report estimates that there will be 10 million deaths globally each year due to antibiotic-resistant bacteria by 2050 (O’Neill’s AMR report, 2016). This impending crisis has reinforced the need for new, rapid, economical and user-friendly bacterial detection and identification methods. These tools will prove crucial in the years ahead in order to facilitate antibiotic stewardship, particularly in the selection of the most effective antibiotic to control specific pathogenic bacteria.
Chromogenic and fluorogenic enzymatic substrates are established diagnostic tools used in clinical microbiology for pathogen identification [3,4]. Used alone however, these diagnostic tools face two major problems: i) The presence of abundant commensal bacteria within clinical samples can camouflage the detection of low amounts of pathogenic bacteria, and ii) production of similar enzymes by different strains or species that act upon the same enzymatic substrate can lead to false results. Due to this, incorporation of specific and selective growth inhibitors such as l-alanyl-l-fosfalin 1 (Figure 1) into the growth medium is of clinical interest [5], as such substances may inhibit the growth of interfering microbes while allowing a pathogen of interest to grow unhindered. Once inside bacterial cells, cleavage of the peptide bond of l-alanyl-l-fosfalin occurs, releasing the active substance l-fosfalin 2-L (Figure 1). This in turn binds to alanine racemase (AlaR), a bacterial enzyme that catalyzes the isomerization of l-alanine to D-alanine 3-D (Figure 1) utilizing a pyridoxal 5′-phosphate cofactor [6]. As D-alanine is a crucial building block for bacterial cell wall formation [7], bacterial growth is hindered if the enzyme is inhibited. The expression of l-alanine aminopeptidase for peptide bond cleavage is found more readily in Gram-negative than Gram-positive bacteria [8]. In addition, some bacteria either do not take in the pseudo-dipeptide or it is removed rapidly by efflux pumps [9]. The combination of these factors provides a basis for selective inhibition of bacterial cell growth in culture.
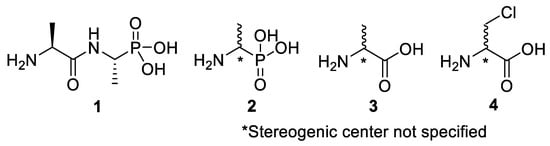
Figure 1.
Structures of l-alanyl-l-fosfalin 1, fosfalin 2, alanine 3, β-chloroalanine 4.
Although fosfalin is a known inhibitor, its use as a single pseudo-amino acid residue is limited due to the multiply ionised forms of fosfalin found at physiologically relevant pH values. This factor reduces its ability to penetrate the cell membrane by passive diffusion. However, dipeptide inhibitors, such as l-alanyl-l-fosfalin, can bypass the cell membrane by hijacking di/tripeptide transporter systems found within the cell membrane [10].
Low minimum inhibitory concentrations (MICs) were observed upon replacing the l-alanine with different hydrophobic l-amino acids, especially l-methionine and l-norvaline at the N-terminus of phosphonodipeptide derivatives containing fosfalin [11,12]. Moreover, selective inhibition against Escherichia coli, Klebsiella species and Enterococcus faecalis has previously been found by replacing the l-alanine at the N-terminus of phosphonotripeptides with sarcosine [13].
Elsewhere, it has been shown that the alanine analogue β-chloroalanine 4 (Figure 1) is known to display irreversible inhibition towards alanine racemase [14]. In studies performed and reviewed by Atherton et al. [11,12,13] and Cheung et al. [15,16] it has been shown that β-chloroalanine exhibits good antibacterial activity alone as well as upon coupling with other peptides or amino acids.
Although many studies have been performed in the past based on fosfalin or β-chloroalanine peptide derivatives, to the best of our knowledge, no studies on the antimicrobial effects of molecules incorporating both of these inhibitors into a single peptide-like derivative have been reported. Herein, a range of phosphonotripeptides based either on a single inhibitor, d/l-fosfalin, or containing dual inhibitors, d/l-fosfalin and β-chloro-l-alanine, coupled to a variety of different N-terminal units in the form of sarcosine, l-norvaline or l-methionine were synthesized. The antibacterial activity of the resulting phosphonotripeptides was evaluated against 19 different strains of clinically relevant bacteria. Although l-fosfalin is most commonly studied version of this particular inhibitory unit, it is costly to produce. This factor may limit its use in the developing nations which lie on the front-line of the battle against emergent drug resistant bacteria. With this in mind, the racemic version of this unit, d/l-fosfalin was utilized in our studies, due to its relatively facile, and thus economical, production.
2. Results and Discussion
2.1. Synthesis of Inhibitors: d/l-Fosfalin and β-Chloro-l-alanine Derivatives
Several synthetic methodologies producing fosfalin are known, including production of its enantiomerically pure forms [17,18,19,20]. Here however, synthesis of racemic fosfalin, i.e., d/l-fosfalin, was utilized due to its more economical synthesis. In our previous unpublished investigations, we have seen that whenever the enantiomerically pure form of fosfalin has been utilized within a peptide sequence the activity is always twice that of the analogous compound containing the racemic version of fosfalin. This is in keeping with the notion that one isomer of fosfalin is active and the other is completely inactive. As the intended use in the present work is to produce economically viable diagnostic tools rather than potent antimicrobial agents, we elected to utilize the racemic version of this warhead within this study. To that end, a good yield of d/l-fosfalin was obtained in one-pot, using N-phenylthiourea 5, acetaldehyde 6 and triphenyl phosphite 7 under acidic conditions (Scheme 1). From there, two different protecting groups, N-trifluoroacetyl and an O-phosphonodiethyl ester group were introduced orthogonally at the N-terminus and O-terminus of racemic fosfalin respectively. Upon removal of N-trifluoroacetyl protecting group 8 by sodium borohydride, d/l-fosfalin diethyl ester 9 was isolated.
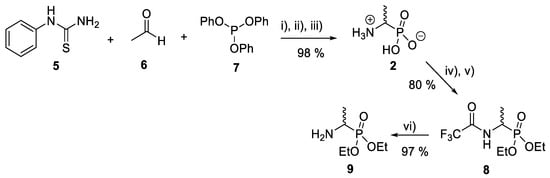
Scheme 1.
Synthesis of d/l-fosfalin diethyl ester. Reagents and conditions: (i) AcOH, reflux, 1 h; (ii) AcOH/HCl (1:10), reflux 24 h; (iii) EtOH; (iv) CF3COOH/(CF3CO)2O (1:5), reflux 1 h; (v) CH(OC2H5)3, reflux 2 h; (vi) NaBH4, reflux 4 h.
Producing the enantiomerically pure β-chloro-l-alanine derivatives, Boc-β-chloro-l-alanine 13 and β-chloro-l-alanine benzyl ester hydrochloride salt 14 was a more economical proposition than the fosfalin unit. Thus, these materials were prepared in their enantiomerically pure forms from an inexpensive and readily available chiral precursor, Boc-l-serine 10. To produce the required molecules, the precursor was subjected to base-catalyzed esterification, followed by chlorination and then chemoselective deprotection (Scheme 2). The corresponding intermediates, Boc-l-Ser-OBzl 11 and Boc-β-Cl-l-Ala-OBzl 12 were isolated in good yield.
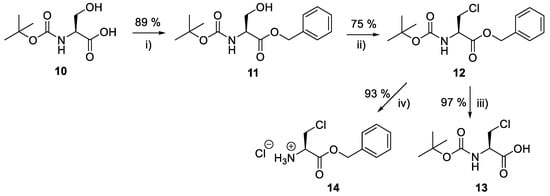
Scheme 2.
Synthesis of β-chloro-l-alanine derivatives, Boc-β-chloro-l-alanine 13 and β-chloro-l-alanine benzyl ester hydrochloride salt 14. Reagents and conditions: (i) DBU, benzyl bromide, dry benzene, r.t., 24 h; (ii) trichloroacetonitrile, PPh3, dry DCM, r.t., 24 h; (iii) MeOH, 10% Pd/C, 3.5 bar H2, r.t., 24 h; (iv) 2M HCl in diethyl ether, r.t., 48 h. Caution: Benzene is a known carcinogen.
2.2. Synthesis of Phosphonotripeptide Derivatives
d/l-fosfalin diethyl ester 9 was coupled to three dipeptides producing a series of tripeptides with a central alanine residue and a variable N-terminal residue (Scheme 3). For this position we chose the amino acids that Atherton et al. [11,12,13] had indicated produced interesting effects in terms of bacterial cell permeation and MIC enhancement, namely sarcosine (Sar), methionine (Met), and norvaline (Nva). In a second series, l-alanine was replaced by β-chloro-l-alanine at the central position thus producing a series that contained two inhibitors within each tripeptide strand. In total, this resulted in six different phosphonotripeptide derivatives, l-Nva-l-Ala-d/l-Fos 21a, Sar-l-Ala-d/l-Fos 21b, l-Met-l-Ala-d/l-Fos 21c, l-Nva-β-chloro-l-Ala-d/l-Fos 25a, Sar-β-chloro-l-Ala-d/l-Fos 25b, l-Met-β-chloro-l-Ala-d/l-Fos 25c. The ratio of diastereoisomers and enantiomers can be found within the Supplementary Materials.
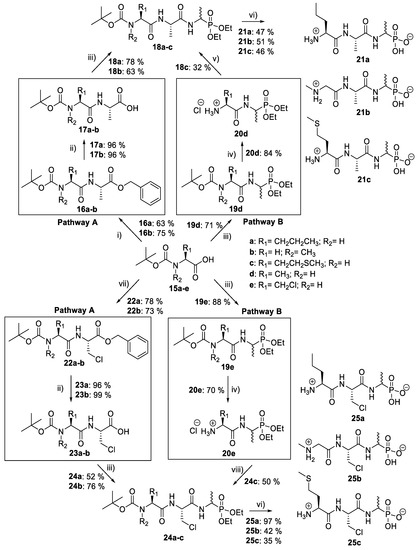
Scheme 3.
Synthesis of phosphonotripeptide derivatives. Reagents and conditions: (i) L-Ala-OBzl, DIPEA, IBCF, NMM, THF/DCM, −5 °C to r.t., 16–18 h; (ii) H2, Pd/C, MeOH, 24 h; (iii) 9, IBCF, NMM, THF, −5 °C to r.t., 16–18 h; (iv) 2 M HCl in diethyl ether, r.t., 48 h; (v) 15c, IBCF, NMM, DIPEA, THF/DCM, −5 °C to r.t., 16–18 h; (vi) HBr, AcOH, 16–18 h then propylene oxide; (vii) 14, IBCF, NMM, THF/DCM, −5 °C to r.t., 16–18 h; (viii) 15c, IBCF, NMM, THF/DCM, −5 °C to r.t., 16–18 h.
A different coupling strategy was utilized where the reactive amino acid methionine, 15c, was employed (Scheme 3, Pathway B). This amino acid displays a sensitive thioether side chain, which is prone to oxidation and cyclisation reactions [21]. Therefore, in order minimize these unwanted side reactions, methionine was introduced at the last stage of the synthesis of tripeptides (20d and 20e).
2.3. Microbiological Evaluation
The inhibitory action of these compounds requires efficient uptake of these molecules across the bacterial cell membrane, hydrolysis of specific peptide bonds by aminopeptidases to release the inhibitor(s), and binding of inhibitor(s) to AlaR. As different amino acids are utilized at the N-terminus of the tripeptide derivatives, differences in uptake may result, and different intracellular aminopeptidases may be required for the liberation of d/l-fosfalin and β-chloro-l-alanine (where incorporated), in order to act on AlaR. The rationale for varying the N-terminal amino acid was that as different species and strains of bacteria produce the transporters and enzymes involved with these processes to differing extents, differences in antibacterial activity were anticipated. The antimicrobial activity of phosphonotripeptide derivatives was evaluated as their minimum inhibitory concentration (MIC), Table 1.

