3.1.2. Experimental Procedures and Compound Data
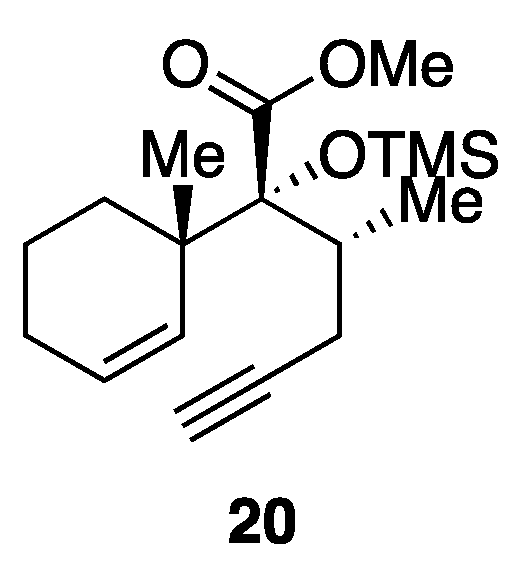
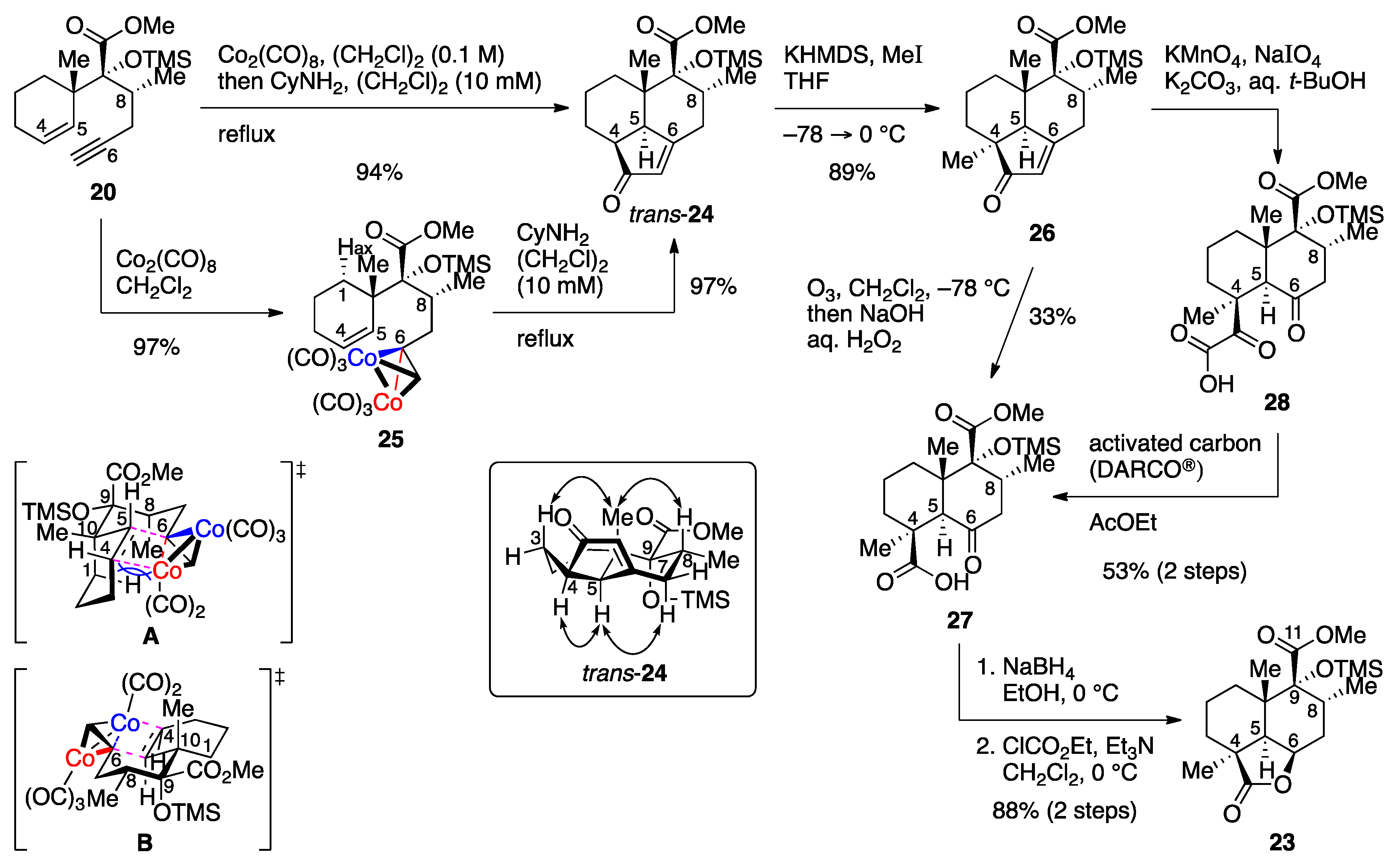
Methyl [2R,2(1S),3R]-3-methyl-2-(1-methylcyclohex-2-en-1-yl)-2-(trimethylsilyl)oxyhex-5-ynoate dicobalt hexacarbonyl complex (25). Co2(CO)8 (663 mg, 1.94 mmol) was added to an ice-cooled (0 °C) solution of enyne 20 (522 mg, 1.62 mmol) in CH2Cl2 (16 mL). After 1.5 h of stirring at room temperature, the reaction mixture was concentrated in vacuo, and the reddish brown residue was chromatographed twice (silica gel 38 g, 20:1 n-hexane/AcOEt) to give dicobalt complex 25 (958 mg, 97%) as a dark red oil. Rf 0.51 (20:1 n-hexane/AcOEt); IR (neat) 2951, 2091, 2048, 2016, 1748, 1250, 1175, 1138, 841 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.19 (s, 9H, Si(CH3)3), 0.87 (d, J = 6.6 Hz, 3H, C8-CH3), 1.00 (s, 3H, C10-CH3), 1.42 (br d, J = 13.8 Hz, 1H, one of C1-H2), 1.63 (m, 1H, one of C2-H2), 1.71 (m, 1H, one of C2-H2), 1.89−2.03 (m, 3H, one of C1-H2, C3-H2), 2.28 (ddq, J = 2.7, 11.4, 6.6 Hz, 1H, C8-H), 2.60 (ddd, J = 1.0, 11.4, 14.7 Hz, 1H, one of C7-H2), 3.49 (br d, J = 14.7 Hz, 1H, one of C7-H2), 3.71 (s, 3H, CO2CH3), 5.71 (ddd, J = 1.7, 5.2, 10.2 Hz, 1H, C4-H), 5.82 (br d, J = 10.2 Hz, 1H, C5-H), 6.01 (br t, J = 1.0 Hz, 1H, C6-CH); 13C-NMR (125.7 MHz, CDCl3) δ 2.8 (CH3), 16.1 (CH3), 20.0 (CH2), 24.4 (CH2), 25.1 (CH3), 31.5 (CH2), 36.9 (CH2), 41.3 (CH), 42.4 (C), 51.5 (CH3), 74.2 (C), 88.5 (C), 95.8 (CH), 126.8 (CH), 132.8 (CH), 175.1 (C), 200.0 (C).