Table 1.
Minimum inhibitory concentration of diastereoisomeric l-Nva-l-Ala-d/l-fosfalin 21a, Sar-l-Ala-d/l-fosfalin 21b, l-Met-l-Ala-l-fosfalin 21c, l-Nva-β-chloro-l-Ala-d/l-fosfalin 25a, Sar-β-chloro-l-Ala-d/l-fosfalin 25b and l-Met-β-chloro-l-Ala-l-fosfalin 25c against pathogenic bacteria.
Broadly speaking, all six phosphonopeptide derivatives produced similar inhibitory profiles to one another against most bacterial species and strains. However, the extent of activity for the collection of inhibitors varied markedly depending on the bacterium in question in a manner that does not appear to be specifically tied to the Gram classification of the organism. For instance, consistently high activity is seen for all inhibitors for two of the Gram-negative species S. marcescens and Y. enterocolitica, with all inhibitors displaying MIC values of 2 mg/L or less. In contrast, no activity against six of the remaining Gram-negative organisms tested was observed at the highest inhibitor concentration tested, which was 8 mg/L. Turning attention to the Gram-positive organisms low or no activity was displayed at the maximum concentrations tested for L. monocytogenes, while very high activity (0.063 mg/L or less) was seen against E. faecalis for all inhibitors tested except for 21b and 25b, which still retained activity, albeit at the 4 mg/L level.
In general, compounds 21b and 25b appear to be the least effective in terms of overall inhibitory profile. In both instances, the N-terminal amino acid is sarcosine, thus the presence of this amino acid in this position of these tripeptide derivatives appears to be detrimental to activity. However, the current data set does not definitively explain whether these poor activity profiles derive from poor uptake, or from slow liberation of the active units once inside the bacterial cell. In many instances these two poorest performing inhibitors have identical MIC values, and when differences do exist they are displayed only to the nearest increment that our tests allow. For example, for S. marcecsens the substance with a simple alanine 21b produces the greater inhibition (1 mg/L) over its β-chloro analogue 25b (2 mg/L). In contrast, for E. coli NCTC 12,241 the reverse behavior is seen with the β-chloro substance 25b exhibiting MIC = 4 mg/L, whereas in this case the simple alanine containing tripeptide 21b produces a weaker MIC of 8 mg/L. Essentially, this indicates that these two poorly performing inhibitors display very similar levels of activity over a wide range of organisms.
With regards to the remaining four inhibitors, there is a subtle yet clear difference in activity for the central β-Cl-Ala series vs. the series of compounds with a standard alanine residue at the central position. Specifically, 25a and 25c are the most consistently active pair of inhibitors, and contain β-Cl-Ala at the center position of each tripeptide. Thus, this indicates that the presence of the β-chloro unit in the center of the tripeptide is indeed beneficial to overall activity. While not definitive, this would indicate that the β-chloro alanine unit is liberated, and becomes active against the bacteria, acting in a synergistic manner with the fosfalin unit, which is common to all six inhibitors.
l-Alanine aminopeptidase is thought to be found more readily in Gram-negative bacteria [8]. However, in our data some inhibitory effects extended to certain Gram positive bacteria, namely E. faecalis, E. faecium, S. epidermidis, S. aureus, and methicillin resistant S. aureus (MRSA). This suggests the likely presence of l-alanine aminopeptidase in these selected Gram-positive bacteria. This finding was consistent with results recently reported by Cellier et al. [23], who demonstrated a chromogenic response upon hydrolysis of l-alanyl aminopeptidase substrates by E. faecalis and E. faecium. The presence of this enzymatic activity in Staphylococcus spp., Streptococcus spp. and E. faecalis has also been reported by Hoosain and Lastovica [24].
As intimated earlier, the eventual purpose of these compounds was to be incorporated into selective culture media for the clinical diagnosis of bacterial infections. Thus, we seek to identify substrates that have a wide range of inhibitory activities but leave specific pathogens of interest free to grow. Looking at our current data set, this suggests a potential application for two of our β-chloro-l-alanyl phosphonotripeptides (25a and 25c) in the selective inhibition of specific Gram-negative bacteria. Specifically, E. cloacae, E. coli, K. pneumoniae, and S. marcescens are commonly found in cystic fibrosis samples [25,26,27]. Our results indicate that these can be selectively inhibited, and thus prevent overgrowth of B. cepacia and P. aeruginosa which often cause severe, and sometimes fatal, lung infections in these patients. A second potential application of our work would be for detection of Salmonella spp. in the clinic. This is due to the relatively weak inhibitory levels (>8 mg/L) displayed by our compounds against these organisms in contrast to the more effective inhibition against the growth of E. coli (<4 mg/L). This is clinically useful as E. coli often overgrow Salmonella spp. when isolated from stool samples [5]. A further potential application of such compounds is the inhibition of Enterococcus spp. facilitating the isolation of Listeria monocytogenes and pathogenic streptococci e.g., Streptococcus pyogenes.
In this paper we present preliminary microbiology data to identify novel compounds that may prove useful as novel selective agents in diagnostic culture media. A major limitation of our data is that only 1 or 2 examples of any particular species have been tested and susceptibility (or resistance) may not be uniformly demonstrated across all strains of a particular species. The utility of these compounds can only be proven in subsequent studies by testing large numbers of strains from each relevant species and large numbers of clinical samples to see if the isolation of target pathogens may be enhanced. However, the fact that such utility as selective agents has been demonstrated for other peptide mimetics based on fosfalin provides encouragement that such agents may ultimately prove useful [5,28].
3. Materials and Methods
All commercially available reagents and solvents were acquired from Sigma-Aldrich, Fluka, Alfa-aesar, Fluorochem, Fischer Scientific and Bachem and used without further purification unless otherwise stated. All deuterated solvents were purchased from Goss Scientific and Apollo Scientific. Solvents were dried using the solvent purification system, PureSolv 400-5-MD, which was purchased from Innovative Technology (currently known as Inert) (Amesbury, MA, USA). Hydrogenation was performed using series 4560 mini reactors, acquired from Parr Instruments (Moline, Il, USA). Column chromatography was performed using Silica 60A (35–70 or 70–200 micron) Davisil® chromatography grade (Merck, Loughborough, UK). Thin layer chromatography was performed on Merck TLC Silica Gel 60 F254 (Loughborough, UK) or Fluka Silca (Dorset, UK) on TLC Alu foils. Melting points were recorded using a Reichart-Kofler (Staffordshire, UK) hot stage microscope apparatus and are uncorrected. Infrared spectra were recorded in the range 4000–550 cm−1 using a Perkin Elmer Spectrum BX FT-IR spectrophotometer (Waltham, MA, USA). NMR spectra were acquired using a Bruker Ultrashield 300 spectrometer (Billerica, MA, USA) at 300 MHz for 1H spectra, 75 MHz for 13C spectra, 121 MHz for 31P-1H decoupled spectra and 282 MHz for 19F-1H decoupled spectra. NMR data was analyzed by MestReNova (Mestrelab Research, Santiago de Compostela, Spain). Low-resolution mass spectra were obtained from a Bruker Esquire 3000plus analyzer (Billerica, MA, USA) using an electrospray source in either positive or negative ion mode. High-resolution mass spectra were acquired from Thermo Scientific LTQ Orbitrap XL (Waltham, MA, USA) using an electrospray source in either positive or negative ion mode. Elemental analyses were performed using an Exeter Analytical CE-440 Elemental Analyzer (Coventry, UK). The LC column, ACE Excel 5 Super C18 (150 × 4.6 mm) and Eclipse Plus C18 (50 × 2.1 mm) were acquired from HICHROM (Reading, UK) and Agilent (Waldbronn, Germany), respectively. Elemental and purity by LC-MS were performed on final compounds or compounds which analytical data are not available in literature.
3.1. Synthetic Procedures
3.1.1. General IBCF/NMM Peptide Coupling Method
In 100 mL one neck round-bottomed flask A, Cα-protected amine salt (1.0 equiv) or phosphonate ester protected amine salt (when l-Met was used) was dissolved in dry DCM or dry THF (25–50 mL) and cooled to −5 °C in an ice/brine bath. To this was added N,N-diisopropylethylamine, DIPEA (1.5 equiv) or N-methyl morpholine, NMM (1.0 equiv) (when β-Cl-l-Ala was used). In 250 mL one neck round-bottomed flask B, Nα-protected amino acid (1.0 equiv) was dissolved in dry THF (25–50 mL) and to this was added N-methyl morpholine, NMM (1.5 equiv or 1.0 equiv when β-Cl-l-Ala moiety was involved) and isobutyl chloroformate (1.0 equiv) while stirring at −5 °C. The Flask B solution was stirred for 2–3 h at −5 °C, after which time the neutralized Cα-protected or phosphonate ester protected amine solution in Flask A was then added. The resulting mixture was stirred at −5 °C for 10mins and then room temperature overnight (16–18 h). Work-up procedure: the mixture was filtered and filtrate was concentrated in vacuo to afford a crude product, which was dissolved in DCM and washed with 10 %w/v citric acid (2 × 30 mL), followed by 10 %w/v potassium carbonate (30 mL) and finally water (30 mL). The organic layer was dried over magnesium sulfate, filtered, concentrated in vacuo, and purified by column chromatography using an appropriate solvent system to afford the product of interest.
3.1.2. Removal of Benzyl Ester Protecting Group
Cα-Benzyl ester amino acid or peptide was dissolved in methanol and to this was added 5%–10% palladium on carbon (10 mol %). The mixture was stirred at 3.5 bar pressure of hydrogen at room temperature overnight (16–18 h). The catalyst was filtered using Celite and the filtrate was concentrated in vacuo to afford the product of interest.
3.1.3. Removal of N-tert-Butoxycarbonyl Protecting Group
N-tBoc amino acid or peptide was dissolved in excess of a solution of dry 2M HCl in diethyl ether. The resulting solution was stirred at room temperature for 2 days to afford a crude solid, which was collected by filtration and washed with excess dry diethyl ether to afford the product of interest.
3.1.4. Removal of tert-Butoxycarbonyl and Diethyl Phosphonate Ester Protecting Groups
Phosphonopeptide containing N-tert-butoxycarbonyl and O,O-diethyl phosphonate ester was dissolved in 35 %wt HBr in acetic acid (10 equiv) and stirred at room temperature overnight (16–18 h). Dry diethyl ether was added to afford a precipitation. The resulting mixture was stored in the freezer overnight (16–18 h) to result in more precipitation. The solvent was decanted and remaining crude solid was triturated (×3) with dry diethyl ether. The resulting solid was dissolved in dry methanol and propylene oxide was added in excess to afford a crude precipitation, which was recrystallized from the appropriate solvent system to afford the product of interest.

1-Aminoethylphosphonic Acid or d/l-fosfalin (2-DL). To suspension of N-phenylthiourea (100.0 mmol, 15.2 g) in glacial acetic acid (50 mL), acetaldehyde (130.0 mmol, 7.40 mL) was added dropwise, followed by triphenyl phosphite (100.0 mmol, 27.0 mL). The mixture was stirred at room temperature for 5 mins, then refluxed at 80 °C for 1 h until a clear solution was obtained. A mixture of glacial acetic acid (5 mL) and hydrochloric acid (37%, 50 mL) was added and the reaction was refluxed overnight. The solution was cooled to room temperature and concentrated in vacuo to afford a brown slurry. Absolute ethanol (150 mL) was added while stirring and the resulting off-white solid was collected by filtration and dried in a desiccator containing phosphorus(V) oxide. The crude solid was recrystallized from hot water/ethanol to afford 2-DL as white crystals, as a mixture of enantiomers (12.2 g, 98 mmol, 98%); m.p. 271–274 °C (sublim); ῡmax/cm−1 2910 (br OH), 1532 (NH bend), 1143 (P=O), 1035 (P-O-C), 930 (P-OH); 1H NMR (300 MHz, D2O) δH 1.40 (3H, dd, 3JH-P = 14.7 Hz, 3JH-H = 7.2 Hz, CH3), 3.33 (1H, m, CH); 13C NMR (75 MHz, D2O) δC 13.5 (d, 2JC-P = 2.6 Hz, CH3), 44.7 (d, 1JC-P = 144.2 Hz, CH); 31P-1Hdecoup NMR (121 MHz, D2O) δP 14.2; m/z (ESI) calcd for (C2H9NO3P)+, MH+: 126.0, found 126.1; CHN (Found: C, 19.45; H, 6.48; N, 11.18. C2H8NO3P requires C, 19.21; H, 6.45; N, 11.20%). (The 1H- and 13C-NMR spectra may be found within the Supplementary Materials).

Diethyl (1-(2,2,2-trifluoroacetamido)ethyl)phosphonate or trifluoroacetyl-d/l-Fos diethyl ester (8). 1-Aminoethylphosphonic acid (2-DL) (51.7 mmol, 6.5 g) was added to a mixture of trifluoroacetic acid (65.3 mmol, 5 mL) and trifluoroacetic anhydride (177.4 mmol, 25 mL). The solution was stirred and refluxed at 60 °C for 1 h, then cooled to room temperature and triethyl orthoformate (901.8 mmol, 150 mL) was added dropwise. The solution was refluxed at 110 °C for 2 h, then cooled to room temperature. The solution was concentrated in vacuo to afford a brown solid, which was re-dissolved in DCM and purified by column chromatography using a gradient elution (DCM (100) to DCM/MeOH (95:5)) to give 8 as an off-white solid, a mixture of enantiomers (11.4 g, 41.0 mmol, 80%); m.p. 101–103 °C (sublim) (lit. m.p. 101–102 °C) [29]; ῡmax/cm−1 3202 (NH), 1715 (C=O), 1565 (NH bend), 1210 (P=O), 1011 (P-O-C), 968 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.24 (3H, t, 3JH-H = 7.2 Hz, OCH2CH3), 1.27 (3H, t, 3JH-H = 7.2 Hz, OCH2CH3), 1.38 (3H, dd, 3JH-P = 16.5 Hz, 3JH-H = 7.2 Hz, CH3-2), 4.06 (4H, m, 2 × OCH2CH3), 4.39 (1H, m, CH-1), 8.00 (1H, d, 3JH-H = 6.0 Hz, NH); 13C NMR (75 MHz, CDCl3) δC 14.8 (CH3-2), 16.2 (d, 3JC-P = 2.3 Hz, OCH2CH3), 16.3 (d, 3JC-P = 2.3 Hz, OCH2CH3), 41.8 (d, 1JC-P = 159.1 Hz, CH-1), 62.8 (d, 2JC-P = 7.0 Hz, OCH2CH3), 63.2 (d, 2JC-P = 7.1 Hz, OCH2CH3), 115.9 (q, 1JC-F = 285.8 Hz, CF3), 156.9 (q, 2JC-F = 5.8 Hz, C=O); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 23.0; 19F-1Hdecoup NMR (282 MHz, CDCl3) δP −75.5; m/z (ESI) calcd for (C8H16F3NO4P)+, MH+: 278.1, found 278.1.

Diethyl 1-aminoethylphosphonate or d/l-Fos diethyl ester (9). Diethyl (1-(2,2,2-trifluoroacetamido)ethyl)phosphonate (8) (20.0 mmol, 5.6 g) was dissolved in ethanol (200 ml) and excess sodium borohydride (200.0 mmol, 7.7 g) was added slowly with stirring. The resulting mixture was stirred at room temperature for 1 h, then heated at reflux for 4 h. The mixture was cooled to room temperature and the solvent was removed in vacuo to afford a white solid, which was dissolved in saturated NaHCO3 (96 g/L) (60 mL) with the addition of 10% aqueous K2CO3 (20 mL). The product was extracted into DCM (6 × 30 mL) and dried over MgSO4. The filtrate was concentrated in vacuo to afford a pale yellow liquid and purified by column chromatography using a gradient elution (DCM (100) to DCM/MeOH (90:10)) to afford 9 as a yellow liquid, a mixture of enantiomers (3.5 g, 19.3 mmol, 97%); ῡmax/cm−1 3431 (NH), 1215 ( P=O), 1020 (P-O-C), 967 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.26 (6H, t, 3JH-H = 7.2 Hz, 2 × OCH2CH3), 1.34 (3H, dd, 3JH-P = 17.7 Hz, 3JH-H = 7.2 Hz, CH3-2), 1.68 (2H, br, NH2), 3.02–3.12 (1H, m, CH-1), 4.06–4.17 (4H, m, 2 × OCH2CH3); 13C NMR (75 MHz, CDCl3) δC 16.4 (OCH2CH3), 16.5 (OCH2CH3), 17.2 (CH3-2), 44.2 (d, 1JC-P = 148.5 Hz, CH-1), 62.1 (d, 2JC-P = 7.5 Hz, OCH2CH3), 62.1 (d, 2JC-P = 7.5 Hz, OCH2CH3); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 29.6; HRMS (NSI) calcd for (C6H17NO3P)+, MH+: 204.0760, found 204.0762. LCMS purity >95% (C-18 reversed phase, MeOH–H2O).

(S)-Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido) propanoate or Boc-l-Nva-l-Ala-OBzl (16a). General peptide coupling method was followed, using Boc-l-Nva-OH (15a) (10.0 mmol, 2.17 g) in dry THF and l-alanine benzyl ester p-tosylic acid (10.0 mmol, 3.52 g) in dry DCM. The yellow crude liquid was purified by column chromatography (40–60 petrol/ethyl acetate (7:3)) to give 16a as an off-white solid (2.40 g, 6.3 mmol, 63%); m.p. 60–63 °C; ῡmax/cm−1 3332 (NH), 1743 (C=O), 1655 (br C=O), 1527 (NH bend), 1245 (C-O), 1162 (C-O); 1H NMR (300 MHz, CDCl3) δH 0.83 (3H, t, 3JH-H = 9.0 Hz, CH3-7″), 1.25–1.31 (2H, m, CH2-7′), 1.34 (3H, d, 3JH-H = 6.0 Hz, CH3-3), 1.36 (9H, s, C(CH3)3), 1.42–1.54 (1H, m, CHa/b-7), 1.64–1.73 (1H, m, CHa/b-7), 4.02 (1H, m, CH-6), 4.54 (1H, pentet, 3JH-H = 6.0 Hz, CH-2), 4.96 (1H, d, 3JH-H = 9.0 Hz, NH-8), 5.07 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 5.12 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 6.56 (1H, d, 3JH-H = 6.0 Hz, NH-4), 7.27 (5H, m, 5 × CHAr); 13C NMR (75 MHz, CDCl3) δC 12.7 (CH3-7″), 17.3 (CH3-3), 17.8 (CH2-7′), 27.3 (C(CH3)3), 33.7 (CH2-7), 47.1 (CH-2), 53.4 (CH-6), 66.1(OCH2Ar), 79.0 (C(CH3)3), 127.1-127.6 (CHAr), 134.3 (CHAr quat.), 154.6 (C=O-9), 170.8 (C=O-5), 171.5 (C=O-1); HRMS (NSI) calcd for (C20H31N2O5)+, MH+: 379.2227, found 379.2222; CHN (Found: C, 63.75; H, 8.37; N, 7.86. C20H30N2O5 requires C, 63.47; H, 7.99; N, 7.40%).

(S)-Benzyl 2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido) propanoate or Boc-Sar-l-Ala-OBzl (16b). General peptide coupling method was followed, using Boc-Sar-OH (15b) (15.0 mmol, 2.84 g) in dry THF and l-alanine benzyl ester p-tosylic acid (15.0 mmol, 5.27 g) in dry DCM. The yellow crude liquid was purified by column chromatography (40–60petrol/ethyl acetate (1:1)) to give 16b as a colourless liquid (3.93 g, 11 mmol, 75%); ῡmax/cm−1 3311 (NH), 1742 (C=O), 1670 (C=O), 1666 (C=O), 1536 (NH bend), 1242 (C-O), 1145 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.35 (3H, t, 3JH-H = 6.0 Hz, CH3-3), 1.39 (9H, s, C(CH3)3), 2.85 (3H, s, CH3-8), 3.72 (1H, d, 2JH-H = 15.0 Hz, CHa/b-6), 3.88 (1H, d, 2JH-H = 15.0 Hz, CHa/b-6), 4.58 (1H, pentet, 3JH-H = 6.0 Hz, CH-2), 5.08 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 5.13 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 6.51 (1H, br, NH-4), 7.25-7.29 (5H, m, 5 × CHAr); 13C NMR (75 MHz, CDCl3) δC 17.5 (CH3-3), 27.3 (C(CH3)3), 34.7 (CH3-8), 47.0 (CH-2), 52.1 (CH2-6), 66.2 (OCH2Ar), 79.8 (C(CH3)3), 127.1–127.6 (CHAr), 134.3 (CHAr quat.), 155.0 (C=O-9), 167.9 (C=O-5), 171.5 (C=O-1); HRMS (NSI) calcd for (C18H27N2O5)+, MH+: 351.1914, found 351.1916. LCMS purity >95% (C-18 reversed phase, MeOH–H2O).

Tert-butyl ((2S)-1-((-1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)carbamate or Boc-l-Ala-d/l-Fos diethyl ester (19d). General peptide coupling method was followed, using Boc-l-Ala-OH (15d) (10.0 mmol, 1.90 g) in dry THF and diethyl 1-aminoethylphosphonate (9) (10.0 mmol, 1.84 g) in dry THF. The pale yellow crude syrup was purified by column chromatography, using 100% DCM and increasing to 95:5 DCM/methanol, to afford 19d as an off-white solid composed of 2 diastereoisomers, Boc-l-Ala-l-Fos diethyl ester and Boc-l-Ala-D-Fos diethyl ester (2.49 g, 7.1 mmol, 71%); m.p. 102–105 °C; ῡmax/cm−1 3280 (NH), 1710 (C=O), 1652 (C=O), 1556 (NH bend), 1229 (P=O), 1173 (C-O), 1013 (P-O-C), 973 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.23–1.43 (12H, m, CH3-2, CH3-6, 2 × OCH2CH3), 1.44 (9H, s, C(CH3)3), 4.06–4.23 (5H, m, 2 × OCH2CH3, CH-5), 4.40–4.52 (1H, m, CH-1), 5.12 (0.5H, d, 3JH-H = 1.5 Hz, NH-7), 5.14 (0.5H, d, 3JH-H = 1.5 Hz, NH-7), 6.72 (0.5H, d, 3JH-H = 2.3 Hz, NH-3), 6.74 (0.5H, d, 3JH-H = 2.3 Hz, NH-3); 13C NMR (75 MHz, CDCl3) δC 15.6 (CH3-2), 16.3 (d, 3JC-P = 3.0 Hz, OCH2CH3), 16.4 (d, 3JC-P = 2.3 Hz, OCH2CH3), 16.5 (d, 3JC-P = 3.0 Hz, OCH2CH3), 16.6 (d, 3JC-P = 2.3 Hz, OCH2CH3), 18.4 (CH3-6), 28.3 (C(CH3)3), 40.8 (d, 1JC-P = 156.8 Hz, CH-1), 41.0 (d, 1JC-P = 156.8 Hz, CH-1), 50.0 (CH-5), 62.4 (d, 2JC-P = 6.8 Hz, OCH2CH3), 62.5 (d, 2JC-P = 6.8 Hz, OCH2CH3), 62.6 (d, 2JC-P = 6.8 Hz, OCH2CH3), 62.8 (d, 2JC-P = 6.8 Hz, OCH2CH3), 80.0 (C(CH3)3), 155.2 (C=O-8), 172.1 (C=O-4); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 25.2; HRMS (NSI) calcd for (C17H35N3O7P)+, MH+: 424.2207, found 424.2200; CHN (Found: C, 48.22; H, 8.58; N, 7.87. C14H29N2O6P requires C, 47.92; H, 8.30; N, 7.95%).

(S)-2-((S)-2-((Tert-butoxycarbonyl)amino)pentanamido) propanoic acid or Boc-l-Nva-l-Ala-OH (17a). Deprotection of benzyl ester was followed, using (S)-benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido)propanoate (16a) (6.0 mmol, 2.27 g) to afford 17a as a white solid (1.66 g, 5.7 mmol, 96.0%); m.p. 55–58 °C (decomp.); ῡmax/cm−1 3500–3000 (br, OH), 3300 (NH), 1688 (br C=O), 1655 (C=O), 1522 (NH bend), 1245 (C-O), 1164 (C-O); 1H NMR (300 MHz, CDCl3) δH 0.85 (3H, t, 3JH-H = 9.0 Hz, CH3-7″), 1.27–1.31 (5H, m, CH3-3, CH2-7′), 1.39 (9H, s, C(CH3)3), 1.48–1.53 (1H, m, CHa/b-7), 1.67–1.71 (1H, m, CHa/b-7), 4.10 (1H, m, CH-6), 4.50 (1H, m, CH-2), 5.27 (1H, m, NH-8), 6.93 (1H, m, NH-4), 8.87 (1H, br, OH); 13C NMR (75 MHz, CDCl3) δC 13.7 (CH3-7″), 18.0 (CH3-3), 18.8 (CH2-7′), 28.3 (C(CH3)3), 34.5 (CH2-7), 48.1 (CH-2), 54.3 (CH-6), 80.4 (C(CH3)3), 156.0 (C=O-9), 172.5 (C=O-5), 175.5 (C=O-1); HRMS (NSI) calcd for (C13H25N2O5)+, MH+: 289.1758, found 289.1758; CHN (Found: C, 54.18; H, 8.78; N, 9.62. C13H24N2O5 requires C, 54.15; H, 8.39; N, 9.72%).

(S)-Benzyl 2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido) propanoic acid or Boc-Sar-l-Ala-OH (17b). Deprotection of benzyl ester was followed, using (S)-benzyl 2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido) propanoate (16b) (10.0 mmol, 3.51 g) to afford 17b as a colorless syrup (2.50 g, 9.6 mmol, 96%); ῡmax/cm−1 3301 (NH), 2961 (broad OH), 1736 (C=O), 1664 (br C=O), 1542 (NH bend), 1241 (C-O), 1147 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.36 (3H, t, 3JH-H = 6.0 Hz, CH3-3), 1.39 (9H, s, C(CH3)3), 2.89 (3H, s, CH3-8), 3.72 (1H, d, 2JH-H = 18.0 Hz, CHa/b-6), 3.98 (1H, d, 2JH-H = 18.0 Hz, CHa/b-6), 4.57 (1H, m, CH-2), 6.96 (1H, m, NH-4), 7.26 (1H, br, OH); 13C NMR (75 MHz, CDCl3) δC 17.2 (CH3-3), 27.3 (C(CH3)3), 46.8 (CH-2), 49.6 (CH3-8), 52.1 (CH2-6), 80.6 (C(CH3)3), 155.5 (C=O-9), 168.3 (C=O-5), 174.1 (C=O-1); HRMS (NSI) calcd for (C11H19N2O5)-, MH−: 259.1299, found 259.1295. LCMS purity >95% (C-18 reversed phase, MeOH-H2O).

(2S)-1-((1-(Diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-aminium chloride or l-Ala-d/l-Fos diethyl ester hydrochloride (20d). Deprotection of tert-butoxycarbonyl was followed, using tert-butyl ((2S)-1-((-1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)carbamate (19d) (6.0 mmol, 2.13 g). The off-white hygroscopic crude solid was washed with petrol to afford 20d as a pale green solid composed of 2 diastereoisomers, l-Ala-l-Fos diethyl ester hydrochloride and l-Ala-D-Fos diethyl ester hydrochloride (1.46 g, 5.1 mmol, 84%); m.p. 60–63 °C; ῡmax/cm−1 2986 (NH+), 1673 (C=O), 1555 (NH bend), 1212(P=O), 1017 (P-O-C), 970 (P-O-C); 1H NMR (300 MHz, CD3OD) δH 1.29-1.44 (9H, m, 2 × OCH2CH3, CH3-2), 1.51 (3H, d, 3JH-H = 6.0 Hz, CH3-6), 3.90-3.98 (1H, m, CH-5), 4.08-4.22 (4H, m, 2 × OCH2CH3), 4.28-4.47 (1H, m, CH-1); 13C NMR (75 MHz, CD3OD) δC 13.7 (CH3-2), 14.0 (CH3-2), 15.4 (2 × OCH2CH3), 16.3 (CH3-6), 41.1 (d, 1JC-P = 158.3 Hz, CH-1), 41.4 (d, 1JC-P = 158.3 Hz, CH-1), 48.8 (CH-5), 48.9 (CH-5), 62.7–63.0 (2 × OCH2CH3), 169.0 (C=O-4); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 29.0, 29.1; HRMS (NSI) calcd for (C9H22N2O4P)+, M+: 253.1312, found 253.1316. LCMS purity >95% (C-18 reversed phase, MeOH-H2O).