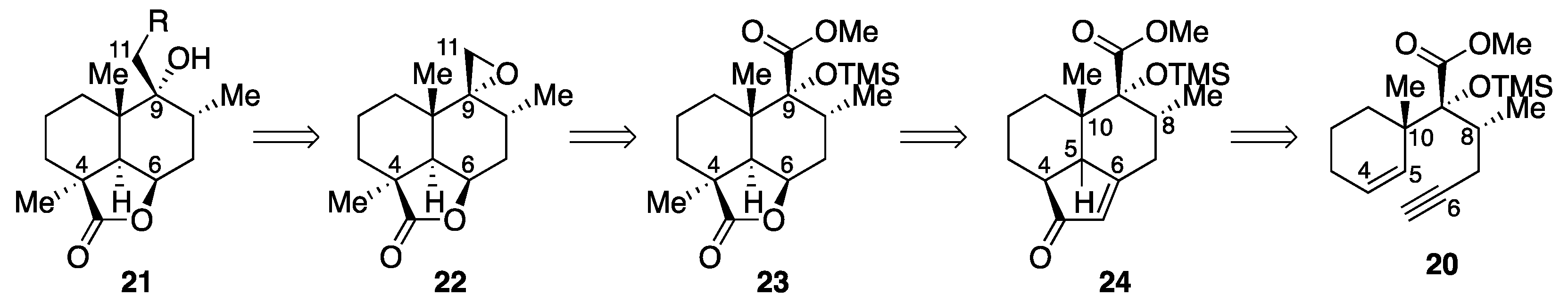
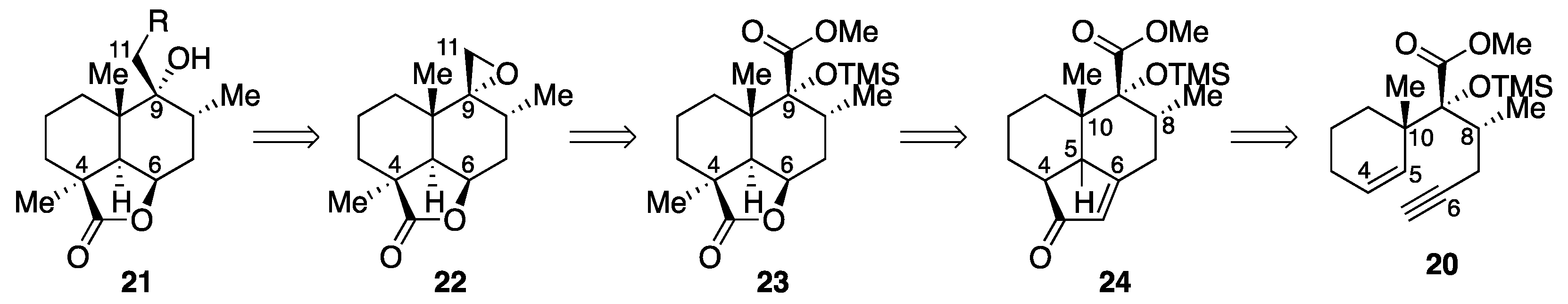
Methyl (1S,6R,7R,8S,12R)-6,8-dimethyl-2-oxo-7-(trimethylsilyl)oxytricyclo[6.3.1.04,12]dodec-3-ene-7-carboxylate (trans-24). A solution of dicobalt complex 25 (150 mg, 0.247 mmol) in 1,2-dichloroethane (5 mL plus 2 × 2.5 mL rinse) was added to a refluxing solution of cyclohexylamine (0.17 mL, 1.49 mmol) in 1,2-dichloroethane (15 mL), and the resulting mixture was refluxed for 1.5 h. After cooling, the mixture was filtered through a Celite pad, and the filtrate was concentrated in vacuo. Purification of the residue (503 mg) by column chromatography (silica gel 30 g, 3:1 n-hexane/AcOEt) gave enone trans-24 (84.0 mg, 97%) as a white solid. Rf 0.42 (3:1 n-hexane/AcOEt); mp 75−76 °C (colorless plates from n-hexane); [α + 62.1 (c 1.34, CHCl3); IR (KBr) 2945, 1736, 1694, 1620, 1458, 1260, 1184, 1096, 1034, 982, 843 cm−1; 1H-NMR (500 MHz, CD2Cl2) δ 0.20 (s, 9H, Si(CH3)3), 0.62 (s, 3H, C10-CH3), 0.91 (d, J = 6.2 Hz, 3H, C8-CH3), 1.01 (dt, J = 13.4, 7.4 Hz, 1H, one of C1-H2), 1.51−1.63 (m, 2H, one of C2-H2, one of C3-H2), 1.69 (m, 1H, one of C2-H2), 1.80 (m, 1H, one of C3-H2), 1.89 (ddd, J = 4.5, 8.9, 13.4 Hz, 1H, one of C1-H2), 2.16 (t, J = 13.9 Hz, 1H, one of C7-H2), 2.47 (ddd, J = 7.3, 8.5, 9.5 Hz, 1H, C4-H), 2.56−2.61 (m, 2H, one of C7-H2, C8-H), 3.16 (d, J = 7.3 Hz, 1H, C5-H), 3.70 (s, 3H, CO2CH3), 5.80 (s, 1H, =CHCO); 13C-NMR (125.7 MHz, CD2Cl2) δ 2.6 (CH3), 18.3 (CH3), 19.7 (CH2), 19.9 (CH2), 21.1 (CH3), 28.8 (CH2), 34.9 (CH2), 37.0 (CH), 43.7 (CH), 45.0 (CH), 45.8 (C), 51.8 (CH3), 86.1 (C), 126.0 (CH), 172.9 (CH), 179.4 (C), 211.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C19H30O4SiNa 373.1806; found 373.1823.
One-pot reaction. Co2(CO)8 (4.08 g, 11.9 mmol) was added to an ice-cooled (0 °C) solution of enyne 20 (3.20 g, 9.93 mmol) in 1,2-dichloroethane (100 mL), and the mixture was stirred at room temperature for 3 h. The resulting dark red suspension was diluted with 1,2-dichloroethane (900 mL), and cyclohexylamine (8.0 mL, 70.7 mmol) was added. After 5 h of heating at reflux, the reaction mixture was cooled to room temperature, and the volatile elements were removed in vacuo. The residue was suspended in 4:1 n-hexane/AcOEt and filtered through a Celite pad. Evaporation of the filtrate in vacuo furnished the crude product (5.75 g), which was purified by column chromatography (silica gel 200 g, 25:1 → 10:1 → 4:1 → 1:1 n-hexane/AcOEt) to give enone trans-24 (3.26 g, 94%) as a white solid.