Tert-butyl ((2S)-1-(((2S)-1-((1-(diethoxyphosphoryl)ethyl) amino)-1-oxopropan-2-yl)amino)-1-oxopentan-2-yl)carbamate or Boc-l-Nva-l-Ala-d/l-Fos diethyl ester (18a). General peptide coupling method was followed, using (S)-2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido)propanoic acid (17a) (5.0 mmol, 1.45 g) in dry THF and diethyl 1-aminoethylphosphonate (9) (4.8 mmol, 0.87 g) in dry THF. The white crude solid was purified by column chromatography using 100% DCM, increasing to 90:10 DCM/methanol, to afford 18a as a white solid composed of 2 diastereoisomers, Boc-l-Nva-l-Ala-l-Fos diethyl ester and Boc-l-Nva-l-Ala-D-Fos diethyl ester (1.70 g, 3.8 mmol, 78%); m.p. 165–168 °C; ῡmax/cm−1 3267 (NH), 1708 (C=O), 1638 (br C=O), 1537 (NH bend), 1227 (P=O), 1165 (C-O), 1019 (P-O-C), 966 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 0.85 (3H, t, 3JH-H = 9.0 Hz, CH3-10″), 1.18–1.34 (14H, m, 2 × OCH2CH3, CH3-2, CH3-6, CH2-10′), 1.37 (9H, s, C(CH3)3), 1.47–1.54 (1H, m, CHa/b-10), 1.65–1.73 (1H, m, CHa/b-10), 3.98–4.12 (5H, m, 2 × OCH2CH3, CH-9), 4.33–4.44 (1H, m, CH-1), 4.48–4.54 (1H, m, CH-5), 5.19 (0.5H, d, 3JH-H = 6.0 Hz, NH-11), 5.23 (0.5H, d, 3JH-H = 6.0 Hz, NH-11), 6.78 (0.5H, d, 3JH-H = 6.0 Hz, NH-7), 6.87 (0.5H, d, 3JH-H = 6.0 Hz, NH-7), 7.15 (0.5H, d, 3JH-H = 9.0 Hz, NH-3), 7.23 (0.5H, d, 3JH-H = 9.0 Hz, NH-3); 13C NMR (75 MHz, CDCl3) δC 12.7 (CH3-10″), 14.4 (CH3-2), 14.5 (CH3-2), 15.3 (OCH2CH3), 15.4 (OCH2CH3), 15.5 (OCH2CH3), 15.6 (OCH2CH3), 17.6 (CH3-6), 17.7 (CH3-6), 17.8 (CH2-10′), 17.9 (CH2-10′), 27.3 (C(CH3)3), 33.8 (CH2-10), 33.9 (CH2-10), 39.9 (d, 1JP-C = 157.5 Hz, CH-1), 40.0 (d, 1JP-C = 156.8 Hz, CH-1), 47.7 (CH-5), 47.9 (CH-5), 53.5 (CH-9), 53.5 (CH-9), 61.5 (d, 2JC-P = 7.5 Hz, OCH2CH3), 61.6 (d, 2JC-P = 7.5 Hz, OCH2CH3), 61.7 (d, 2JC-P = 7.5 Hz, OCH2CH3), 61.9 (d, 2JC-P = 7.5 Hz, OCH2CH3), 78.9 (C(CH3)3), 154.7 (C=O-12), 170.6 (C=O-4 or C=O-8), 170.7 (C=O-4 or C=O-8), 171.0 (C=O-4 or C=O-8), 171.1 (C=O-4 or C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 25.0, 25.1; HRMS (NSI) calcd for (C19H39N3O7P)+, MH+: 452.2520, found 452.2518; CHN (Found: C, 50.74; H, 8.55; N, 9.51. C19H38N3O7P requires C, 50.54; H, 8.48; N, 9.31%).

Tert-butyl (2-(((2S)-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)amino)-2-oxoethyl)(methyl)carbamate or Boc-Sar-l-Ala-d/l-Fos (18b). General peptide coupling method was followed, using (S)-benzyl 2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido)propanoic acid (17b) (6.0 mmol, 1.57 g) in dry THF and diethyl 1-aminoethylphosphonate (9) (6.0 mmol, 1.10 g) in dry THF. The yellow crude liquid was purified by column chromatography, using 100% DCM and increasing to 90:10 DCM/methanol, to afford 18b as a colorless liquid composed of 2 diastereoisomers, Boc-Sar-l-Ala-l-Fos diethyl ester and Boc-Sar-l-Ala-D-Fos diethyl ester (1.60 g, 3.8 mmol, 63%); ῡmax/cm−1 3270 (NH), 1700 (br C=O), 1655 (C=O), 1545 (NH bend), 1225 (P=O), 1149 (C-O), 1018 (P-O-C), 966 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.19–1.34 (12H, m, CH3-2, CH3-6, 2 × OCH2CH3), 1.40 (9H, s, C(CH3)3), 2.87 (3H, s, CH3-11), 3.72 (0.5H, d, 2JH-H = 15.0 Hz, CHa/b-9), 3.78 (0.5H, d, 2JH-H = 15.0 Hz, CHa/b-9), 3.81 (0.5H, d, 2JH-H = 15.0 Hz, CHa/b-9), 3.87 (0.5H, d, 2JH-H = 15.0 Hz, CHa/b-9), 4.00–4.11 (4H, m, 2 × OCH2CH3), 4.35-4.43 (1H, m, CH-1), 4.47–4.52 (1H, m, CH-5), 6.67 (1H, d, 3JH-H = 9.0 Hz, NH-7), 6.98 (0.5H, d, 3JH-H = 9.0 Hz, NH-3), 7.15 (0.5H, d, 3JH-H = 9.0 Hz, NH-3); 13C NMR (75 MHz, CDCl3) δC 15.5 (CH3-2), 15.5 (CH3-2), 16.3 (d, 3JP-C = 3.0 Hz, OCH2CH3), 16.4 (d, 3JP-C = 3.0 Hz, OCH2CH3), 18.7 (CH3-6), 28.3 (C(CH3)3), 35.8 (CH3-11), 41.0 (d, 1JP-C = 157.5 Hz, CH-1), 48.5 (CH-5), 53.0 (CH2-9), 62.5 (d, 2JP-C = 6.8 Hz, OCH2CH3), 62.6 (d, 2JP-C = 6.8 Hz, OCH2CH3), 62.7 (d, 2JP-C = 6.8 Hz, OCH2CH3), 62.9 (d, 2JP-C = 6.8 Hz, OCH2CH3), 80.7 (C(CH3)3), 156.0 (C=O-12), 171.5 (C=O-4 or C=O-8), 171.6 (C=O-4 or C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 25.0, 25.1; HRMS (NSI) calcd for (C17H35N3O7P)+, MH+: 424.2207, found 424.2203. LCMS purity >95% (C-18 reversed phase, MeOH–H2O).

Tert-butyl ((2S)-1-(((2S)-1-((1-(diethoxyphosphoryl)ethyl) amino)-1-oxopropan-2-yl)amino-4-(methylthio)-1-oxobutan-2-yl)carbamate or Boc-l-Met-l-Ala-d/l-Fos diethyl ester (18c). General peptide coupling method was followed, using Boc-l-Ala-Met-OH (15c) (3.4 mmol, 0.88 g) in dry THF and (2S)-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-aminium chloride (20d) (3.4 mmol, 0.97 g) in dry DCM. The yellow crude solid was purified by column chromatography (DCM/MeOH (95:5)) to give 18c as an off-white solid composed of 2 diastereoisomers, Boc-l-Met-l-Ala-l-Fos diethyl ester and Boc-l-Met-l-Ala-D-Fos diethyl ester (0.53 g, 1.1 mmol, 32%); m.p. 172–176 °C; ῡmax/cm−1 3272 (NH), 1708 (C=O), 1673 (C=O), 1637 (C=O), 1530 (NH bend), 1226 (P=O), 1165 (C-O), 1020 (P-O-C), 976 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.16–1.36 (12H, m, CH3-2, CH3-6, 2 × OCH2CH3), 1.36 (9H, s, C(CH3)3), 1.82–2.01 (2H, m, CH2-10), 2.04 (3H, s, CH3-10″), 2.49 (2H, dd, 3JH-H = 9.0 Hz, 3.0 Hz, CH2-10′), 4.00–4.12 (4H, m, 2 × OCH2CH3), 4.16–4.26 (1H, m, CH-9), 4.33–4.43 (1H, m, CH-1), 4.45–4.53 (1H, m, CH-5), 5.40 (0.5H, d, 3JH-H = 9.0 Hz, NH-11), 5.44 (0.5H, d, 3JH-H = 6.0 Hz, NH-11), 6.85 (0.5H, d, 3JH-H = 6.0 Hz, NH-7), 6.92 (0.5H, d, 3JH-H = 6.0 Hz, NH-7), 7.07 (0.5H, d, 3JH-H = 9.0 Hz, NH-3), 7.16 (0.5H, d, 3JH-H = 9.0 Hz, NH-3); 13C NMR (75 MHz, CDCl3) δC 14.2 (CH3-2), 14.3 (CH3-2), 14.5 (CH3-10″), 14.6 (CH3-10″), 15.4 (d, 3JC-P = 3.0 Hz, OCH2CH3), 15.4 (d, 3JC-P = 2.3 Hz, OCH2CH3), 15.5 (d, 3JC-P = 3.0 Hz, OCH2CH3), 15.5 (d, 3JC-P = 2.3 Hz, OCH2CH3), 17.7 (CH3-6), 27.3 (C(CH3)3), 29.2 (CH2-10′), 29.3 (CH2-10′), 30.8 (CH2-10), 30.9 (CH2-10), 39.9 (d, 1JC-P = 156.8 Hz, CH-1), 40.0 (d, 1JC-P = 156.8 Hz, CH-1), 47.9 (CH-5), 48.0 (CH-5), 52.6 (CH-9), 61.5 (d, 2JC-P = 6.8 Hz, OCH2CH3), 61.6 (d, 2JC-P = 6.8 Hz, OCH2CH3), 61.7 (d, 2JC-P = 6.8 Hz, OCH2CH3), 61.9 (d, 2JC-P = 6.8 Hz, OCH2CH3), 79.1 (C(CH3)3), 154.6 (C=O-12), 170.3 (C=O-4 or C=O-8), 170.4 (C=O-4 or C=O-8), 170.5 (C=O-4 or C=O-8), 170.6 (C=O-4 or C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 25.0, 25.1; HRMS (NSI) calcd for (C19H39N3O7PS)+, MH+: 484.2241, found 484.2228. LCMS purity >95% (C-18 reversed phase, MeOH–H2O).

(1-((S)-2-((S)-2-Aminopentanamido)propanamido)ethyl) phosphonic acid or l-Nva-l-Ala-d/l-Fos (21a). The tert-butoxycarbonyl and diethyl ester protecting groups of tert-butyl ((2S)-1-(((2S)-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)amino)-1-oxopentan-2-yl)carbamate (18a) (1.6 mmol, 0.72 g) were removed. The pale green crude solid was recrystallised from hot water/acetone to give 21a as a pale green solid composed of 2 diastereoisomers, l-Nva-l-Ala-l-Fos and l-Nva-l-Ala-D-Fos (0.22 g, 0.75 mmol, 47%); m.p. 207–210 °C (decomp.); ῡmax/cm−1 3280 (NH+), 3500-2900 (br OH), 1643 (br C=O), 1552 (NH bend), 1149 (P=O), 1037 (P-O-C), 922 (P-OH); 1H NMR (300 MHz, D2O) δH 0.96 (3H, t, 3JH-H = 7.1 Hz, CH3-10″), 1.27-1.32 (3H, d, 3JH-H = 6.8 Hz, CH3-2), 1.40-1.42 (3H, m, CH3-6), 1.40–1.42 (2H, m, CH2-10′), 1.88–1.86 (2H, m, CH2-10), 4.00–4.02 (2H, m, CH-1, CH-9), 4.34–4.39 (1H, m, CH-5); 13C NMR (75 MHz, D2O) δC 13.4 (CH3-10″), 16.0 (CH3-2), 17.1 (CH3-6), 17.2 (CH3-6), 18.1 (CH2-10′), 18.2 (CH2-10′), 33.5 (CH2-10), 33.6 (CH2-10), 50.5 (CH-5), 50.8 (CH-5), 53.5 (CH-1, CH-9), 170.4 (C=O-8), 170.6 (C=O-8),174.7 (C=O-4); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 18.5; HRMS (NSI) calcd for (C10H23N3O5P)+, MH+: 296.1370, found 296.1373. LCMS purity >95% (C-18 reversed phase, MeOH-H2O). (The LCMS chromatogram and conditions may be found within the Supplementary Materials).

(1-((S)-2-(2-(Methylamino)acetamido)propanamido)ethyl) phosphonic acid or Sar-l-Ala-d/l-Fos (21b). The tert-butoxycarbonyl and diethyl ester protecting groups of (1-((S)-2-((S)-2-aminopropanamido)propanamido)ethyl)phosphonic acid (18b) (3.3 mmol, 1.40 g) were removed. The pale green crude solid was recrystallised from hot water/ethanol to give 21b as a pale green solid composed of 2 diastereoisomers, Sar-l-Ala-l-Fos and Sar-l-Ala-D-Fos (0.45 g, 1.7 mmol, 51%); m.p. 241–245 °C (decomp.); ῡmax/cm−1 3289 (NH+), 3500–2900 (br OH), 1632 (br C=O), 1556 (NH bend), 1174 (P=O), 1059 (P-O-C), 919 (P-OH); 1H NMR (300 MHz, D2O) δH 1.14–1.57 (6H, m, CH3-2, CH3-6), 2.74 (3H, s, CH3-11), 3.84–4.07 (3H, m, CH2-9, CH-1), 4.32–4.58 (1H, m, CH-5); 13C NMR (75 MHz, D2O) δC 15.4 (CH3-2), 16.8 (CH3-6), 32.9 (CH3-11), 43.9 (d, 1JP-C = 148.5 Hz, CH-1), 49.4 (CH2-9), 50.0 (CH-5), 166.0 (C=O-8), 173.7 (C=O-4); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 19.2; HRMS (NSI) calcd for (C8H19N3O5P)+, MH+: 268.1057, found 268.1016; LCMS purity >95% (C-18 reversed phase, MeOH–H2O). (The LCMS chromatogram and conditions may be found within the Supplementary Materials).

(1-((S)-2-((S)-2-Amino-4-(methylthio)butanamido) propanamido)ethyl)phosphonic acid or l-Met-l-Ala-d/l-Fos (21c). The tert-butoxycarbonyl and diethyl ester protecting groups of tert-butyl ((2S)-1-(((2S)-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)amino-4-(methylthio)-1-oxobutan-2-yl)carbamate (18c) (0.9 mmol, 0.43 g) were removed. The green crude solid was recrystallised from hot water/ethanol to give 21c as a pale green solid composed of 2 diastereoisomers, l-Met-l-Ala-l-Fos and l-Met-l-Ala-D-Fos (0.13 g, 0.41 mmol, 46%); m.p. 214–217 °C (decomp.); ῡmax/cm−1 3263 (NH+), 2834 (broad OH), 1641 (br C=O), 1552 (NH bend), 1150 (P=O), 1041 (P-O-C), 919 (P-OH); 1H NMR (300 MHz, D2O) δH 1.29–1.33 (3H, m, CH3-2), 1.42 (3H, d, 3JH-H = 6.0 Hz, CH3-6), 2.15 (3H, s, CH3-10″), 2.20 (2H, m, CH2-10), 2.62 (2H, m, CH2-10′), 4.05 (1H, m, CH-1), 4.14 (1H, m, CH-9), 4.39–4.41 (1H, m, CH-5); 13C NMR (75 MHz, D2O) δC 16.9 (CH3-10″), 18.4 (CH3-2), 19.5 (CH3-6), 19.6 (CH3-6), 32.8 (CH2-10′), 33.0 (CH2-10′), 30.7 (CH2-10), 31.0 (CH2-10), 44.4 (CH-1), 52.9 (CH-5), 53.0 (CH-5), 55.0 (CH-9), 176.1 (C=O-4, C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 20.7; HRMS (NSI) calcd for (C10H23N3O5PS)+, MH+: 328.1091, found 328.1094; LCMS purity >95% (C-18 reversed phase, MeOH-H2O). (The LCMS chromatogram and conditions may be found within the Supplementary Materials).

(S)-Benzyl 2-((tert-butoxycarbonyl)amino)-3-hydroxy propanoate or Boc-l-Ser-OBzl (11). Boc-l-Serine (10) (20 mmol, 4.10 g) and 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) (30 mmol, 4.5 mL) were dissolved in a round-bottom flask containing dry benzene (80 mL), followed by the addition of benzyl bromide (30 mmol, 3.60 mL). Caution: Benzene is a known carcinogen. The solution was stirred overnight at room temperature under nitrogen and later the solvent was removed under reduced pressure to afford an off-white residue. Ethyl acetate (100 mL) was added, the flask contents were sonicated and then washed with 1M HCl (50 mL) and brine (2 × 50 mL). The organic layer was dried over MgSO4, filtered, concentrated in vacuo and purified by column chromatography (petrol/ethyl acetate (1:1)) to give 11 as a white solid (5.24 g, 17.7 mmol, 89%); m.p. 61–63 °C (lit. m.p. 59–60 °C) [30]; ῡmax/cm−1 3416 (NH, OH), 1756 (C=O), 1666 (C=O), 1522 (NH bend), 1200 (C-O), 1154 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.36 (9H, s, C(CH3)3), 2.17 (1H, br, OH), 3.82 (1H, dd, 2JH-H = 11.1 Hz, 3JH-H = 3.6 Hz, CHa/b-3), 3.90 (1H, dd, 2JH-H = 11.1 Hz, 3JH-H = 3.9 Hz, CHa/b-3), 4.33 (1H, m, CH-2), 5.11 (1H, d, 2JH-H = 12.3 Hz, OCHa/bAr), 5.16 (1H, d, 2JH-H = 12.3 Hz, OCHa/bAr), 5.40 (1H, br, NH-4), 7.27 (5H, m, 5 × CHAr); 13C NMR (75 MHz, CDCl3) δC 27.1 (C(CH3)3), 54.7 (CH-2), 62.3 (CH2-3), 66.2 (OCH2Ar), 79.1 (C(CH3)3), 127.0 (2 × CHAr), 127.3 (CHAr), 127.4 (2 × CHAr), 134.1 (CHAr quat.), 153.0 (C=O-5), 170.7 (C=O-1); m/z (ESI) calcd for (C15H21NNaO5)+, MNa+: 318.3, found 318.2. (The 1H-NMR spectrum may be found within the Supplementary Materials).

(R)-Benzyl 2-((tert-butoxycarbonyl)amino)-3-chloropropanoate or Boc-β-Cl-l-Ala-OBzl (12). (S)-Benzyl 2-((tert-butoxycarbonyl)amino)-3-hydroxypropanoate (11) (15 mmol, 4.43 g) was dissolved in dry DCM (40 mL), followed by the addition of trichloroacetonitrile (30 mmol, 3 ml). The solution was stirred at room temperature for 2 h. To this solution, triphenylphosphine (30 mmol, 7.87 g) in dry DCM (50 mL) was added slowly. The resulting solution was stirred overnight at room temperature under nitrogen; brine (100 mL) was added to quench the reaction. The organic layer was washed with brine (3 × 100 mL), dried over MgSO4, filtered and concentrated in vacuo to afford an orange residue. The residue was purified by column chromatography (petrol/ethyl acetate (7:3)) to give 12 as a white solid (3.53 g, 11.2 mmol, 75%); m.p. 55–58 °C; ῡmax/cm−1 3364 (NH), 1725 (C=O), 1680 (C=O), 1519 (NH bend), 1208 (C-O), 1158 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.38 (9H, s, C(CH3)3), 3.78 (1H, dd, 2JH-H = 11.2 Hz, 3JH-H = 3.2 Hz, CHa/b-3), 3.92 (1H, dd, 2JH-H = 11.3 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 4.67 (1H, m, CH-2), 5.13 (1H, d, 2JH-H = 12.2 Hz, OCHa/bAr), 5.18 (1H, d, 2JH-H = 12.2 Hz, OCHa/bAr), 5.37 (1H, d, 3JH-H = 7.5 Hz, NH-4), 7.29 (5H, m, 5 × CHAr); 13C NMR (75 MHz, CDCl3) δC 28.3 (C(CH3)3), 45.5 (CH2-3), 54.5 (CH-2), 67.8 (OCH2Ar), 80.5 (C(CH3)3), 128.4 (CHAr), 128.6 (CHAr), 128.7 (CHAr), 134.9 (CHAr quat.), 155.0 (C=O-5), 169.0 (C=O-1); m/z (ESI) calcd for (C15H20ClNNaO4)+, MNa+: 336.1 (35Cl), 338.1 (37Cl), found 336.2 (35Cl), 338.2 (37Cl); CHN (Found: C, 57.71; H, 6.46; N, 4.38. C15H20ClNO4 requires C, 57.42; H, 6.42; N, 4.46%). (The mass spectrum may be found within the Supplementary Materials).

(R)-2-((Tert-butoxycarbonyl)amino)-3-chloropropanoic acid or Boc-β-Cl-l-Ala-OH (13). Deprotection of benzyl ester was followed, using (R)-benzyl 2-((tert-butoxycarbonyl)amino)-3-chloropropanoate (12) (7.0 mmol, 2.20 g) to afford 13 as an off-white solid (1.52 g, 6.78 mmol, 97%); m.p. 125–128 °C (lit. m.p. 127–129 °C) [15]; ῡmax/cm−1 3434 (NH), 2973 (br OH), 1752 (C=O), 1735 (C=O), 1519 (NH bend), 1159 (C-O), 1148 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.40 (9H, s, C(CH3)3), 3.80 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 3.95 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 4.70 (1H, m, CH-2), 5.42 (1H, d, 3JH-H = 7.2 Hz, NH-4), 9.03 (1H, br, OH); 13C NMR (75 MHz, CDCl3) δC 27.1 (C(CH3)3), 44.0 (CH2-3), 53.1 (CH-2), 79.8 (C(CH3)3), 154.2 (C=O-5), 172.1 (C=O-1); m/z (ESI) calcd for (C8H14ClNNaO4)+, MNa+: 246.1 (35Cl), 248.1 (37Cl), found 246.1 (35Cl), 248.1 (37Cl).

(R)-1-(Benzyloxy)-3-chloro-1oxopropan-2-aminium chloride or β-Cl-l-Ala-OBzl hydrochloride (14). Deprotection of tert-butoxycarbonyl was followed, using (R)-benzyl 2-((tert-butoxycarbonyl)amino)-3-chloropropanoate (12) (15 mmol, 4.71 g). The white crude solid was filtered and washed by diethyl ether to give 14 as a white solid (3.47 g, 13.4 mmol, 93%); m.p. 145 °C (sub); ῡmax/cm−1 2841 (NH+), 1750 (C=O), 1231 (C-O); 1H NMR (300 MHz, D2O) δH 4.06 (1H, dd, 2JH-H = 15.0 Hz, 3JH-H = 6.0 Hz, CHa/b-3), 4.20 (1H, dd, 2JH-H = 15.0 Hz, 3JH-H = 6.0 Hz, CHa/b-3), 4.70 (1H, t, 3JH-H = 6.0 Hz, CH-2), 5.29 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 5.37 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 7.42-7.47 (5H, m, 5 × CHAr); 13C NMR (75 MHz, D2O) δC 41.8 (CH2-3), 54.0 (CH-2), 69.1 (OCH2Ar), 128.6–129.1 (CHAr), 134.5 (CHAr quart.), 167.0 (C=O-1); m/z (ESI) calcd for (C10H13ClNO2), M+: 214.1 (35Cl), 216.1 (37Cl), found 214.1 (35Cl), 216.1 (37Cl); CHN (Found: C, 47.16; H, 5.43; N, 5.43. C10H13Cl2NO2∙0.2H2O requires C, 47.34; H, 5.32; N, 5.52%).

(R)-Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido)-3-chloropropanoate or Boc-l-Nva-β-chloro-l-Ala-OBzl (22a). General peptide coupling method was followed, using Boc-l-Nva-OH (15a) (6.0 mmol, 1.31 g) in dry THF and (R)-1-(benzyloxy)-3-chloro-1oxopropan-2-aminium chloride (14) (5.4 mmol, 1.36 g) in dry DCM. The yellow crude liquid was purified by column chromatography (40-60 petrol/ethyl acetate (7:3)) to give 22a as a white solid (1.74 g, 4.2 mmol, 78%); m.p. 95–98 °C; ῡmax/cm−1 3327 (NH), 1743 (C=O), 1688 (C=O), 1653 (C=O), 1518 (NH bend), 1206 (C-O), 1169 (C-O); 1H NMR (300 MHz, CDCl3) δH 0.92 (3H, t, 3JH-H = 9.0 Hz, CH3-7″), 1.32-1.43 (2H, m, CH2-7′), 1.45 (9H, s, C(CH3)3), 1.52–1.65 (1H, m, CHa/b-7), 1.75-1.82 (1H, m, CHa/b-7), 3.89 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 3.99 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 4.11–4.15 (1H, m, CH-6), 4.96–5.00 (2H, m, CH-2, NH-8), 5.20 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 5.25 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 6.97 (1H, d, 3JH-H = 6.0 Hz, NH-4), 7.33–7.37 (5H, m, 5 x CHAr); 13C NMR (75 MHz, CDCl3) δC 12.7 (CH3-7″), 17.8 (CH2-7′), 27.3 (C(CH3)3), 33.4 (CH2-7), 43.8 (CH2-3), 52.2 (CH-2), 53.4 (CH-6), 67.0 (OCH2Ar), 79.3 (C(CH3)3), 127.4 (CHAr), 127.6 (CHAr), 127.7 (CHAr), 133.8 (CHAr quat.), 154.5 (C=O-9), 167.5 (C=O-1), 171.2 (C=O-5); HRMS (NSI) calcd for (C20H30ClN2O5)+, MH+: 413.1838 (35Cl), 415.1809 (37Cl), found 413.1837 (35Cl), 415.1807 (37Cl); CHN (Found: C, 58.49; H, 7.22; N, 6.81. C20H29ClN2O5 requires C, 58.18; H, 7.08; N, 6.78%).

(R)-Benzyl 2-(2-((tert-butoxycarbonyl)(methyl)amino) acetamido)-3-chloropropanoate or Boc-Sar-β-chloro-l-Ala-OBzl (22b). General peptide coupling method was followed, using Boc-Sar-OH (15b) (13.0 mmol, 2.46 g) in dry THF and (R)-1-(benzyloxy)-3-chloro-1oxopropan-2-aminium chloride (14) (13.3 mmol, 3.34 g) in dry DCM. The yellow crude liquid was purified by column chromatography (40–60 petrol/ethyl acetate (7:3)) to give 22b as a light yellow syrup (3.63 g, 9.4 mmol, 73%); ῡmax/cm−1 3302 (NH), 1747 (C=O), 1686 (br C=O), 1522 (NH bend), 1175 (C-O), 1148 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.40 (9H, s, C(CH3)3), 2.87 (3H, s, NCH3-8), 3.80 (1H, d, 2JH-H = 15.0 Hz, CHa/b-6), 3.82 (1H, d, 2JH-H = 15.0 Hz, CHa/b-6), 3.83 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 3.94 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 3.0 Hz, CHa/b-3), 4.91–4.96 (1H, m, CH-2), 5.13 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 5.18 (1H, d, 2JH-H = 12.0 Hz, OCHa/bAr), 6.97 (1H, d, 3JH-H = 6.0 Hz, NH-4), 7.26–7.30 (5H, m, 5 x CHAr); 13C NMR (75 MHz, CDCl3) δC 28.2 (C(CH3)3), 35.6 (NCH3-8), 44.9 (CH2-3), 53.0 (CH2-6), 53.0 (CH-2), 68.0 (OCH2Ar), 81.0 (C(CH3)3), 128.4 (CHAr), 128.6 (CHAr), 128.7 (CHAr), 134.8 (CHAr quat.), 154.5 (C=O-9), 168.4 (C=O-1), 169.4 (C=O-5); HRMS (NSI) calcd for (C18H26ClN2O5)+, MH+: 385.1525 (35Cl), 387.1496 (37Cl), found 385.1527 (35Cl), 387.1498 (37Cl). LCMS purity >95% (C-18 reversed phase, MeOH–H2O).

Tert-butyl ((2R)-3-chloro-1-((1-(diethoxyphosphoryl)ethyl) amino)-1-oxopropan-2-yl)carbamate or Boc-β-chloro-l-Ala-d/l-Fos diethyl ester (19e). General peptide coupling method was followed, using (R)-2-((tert-butoxycarbonyl)amino)-3-chloropropanoic acid (13) (6.0 mmol, 1.34 g) in dry THF and diethyl 1-aminoethylphosphonate (9) (6.0 mmol, 1.09 g) in dry THF. The light yellow crude liquid was purified by column chromatography, using 100% petrol and increasing to 100% ethyl acetate, to afford 19e as colorless syrup composed of 2 diastereoisomers, Boc-β-Cl-l-Ala-l-Fos diethyl ester and Boc-β-Cl-l-Ala-D-Fos diethyl ester (2.03 g, 5.2 mmol, 88%); ῡmax/cm−1 3261 (NH), 1713 (C=O), 1670 (C=O), 1517 (NH bend), 1225 (P=O), 1164 (C-O), 1020 (P-O-C), 970 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.15–1.46 (9H, m, CH3-2, 2 × OCH2CH3), 1.47 (9H, s, C(CH3)3), 3.74 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 4.00 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 4.06–4.22 (4H, m, 2 × OCH2CH3), 4.40–4.56 (2H, m, CH-1, CH-5), 5.46 (0.5H, d, 3JH-H = 6.0 Hz, NH-3 or NH-7), 5.48 (0.5H, d, 3JH-H = 9.0 Hz, NH-3 or NH-7), 7.01 (0.5H, m, NH-3 or NH-7), 7.09 (0.5H, m, NH-3 or NH-7); 13C NMR (75 MHz, CDCl3) δC 15.6 (CH3-2), 15.7 (CH3-2), 16.3 (d, 3JC-P = 1.5 Hz, OCH2CH3), 16.4 (d, 3JC-P = 2.3 Hz, OCH2CH3), 16.4 (d, 3JC-P = 1.5 Hz, OCH2CH3), 16.4 (d, 3JC-P = 2.3 Hz, OCH2CH3), 28.2 (C(CH3)3), 41.2 (d, 1JC-P = 157.5 Hz, CH-1), 41.3 (d, 1JC-P = 157.5 Hz, CH-1), 55.2 (CH2-6), 55.3 (CH-5), 62.6 (d, 2JC-P = 6.8 Hz, OCH2CH3), 62.6 (d, 2JC-P = 6.8 Hz, OCH2CH3), 63.0 (d, 2JC-P = 6.8 Hz, OCH2CH3), 63.0 (d, 2JC-P = 6.8 Hz, OCH2CH3), 80.8 (C(CH3)3), 155.0 (C=O-8), 168.3 (C=O-4); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 24.7, 24.8; HRMS (NSI) calcd for (C14H28ClN2O6P), MH+: 409.1266 (35Cl), 411.1237 (37Cl), found 409.1258 (35Cl), 411.1231 (37Cl). LCMS purity >95% (C-18 reversed phase, MeOH-H2O).

(R)-2-((S)-2-((tert-Butoxycarbonyl)amino)pentanamido)-3-chloropropanoic acid or Boc-l-Nva-β-chloro-l-Ala-OH (23a). Deprotection of benzyl ester was followed, using (R)-benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido)-3-chloropropanoate (22a) (5.8 mmol, 2.41 g) to afford 23a as a light yellow solid (1.81 g, 5.61 mmol, 96%); m.p. 60–63 °C; ῡmax/cm−1 3312 (br OH), 2963 (NH), 1655 (br C=O), 1509 (NH bend), 1161 (C-O); 1H NMR (300 MHz, DMSO) δH 0.85 (3H, t, 3JH-H = 9.0 Hz, CH3-7″), 1.24–1.34 (2H, m, CH2-7′), 1.38 (9H. s, C(CH3)3), 1.42–1.52 (1H, m, CHa/b-7), 1.54–1.59 (1H, m, CHa/b-7), 3.34 (1H, br, OH), 3.84 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-3), 3.91 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-3), 3.95–4.02 (1H, m, CH-6), 4.62–4.67 (1H, m, CH-2), 6.92 (1H, d, 3JH-H = 9.0 Hz, NH-8), 8.07 (1H, d, 3JH-H = 9.0 Hz, NH-4); 13C NMR (75 MHz, CDCl3) δC 14.1 (CH3-7″), 19.1 (CH2-7′), 28.6 (C(CH3)3), 34.4 (CH2-7), 45.1 (CH2-3), 53.6 (CH-2), 54.5 (CH-6), 78.5 (C(CH3)3), 155.8 (C=O-9), 170.6 (C=O-5), 173.0 (C=O-1); m/z (ESI) calcd for (C13H23ClN2NaO5)+, MNa+: 345.1 (35Cl), 347.1 (37Cl), found 345.2 (35Cl), 347.2 (37Cl); CHN (Found: C, 48.67; H, 7.51; N, 8.42. C13H23ClN2O5 requires C, 48.37; H, 7.18; N, 8.68%).

(R)-2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido)-3-chloropropanoic acid or Boc-Sar-β-chloro-l-Ala-OH (23b). Deprotection of benzyl ester was followed, using (R)-benzyl 2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido)-3-chloropropanoate (22b) (6.2 mmol, 2.40 g) to afford 23b as an off-white solid (1.81 g, 6.1 mmol, 99%); m.p. 89–91 °C; ῡmax/cm−1 3342 (NH), 2982 (br OH), 1734 (C=O), 1672 (C=O), 1644 (C=O), 1524 (NH bend), 1152 (C-O); 1H NMR (300 MHz, CDCl3) δH 1.47 (9H. s, C(CH3)3), 2.99 (3H, s, NCH3-8), 3.81–4.17 (4H, m, CH2-6, CH2-3), 5.01 (1H, m, CH-2), 7.07 (1H, br, NH-4), 7.45 (1H, br, OH); 13C NMR (75 MHz, CDCl3) δC 28.3 (C(CH3)3), 36.1 (NCH3-8), 44.5 (CH2-3), 53.0 (CH2-6 and CH-2), 81.8 (C(CH3)3), 156.8 (C=O-9), 169.5 (C=O-1 and C=O-5); m/z (ESI) calcd for (C11H19ClN2NaO5)+, MNa+: 317.1 (35Cl), 319.1 (37Cl), found 317.1 (35Cl), 319.1 (37Cl); CHN (Found: C, 43.98; H, 6.69; N, 9.53. C11H19ClN2O5·0.3H2O requires C, 44.02; H, 6.58; N, 9.33%).

(2R)-3-Chloro-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-aminium chloride or β-Cl-l-Ala-d/l-Fos diethyl ester hydrochloride (20e). Deprotection of tert-butoxycarbonyl was followed, using ((2R)-3-chloro-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)carbamate (19e) (6.7 mmol, 2.59 g). The off-white hygroscopic crude solid was washed with petrol to afford 20e as a pale green solid composed of 2 diastereoisomers, β-Cl-l-Ala-l-Fos diethyl ester hydrochloride and β-Cl-l-Ala-D-Fos diethyl ester hydrochloride (1.51 g, 4.7 mmol, 70%); m.p. 129–133 °C (decomp.); ῡmax/cm−1 3204 (NH+), 1687 (C=O), 1562 (NH bend), 1204 (P=O), 1010 (P-O-C), 961 (P-O-C); 1H NMR (300 MHz, D2O) δH 1.28 (3H, t, 3JH-H = 6.0 Hz, OCH2CH3), 1.29 (3H, t, 3JH-H = 6.0 Hz, OCH2CH3), 1.37 (3H, dd, 3JP-H = 18.0 Hz, 3JH-H = 6.0 Hz, CH3-2), 3.92–4.04 (2H, m, CH2-6), 4.07-4.21 (4H, m, 2 × OCH2CH3), 4.38–4.48 (2H, m, CH-1, CH-5); 13C NMR (75 MHz, D2O) δC 13.7 (CH3-2), 14.0 (CH3-2), 15.7 (OCH2CH3), 15.7 (OCH2CH3), 41.7 (d, 1JP-C = 158.3 Hz, CH-1), 42.0 (d, 1JP-C = 157.5 Hz, CH-1), 42.4 (CH2-6), 53.7 (CH-5), 53.8 (CH-5), 64.3 (d, 2JP-C = 6.8 Hz, OCH2CH3), 64.5 (d, 2JP-C = 6.8 Hz, OCH2CH3), 165.7 (C=O-4), 165.8 (C=O-4); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 26.1, 26.2; HRMS (NSI) calcd for (C9H21ClN2O4P)+, M+: 287.0922 (35Cl), 289.0892 (37Cl), found 287.0922 (35Cl), 289.0890 (37Cl). LCMS purity >95% (C-18 reversed phase, MeOH–H2O).

Tert-butyl ((2S)-1-(((2R)-3-chloro-1-((1-(diethoxyphosphoryl)-ethyl)amino)-1-oxopropan-2-yl)amino)-1-oxopentan-2-yl)carbamate or Boc-l-Nva-β-chloro-l-Ala-d/l-Fos diethyl ester (24a). General peptide coupling method was followed, using (R)-2-((S)-2-((tert-butoxycarbonyl)amino)pentanamido)-3-chloropropanoic acid (23a) (1.8 mmol, 0.58 g) in dry THF and diethyl 1-aminoethylphosphonate (9) (1.8 mmol, 0.33 g) in dry THF. The light yellow crude liquid was purified by column chromatography, using ethyl acetate/methanol (96:4), to afford 24a as a white solid composed of 2 diastereoisomers, Boc-l-Nva-β-Cl-l-Ala-l-Fos diethyl ester and Boc-l-Nva-β-Cl-l-Ala-D-Fos diethyl ester (0.45 g, 0.93 mmol, 52%); m.p. 196 °C (decomp); ῡmax/cm−1 3272 (NH), 1709 (C=O), 1680 (C=O), 1644 (C=O), 1530 (NH bend), 1229 (P=O), 1165 (C-O), 1019 (P-O-C), 972 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 0.86 (1.5H, t, 3JH-H = 9.0 Hz, CH3-10″), 0.88 (1.5H, t, 3JH-H = 9.0 Hz, CH3-10″), 1.22–1.34 (11H, m, 2 × OCH2CH3, CH3-2, CH2-10′), 1.38 (9H, s, C(CH3)3), 1.53-1.59 (1H, m, CHa/b-10), 1.70–1.77 (1H, m, CHa/b-10), 3.69 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 3.78-3.81 (1H, m, CH-9), 3.91 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 3.97-4.13 (4H, m, 2 x OCH2CH3), 4.35-4.46 (1H, m, CH-1), 4.73-4.79 (1H, m, CH-5), 4.97–5.03 (1H, m, NH-11), 7.01 (0.5H, d, 3JH-H = 9.0 Hz, NH-7), 7.09 (0.5H, d, 3JH-H = 9.0 Hz, NH-7), 7.25 (0.5H, d, 3JH-H = 9.0 Hz, NH-3), 7.33 (0.5H, d, 3JH-H = 9.0 Hz, NH-3); 13C NMR (75 MHz, CDCl3) δC 12.7 (CH3-10″), 14.5 (CH3-2), 15.3 (OCH2CH3), 15.4 (OCH2CH3), 15.5 (OCH2CH3), 15.6 (OCH2CH3), 17.9 (CH2-10′), 18.0 (CH2-10′), 27.0 (C(CH3)3), 27.3 (C(CH3)3), 33.2 (CH2-10), 40.4 (d, 1JC-P = 157.5 Hz, CH-1), 43.4 (CH2-6), 52.6 (CH-5), 52.8 (CH-5), 61.4 (d, 2JC-P = 6.8 Hz, OCH2CH3), 61.6 (d, 2JC-P = 6.8 Hz, OCH2CH3), 61.7 (d, 2JC-P = 6.8 Hz, OCH2CH3), 61.9 (d, 2JC-P = 6.8 Hz, OCH2CH3), 70.5 (CH-9), 79.4 (C(CH3)3), 154.7 (C=O-12), 166.8 (C=O-4), 171.4 (C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 24.9, 25.0; HRMS (NSI) calcd for (C19H38ClN3O7P)+, MH+: 486.2130 (35Cl), 488.2102 (37Cl), found 486.2124 (35Cl), 488.2098 (37Cl); CHN (Found: C, 46.61; H, 7.76; N, 8.31. C19H37ClN3O7P requires C, 46.96; H, 7.67; N, 8.65%).

Tert-butyl (2-(((2R)-3-chloro-1-((1-(diethoxyphosphoryl)ethyl) amino)-1-oxopropan-2-yl)amino)-2-oxoethyl)(methyl)carbamate or Boc-Sar-β-chloro-l-Ala-d/l-Fos diethyl ester (24b). General peptide coupling method was followed, using (R)-2-(2-((tert-butoxycarbonyl)(methyl)amino)acetamido)-3-chloropropanoic acid (23b) (5.5 mmol, 1.61 g) in dry THF and diethyl 1-aminoethylphosphonate (9) (6.0 mmol, 1.09 g) in dry THF. The light yellow crude liquid was purified by column chromatography, using DCM/methanol (95:5), to afford 24b as a light yellow syrup composed of 2 diastereoisomers, Boc-Sar-β-Cl-l-Ala-l-Fos diethyl ester and Boc-Sar-β-Cl-l-Ala-D-Fos diethyl ester (1.93 g, 4.21 mmol, 76%); ῡmax/cm−1 3218 (NH), 1690 (C=O), 1665 (br C=O), 1518 (NH bend), 1224 (P=O), 1148 (C-O), 1018 (P-O-C), 967 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.11–1.35 (9H, m, 2 × OCH2CH3, CH3-2), 1.41 (9H, s, C(CH3)3), 2.90 (3H, s, CH3-11), 3.70–3.88 (4H, m, CH2-6, CH2-9), 4.02–4.13 (4H, m, 2 × OCH2CH3), 4.36–4.47 (1H, m, CH-1), 4.78–4.82 (1H, m, CH-5), 6.94 (1H, m, NH-7), 7.36 (1H, m, NH-3); 13C NMR (75 MHz, CDCl3) δC 15.2 (CH3-2), 15.6 (CH3-2), 16.3 (OCH2CH3), 16.4 (OCH2CH3), 28.3 (C(CH3)3), 35.9 (CH3-11), 41.2 (d, 1JC-P = 156.8 Hz, CH-1), 44.7 (CH2-6), 53.1 (CH2-9), 53.4 (CH-5), 62.7 (d, 2JC-P = 6.0 Hz, OCH2CH3), 63.0 (d, 2JC-P = 7.5 Hz, OCH2CH3), 81.0 (C(CH3)3), 152.3 (C=O-12), 167.7 (C=O-4 or C=O-8), 169.4 (C=O-4 or C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 24.7, 24.8; HRMS (NSI) calcd for (C16H34ClN3O7P)+, MH+: 480.1637 (35Cl), 482.1608 (37Cl), found 480.1642 (35Cl), 482.1612 (37Cl). LCMS purity >92% (C-18 reversed phase, MeOH-H2O).

Tert-butyl ((2S)-1-(((2R)-3-chloro-1-((1-(diethoxyphosphoryl) ethyl)amino)-1-oxopropan-2-yl)amino)-4-(methylthio)-1-oxobutan-2-yl) carbamate or Boc-l-Met-β-Cl-l-Ala-d/l-Fos diethyl ester (24c). General peptide coupling method was followed, using Boc-l-Met-OH (15c) (3.4 mmol, 0.85 g) in dry THF and (2R)-3-chloro-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-aminium chloride (20e) (3.4 mmol, 1.10 g) in dry DCM. The yellow crude liquid was purified by column chromatography (DCM/MeOH (95:5)) and recrystallized from diethyl ether/petrol to give 24c as a white solid composed of 2 diastereoisomers, Boc-l-Met-β-Cl-l-Ala-l-Fos diethyl ester and Boc-l-Met-β-Cl-l-Ala-D-Fos diethyl ester (0.88 g, 1.7 mmol, 50%); m.p. 96–99 °C; ῡmax/cm−1 3278 (NH), 1709 (C=O), 1687 (C=O), 1639 (C=O), 1523 (NH bend), 1228 (P=O), 1165 (C-O), 1018 (P-O-C), 970 (P-O-C); 1H NMR (300 MHz, CDCl3) δH 1.17-1.36 (9H, m, CH3-2, 2 × OCH2CH3), 1.38 (9H, s, C(CH3)3), 1.87–2.03 (2H, m, CH2-10), 2.04 (3H, s, CH3-10″), 2.48–2.54 (2H, m, CH2-10′), 3.71 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 3.88 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 3.99–4.13 (4H, m, 2 × OCH2CH3), 4.20 (1H, m, CH-9), 4.37–4.47 (1H, m, CH-1), 4.78–4.84 (1H, m, CH-5), 5.39 (0.5H, d, 3JH-H = 6.0 Hz, NH-11), 5.41 (0.5H, d, 3JH-H = 6.0 Hz, NH-11), 7.15 (0.5H, d, 3JH-H = 6.0 Hz, NH-7), 7.24 (0.5H, d, 3JH-H = 6.0 Hz, NH-7), 7.52 (1H, m, NH-3); 13C NMR (75 MHz, CDCl3) δC 14.3 (CH3-2), 14.4 (CH3-2), 14.5 (CH2-10″), 15.3 (OCH2CH3), 15.4 (OCH2CH3), 15.5 (OCH2CH3), 15.6 (OCH2CH3), 27.3 (C(CH3)3), 29.2 (CH2-10′), 29.3 (CH2-10′), 30.2 (CH2-10), 30.4 (CH2-10), 40.3 (d, 1JP-C = 159.0 Hz, CH-1), 43.5 (CH2-6), 43.7 (CH2-6), 52.7 (CH-5), 53.1 (CH-9), 61.6 (d, 2JP-C = 6.8 Hz, OCH2CH3), 61.7 (d, 2JP-C = 6.0 Hz, OCH2CH3), 62.0 (d, 2JP-C = 6.8 Hz, OCH2CH3), 62.1 (d, 2JP-C = 7.5 Hz, OCH2CH3), 79.6 (C(CH3)3), 154.8 (C=O-12), 166.7 (C=O-4), 166.8 (C=O-4), 170.7 (C=O-8), 170.8 (C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 24.5, 24.8; HRMS (NSI) calcd for (C19H3HClN3O7PS)+, MH+: 518.1851 (35Cl), 520.1821 (37Cl), found 518.1842 (35Cl), 520.1814 (37Cl); CHN (Found: C, 44.08; H, 7.47; N, 8.18. C19H37ClN3O7PS requires C, 44.06; H, 7.20; N, 8.11%).

(1-((R)-2-((S)-2-Ammoniopentanamido)-3-chloropropanamido) ethyl)phosphonic acid or l-Nva-β-chloro-l-Ala-d/l-Fos (25a). The tert-butoxycarbonyl and diethyl ester protecting groups of tert-butyl ((2S)-1-(((2R)-3-chloro-1-((1-(diethoxyphosphoryl)-ethyl)amino)-1-oxopropan-2-yl)amino)-1-oxopentan-2-yl)carbamate (24a) (2.0 mmol, 0.99 g) were removed. The pale green crude solid was washed with diethyl ether to give 25a as a pale green solid composed of 2 diastereoisomers, l-Nva-β-Cl-l-Ala-l-Fos and l-Nva-β-Cl-l-Ala-D-Fos (0.64 g, 1.94 mmol, 97%); m.p. 175 °C (sub); ῡmax/cm−1 3294 (NH+), 3000 (br OH), 1668 (C=O), 1645 (C=O), 1538 (NH bend), 1132 (P=O), 1039 (P-O-C), 921 (P-OH); 1H NMR (300 MHz, D2O) δH 1.01 (3H, t, 3JH-H = 9.0 Hz, CH3-10″), 1.30–1.37 (3H, br m, CH3-2), 1.44–1.54 (2H, br m CH2-10′), 1.90–1.98 (2H, br m, CH2-10), 3.91–4.15 (4H, br m, CH2-6, CH-9, CH-1), 4.79 (1H, br m, CH-5); 13C NMR (75 MHz, D2O) δC 12.9 (CH3-10″), 15.7 (CH3-2), 17.6 (CH2-10′), 33.0 (CH2-10), 43.3 (CH2-6), 53.1 (CH-1 and CH-9), 55.0 (CH-5), 170.4 (C=O-4 and C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 18.5; HRMS (NSI) calcd for (C10H20ClN3O5P)-, MH-: 328.0835 (35Cl), 330.0805 (37Cl), found 328.0833 (35Cl), 330.0800 (37Cl). LCMS purity >95% (C-18 reversed phase, MeOH-H2O). (The LCMS chromatogram and conditions may be found within the Supplementary Materials).

1-((R)-3-Chloro-2-(2-(methylammonio)acetamido) propanamido)ethyl)phosphonic acid or Sar-β-chloro-l-Ala-d/l-Fos (25b). The tert-butoxycarbonyl and diethyl ester protecting groups of tert-butyl (2-(((2R)-3-chloro-1-((1-(diethoxyphosphoryl)ethyl)amino)-1-oxopropan-2-yl)amino)-2-oxoethyl)(methyl)carbamate (24b) (3.8 mmol, 1.74 g) were removed. The pale green crude solid was recrystallised from hot water/ethanol to give 25b as an off-white solid composed of 2 diastereoisomers, Sar-β-Cl-l-Ala-l-Fos and Sar-β-Cl-l-Ala-D-Fos (0.49 g, 1.61 mmol, 42%); m.p. 185-188 °C (decomp.); ῡmax/cm−1 3287 (NH+), 3000 (br OH), 1657 (C=O), 1634 (C=O), 1552 (NH bend), 1172 (P=O), 1054 (P-O-C), 919 (P-OH); 1H NMR (300 MHz, CD3OD) δH 1.24 (3H, dd, 3JH-P = 15.0 Hz, 3JH-H = 6.0 Hz, CH3-2), 2.74 (3H, s, NCH3-11), 3.81–3.87 (2H, m, CH2-6), 3.93–3.94 (2H, m, CH2-9), 3.97–4.10 (1H, m, CH-1), 4.79 (1H, m, CH-5); 13C NMR (75 MHz, CD3OD) δC 15.4 (CH3-2), 32.9 (NCH3-11), 43.6 (CH2-6), 44.1 (d, 1JC-P = 148.5 Hz, CH-1), 49.5 (CH2-9), 54.6 (CH-5), 166.4 (C=O-4 or C=O-9), 166.9 (C=O-4 or C=O-9); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 18.8; HRMS (NSI) calcd for (C8H18ClN3O5P)+, MH+: 302.0667 (35Cl), 304.0638 (37Cl), found 302.0670 (35Cl), 304.0640 (37Cl). LCMS purity >95% (C-18 reversed phase, MeOH-H2O). (The LCMS chromatogram and conditions may be found within the Supplementary Materials).

(1-((R)-2-((S)-2-Ammonio-4-(methylthio)butanamido)-3-chloro propanamido)ethyl)phosphonic acid or l-Met-β-Cl-l-Ala-d/l-Fos (25c). The tert-butoxycarbonyl and diethyl ester protecting groups of tert-butyl ((2S)-1-(((2R)-3-chloro-1-((1-(diethoxyphosphoryl) ethyl)amino)-1-oxopropan-2-yl)amino)-4-(methylthio)-1-oxobutan-2-yl)carbamate (24c) (1.4 mmol, 0.71 g) were removed. The green crude solid was recrystallised from hot water/ethanol to give 25c as a pale green solid composed of 2 diastereoisomers, l-Met-β-Cl-l-Ala-l-Fos and l-Met-β-Cl-l-Ala-D-Fos (0.17 g, 0.48 mmol, 35%); m.p. 175–179 °C (decomp.); ῡmax/cm−1 3264 (NH+), 2829 (broad OH), 1666(C=O), 1641 (C=O), 1546 (NH bend), 1149 (P=O), 1041 (P-O-C), 921 (P-OH); 1H NMR (300 MHz, D2O) δH 1.31 (3H, dd, 3JH-P = 15.0 Hz, 3JH-H = 6.0 Hz, CH3-2), 2.13 (3H, s, CH3-10″), 2.18–2.29 (2H, m, CH2-10), 2.63–2.69 (2H, m, CH2-10′), 3.89 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 3.97 (1H, dd, 2JH-H = 12.0 Hz, 3JH-H = 6.0 Hz, CHa/b-6), 4.01–4.13 (1H, m, CH-1), 4.22 (1H, br m, CH-9), 4.75–4.79 (1H, m, CH-5); 13C NMR (75 MHz, D2O) δC 16.9 (CH3-10″), 17.0 (CH3-10″), 18.4 (CH3-2), 31.1 (CH2-10′), 32.9 (CH2-10), 46.2 (CH2-6), 47.0 (d, 1JC-P = 147.0 Hz, CH-1), 52.2 (CH-9), 52.3 (CH-9), 57.8 (CH-5), 58.0 (CH-5), 171.7 (C=O-4), 171.8 (C=O-4), 172.3 (C=O-8); 31P-1Hdecoup NMR (121 MHz, CDCl3) δP 18.7; HRMS (NSI) calcd for (C10H21ClN3O5PS), MNa+: 384.0520 (35Cl), 386.0489 (37Cl), found 384.0523 (35Cl), 386.0491 (37Cl). LCMS purity >95% (C-18 reversed phase, MeOH-H2O). (The LCMS chromatogram and conditions may be found within the Supplementary Materials).
3.2. Microbiological Procedures
3.2.1. Media Constituents
l-Arginine, l-aspartic acid, l-cysteine, glycine, l-histidine, l-isoleucine, l-lysine, l-methionine, l-phenylalanine, l-proline, l-serine, l-threonine, l-tryptophan, l-valine, magnesium sulphate, haemin and d-(+)-glucose were purchased from Sigma Chemical Co. (Poole, UK). l-Tyrosine, uracil, guanine, cytosine, adenine, ammonium sulphate, potassium dihydrogen phosphate, saponin, and dipotassium hydrogen phosphate were acquired from BDH Merck Ltd. (Poole, England). Yeast extract was supplied by bioMérieux (Craponne, France) and bacteriological agar from Oxoid (Basingstoke, UK). Nicotinamide adenine dinucleotide (NAD) was obtained from Merck (Darmstadt, Germany) and heparinized horse blood from TCS Biosciences (Buckingham, UK).
3.2.2. Microbiology Strains
Microbial reference strains were obtained from National Collection of Type Cultures (NCTC) (Colindale, UK) and the American Type Culture Collection (ATCC) (Manassas, US). These included twelve Gram-negative bacteria: A. baumannii, B. cepacia, E. cloacae, E. coli (n = 2), K. pneumoniae, P. rettgeri, P. aeruginosa, S. typhimurium, S. enteritidis, S. marcescens, and Y. enterocolitica; as well as seven Gram positive bacteria: E. faecalis, E. faecium, L. monocytogenes, S. epidermidis, S. aureus, methicillin resistant S. aureus (MRSA), and S. pyogenes. These 19 bacterial strains were maintained on Columbia agar.
3.2.3. Preparation of Antagonist-free (AF) Medium
The methodology utilized, which was reported by Atherton et al. [11], was adopted with slight modification. In this method, 1.5% (15 g/L) bacteriological agar and 0.5% (5 g/L) glucose were added to 880 mL deionized water. The resulting mixture was autoclaved at 121 °C for 15 min, followed by addition of 2 %v/v (20 mL/L) saponin-lysed horse blood, 25 mg/L hemin, 25 mg/L NAD and 100 mL of 10 x strength antagonist-free broth prepared as previously described [11]. The saponin-lysed blood was prepared by incubating 100 mL sterile hose blood at 37 ± 0.5 °C for 15 min, adding 5 mL 10% saponin and re-incubating for 15 min or longer to ensure completely blood lysis. The saponin-lysed blood was stored at 4 °C until used. The pH of medium was 7.0.
3.2.4. Preparation of Media Containing Phosphonotripeptide Derivatives
Each derivative was dissolved in sterile deionized water (SDW) and incorporated with AF-agar at a concentration range of 0.032–8 mg/L. A series of dilutions was performed to achieve the required concentration and then poured into the sterile Petri dishes. Plates containing AF-agar were also prepared without inhibitor. Solidified agar plates were placed in a warm cabinet (37 ± 0.5 °C) for 5 min to dry the surface of the agar and then stored at 4 °C.
3.2.5. Multiple Inoculation of Agar
Each microbial strain was isolated from the respective Columbia agar after 18 h of incubation and suspended in SDW to a density equivalent to 0.5 McFarland units using a densitometer. One hundred microliters of each suspension was transferred into the corresponded wells of a multipoint inoculation device. One microliter of the bacterial suspension, equivalent to 1.5 × 105 organisms was applied as a spot by this instrument on the plates containing different phosphonotripeptide derivatives (as well as inhibit-free growth control plates). Nineteen different bacterial strains were applied on a plate and incubated for 22 h at 37 ± 0.5 °C. Un-inoculated AF-agar plates from the same batch of agar were incubated as a sterility check and a sterility check was also performed on the SDW used to make bacterial suspensions by culture of aliquots on Columbia agar.
3.2.6. Determination of Minimum Inhibitory Concentration (MIC)
After incubation, each plate was inspected for growth or inhibition of the inoculated spots. The minimum inhibitory concentration (MIC) was recorded as the lowest concentration of inhibitor to completely inhibit the growth of the test strain. Tests were performed in duplicate on separate occasions and in the vast majority of cases generated identical results. In a small minority of cases, results differed but were always within one double-dilution and this was always resolved by a third test. MIC values were only interpreted for any particular strain if growth on the inhibitor-free control plate was completely uninhibited and if sterility of the culture medium and diluent were validated.
4. Conclusions
Here we have disclosed the synthesis of six phosphonopeptide based inhibitors, all of which contain the antimicrobial agent fosfalin at their C-terminus. Three of the inhibitors also contain a second antimicrobial agent, β-chloroalanine, at the center of their tripeptide sequence. The identity of the N-terminal amino acid was varied in order to elicit selectivity via exploiting differences in permeability and hydrolysis rates within bacterial cells.
We have shown that the β-chloro containing compounds elicit lower MIC values than their alanine analogues against a wide range of clinically relevant bacteria. This is consistent with release of free β-chloroalanine which then is able to act synergistically with the fosfalin unit.
Finally, we show that the MIC profiles of inhibitors 25a and 25c have potential for two differing real-world clinical applications. These are the detection of lung pathogens in cystic fibrosis patients, and the detection of Salmonella within stool samples for diagnosis of food poisoning, However, further work is required with large numbers of bacterial strains and relevant clinical samples to conclusively demonstrate the utility of these compounds.
5. Patents
Antimicrobial Compounds, 2018, WO2015140481.
Supplementary Materials
The following are available online. Enantiomer and diastereoisomer data—Table S1; NMR spectra—Figure S1–S3; Low resolution MS spectrum—Figure S4; LC-MS conditions and final product chromatograms—Figure S5–S8.
Author Contributions
Conceptualization, J.D.P., M.G., R.J.A. and S.O.; Formal analysis, K.T.N., E.C.L.M. and J.D.P.; Funding acquisition, M.G. and R.J.A.; Investigation, K.T.N. and E.C.L.M.; Methodology, K.T.N. and J.D.P.; Resources, S.O. and J.D.P.; Supervision, J.D.P., M.G. and R.J.A.; Writing—original draft preparation, K.T.N.; Writing—review and editing, M.G., J.D.P. and S.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by bioMérieux (France).
Acknowledgments
Authors would like to acknowledge the NMR facility and HRMS analyses provided by University of Sunderland and EPRSC, for structural elucidation and characterization.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| AcOH | Acetic acid |
| HCl | Hydrochloric acid |
| EtOH | Ethanol |
| CF3COOH | Trifluoroacetic acid |
| (CF3COO)2O | Trifluoroacetic anhydride |
| CH(OC2H5)3 | Triethyl orthoformate |
| NaBH4 | Sodium borohydride |
| tBoc | N-tert-butoxycarbonyl protecting group |
| OEt | Ethyl ester protecting group |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| PPh3 | Triphenylphosphine |
| DCM | Dichloromethane |
| MeOH | Methanol |
| Pd | Palladium |
| C | Carbon |
| H2 | Hydrogen |
| r.t. | Room temperature |
| h | Hour |
| M | Molarity |
| PPEs | Personal protective equipments |
| OBzl | Benzyl ester protecting group |
| DIPEA | N,N-Diisopropylethylamine |
| IBCF | Isobutyl chloroformate |
| NMM | N-methyl morpholine |
| THF | Tetrahydrofuran |
| HBr | Hydrogen bromide |
References
- Center for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2013. Available online: https://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf (accessed on 9 January 2018).
- Mclntosh, J.; MedicalNewsToday. Antibiotic Resistance: What You Need to Know. 2018. Available online: https://www.medicalnewstoday.com/articles/283963.php (accessed on 9 December 2019).
- Perry, J.D.; Freydière, A.M. The application of chromogenic media in clinical microbiology. J. Appl. Microbiol. 2007, 103, 2046–2055. [Google Scholar] [CrossRef] [PubMed]
- Váradi, L.; Luo, J.L.; Hibbs, D.E.; Perry, J.D.; Anderson, R.J.; Orenga, S.; Groundwater, P.W. Methods for the detection and identification of pathogenic bacteria: Past, present, and future. Chem. Soc. Rev. 2017, 46, 4818–4832. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.D.; Riley, G.; Gould, F.K.; Perez, J.M.; Boissier, E.; Ouedraogo, R.T.; Freydiere, A.M. Alafosfalin as a selective agent for isolation of Salmonella from clinical samples. J. Clin. Microbiol. 2002, 40, 3913–3916. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oliveira, E.F.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Ramos, M.J. Mechanism of formation of the internal aldimine in pyridoxal 5′-phosphate-dependent enzymes. J. Am. Chem. Soc. 2011, 133, 15496–15505. [Google Scholar] [CrossRef] [PubMed]
- Stamper, C.G.F.; Morollo, A.A.; Ringe, D. Reaction of alanine racemase with 1-aminoethylphosphonic acid forms a stable external aldimine. Biochemistry 1998, 37, 10438–10445. [Google Scholar] [CrossRef] [PubMed]
- James, A.L.; Perry, J.D.; Rigby, A.; Stanforth, S.P. Synthesis and evaluation of novel chromogenic aminopeptidase substrates for microorganism detection and identification. Bioorg. Med. Chem. Lett. 2007, 17, 1418–1421. [Google Scholar] [CrossRef]
- Kumar, A.; Schweizer, H.P. Bacterial resistance to antibiotics: Active efflux and reduced uptake. Adv. Drug Deliv. Rev. 2005, 57, 1486–1513. [Google Scholar] [CrossRef]
- Verheul, A.; Hagting, A.; Amezaga, M.R.; Booth, I.R.; Rombouts, F.M.; Abee, T. A Di- and tripeptide transport system can supply Listeria Monocytogenes Scott A with amino acids essential for growth. Appl. Environ. Microbiol. 1995, 61, 226–233. [Google Scholar] [CrossRef]
- Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Lambert, R.W.; Ringrose, P.S. Phosphonopeptides as antibacterial agents: Rationale, chemistry, and structure-activity relationships. Antimicrob. Agents Chemother. 1979, 15, 677–683. [Google Scholar] [CrossRef]
- Atherton, F.R.; Hassall, C.H.; Lambert, R.W. Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl)phosphonic acid and (aminomethyl)phosphonic acid. J. Med. Chem. 1986, 29, 29–40. [Google Scholar] [CrossRef]
- Atherton, F.R.; Hall, M.J.; Hassall, C.H.; Holmes, S.W.; Lambert, R.W.; Lloyd, W.J.; Ringrose, P.S. Phosphonopeptide antibacterial agents related to alafosfalin: Design, synthesis, and structure-activity relationships. Antimicrob. Agents Chemother. 1980, 18, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Walsh, C. Suicide substrates for the alanine racemase of Escherichia coli B. Biochemistry 1978, 17, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Wasserman, S.A.; Dudek, E.; Lerner, S.A.; Johnston, M. Chloroalanyl and propargylglycyl dipeptides. suicide-substrate-containing antibacterials. J. Med. Chem. 1983, 26, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Boisvert, W.; Lerner, S.A.; Johnston, M. Chloroalanyl antibiotic peptides: Antagonism of their antimicrobial effects by l-alanine and l-alanyl peptides in gram-negative bacteria. J. Med. Chem. 1986, 29, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Oleksyszyn, J.; Tyka, R.; Mastalerz, P. Direct synthesis of 1-aminoalkanephosphonic and 1-aminoalkanephosphinic acids from phosphorus trichloride or dichlorophosphines. Synthesis 1978, 6, 479–480. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Stec, W.J. Synthesis of 1-aminoalkanephosphonates via thioureidoalkanephosphonates. Synthesis 1978, 6, 469–472. [Google Scholar] [CrossRef]
- Kudzin, Z.H.; Kotynski, A. Synthesis of O,O-Dialkyl 1-Aminoalkanephosphonates. Synthesis 1980, 12, 1028–1032. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Guarneri, M.; Moroder, F.; Pollini, G.P.; Simoni, D. Synthesis of 1-phthalimidoalkanephosphonates. Synthesis 1982, 8, 653–655. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef]
- Cellier, M.; James, A.L.; Orenga, S.; Perry, J.D.; Rasul, A.K.; Robinson, S.N.; Stanforth, S.P. Novel chromogenic aminopeptidase substrates for the detection and identification of clinically important microorganisms. Bioorg. Med. Chem. 2014, 22, 5249–5269. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hossain, N.; Lastovica, A.J. An evaluation of the Oxoid biochemical identification system campy rapid screening test for Campylobacteraceae and Helicobacter spp. Lett. Appl. Microbiol. 2009, 48, 675–679. [Google Scholar] [CrossRef]
- Bonestroo, H.J.C.; de Winter-de Groot, K.M.; van der Ent, C.K.; Arets, H.G.M. Upper and lower airway cultures in children with cystic fibrosis: Do not neglect the upper airways. J. Cyst. Fibros. 2010, 9, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Barillova, P.; Tchesnokova, V.; Dübbers, A.; Küster, P.; Peters, G.; Dobrindt, U.; Sokurenko, E.V.; Kahl, B.C. Prevalence and persistence of Escherichia coli in the airways of cystic fibrosis patients—An unrecognized CF pathogen? Int. J. Med. Microbiol. 2014, 304, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.L.; Emerson, J.; Stapp, J.R.; Yim, D.L.; Krzewinski, J.; Louden, L.; Ramsey, B.W.; Clausen, C.R. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin. Infect. Dis. 1998, 27, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Druggan, P. Improvements in or Relating to Selective Agents for Biological Cultures. PCT International Application Patent No. WO0222785, 21 March 2002. [Google Scholar]
- Kudzin, Z.H.; Luczak, J. A Facile Conversion of Aminoalkanephosphonic Acids Into O,O-Dialkyl N-Acylaminoalkanephosphonate Derivatives. Synthesis 1995, 1995, 509–511. [Google Scholar] [CrossRef]
- Lavielle, S.; Ling, N.C.; Saltman, R.; Guillemn, R.C. Synthesis of a glycotripeptide and a glycosomatostatin containing the 3-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-l-serine residue. Carbohydr. Res. 1981, 89, 229–236. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).