Methyl (1S,6R,7R,8S,12R)-1,6,8-trimethyl-2-oxo-7-(trimethylsilyl)oxytricyclo[6.3.1.04,12]dodec-3-ene-7-carboxylate (26). KHMDS in toluene (0.5 M, 6.2 mL, 3.10 mmol) was added to a cooled (−78 °C) solution of enone trans-24 (988 mg, 2.82 mmol) in THF (30 mL). After 40 min of stirring, iodomethane (0.23 mL, 3.69 mmol) was added, and the resulting mixture was stirred at 0 °C for 40 min. The reaction was quenched with saturated aqueous NH4Cl (30 mL), and the mixture was extracted with AcOEt (3 × 60 mL). The combined organic extracts were washed with brine (2 × 30 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (1.16 g), which was purified by column chromatography (silica gel 30 g, 6:1 n-hexane/AcOEt) to give methylated product 26 (915 mg, 89%) as a yellow oil. Rf 0.40 (4:1 n-hexane/AcOEt); [α +72.2 (c 0.95, CHCl3); IR (neat) 3433, 2953, 1738, 1703, 1624, 1458, 1252, 1186, 1103, 1042, 841 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.20 (s, 9H, Si(CH3)3), 0.71 (s, 3H, C10-CH3), 0.91 (d, J = 6.3 Hz, 3H, C8-CH3), 1.02 (ddd, J = 7.4, 9.2, 13.5 Hz, 1H, one of C1-H2), 1.11 (s, 3H, C4-CH3), 1.43 (m, 1H, one of (CH2)2), 1.60 (m, 1H, one of (CH2)2), 1.64−1.75 (m, 2H, two of (CH2)2), 1.92 (ddd, J = 2.8, 10.2, 13.5 Hz, 1H, one of C1-H2), 2.12 (m, 1H, one of C7-H2), 2.54−2.62 (m, 2H, one of C7-H2, C8-H), 2.67 (br s, 1H, C5-H), 3.73 (s, 3H, CO2CH3), 5.78 (t, J = 1.4 Hz, 1H, =CHCO); 13C-NMR (125.7 MHz, CDCl3) δ 2.5 (CH3), 17.7 (CH2), 18.0 (CH3), 21.1 (CH3), 26.6 (CH2), 27.3 (CH3), 28.0 (CH2), 34.8 (CH2), 36.4 (CH), 45.80 (C), 45.84 (C), 51.5 (CH3), 52.3 (CH), 85.7 (C), 123.8 (CH), 172.6 (C), 178.5 (C), 215.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C20H32O4SiNa 387.1962; found 387.1950.
(1S,4aS,5R,6R,8aR)-5-(Methoxycarbonyl)-1,4a,6-trimethyl-8-oxo-5-(trimethylsilyl)oxydecahydronaphthalene-1-carboxylic acid (27). KMnO4 (4.6 mg, 29.1 μmol) was added to a solution of NaIO4 (270 mg, 1.26 mmol) in H2O (7.0 mL), and the mixture was stirred for 30 min. To the mixture was added K2CO3 (23.0 mg, 0.166 mmol), followed by a solution of enone 26 (50.3 mg, 0.138 mmol) in t-BuOH (1.4 mL plus 2 × 1 mL rinse). After 24 h of stirring, the reaction was quenched with NaHSO3 (507 mg, 4.87 mmol), and the resulting mixture was extracted with AcOEt (3 × 20 mL). The combined organic extracts were washed with brine (2 × 15 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (70.9 mg), which was used without further purification.
Activated carbon (708 mg) was added to an ice-cooled (0 °C) solution of the crude α-ketocarboxylic acid 28 (70.9 mg) in AcOEt (14 mL). After 63 h of stirring at room temperature, the resulting suspension was filtered through a Celite pad, and the filtrate was evaporated in vacuo. Purification of the crude product (74.7 mg) by column chromatography (silica gel 5.2 g, 7:1 n-hexane/AcOEt) gave γ-ketocarboxylic acid 27 (28.3 mg, 53%) as an amorphous solid. Rf 0.60 (1:1 n-hexane/AcOEt); [α + 62.5 (c 1.01, CHCl3); IR (neat) 2953, 1732, 1717, 1674, 1456, 1252, 1182, 1153, 1090, 843 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.24 (s, 9H, Si(CH3)3), 0.82 (dt, J = 3.5, 13.4 Hz, 1H, one of C3-H2), 0.88 (d, J = 6.4 Hz, 3H, C8-CH3), 0.97 (s, 3H, C10-CH3), 1.04 (d, J = 13.2 Hz, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.51 (m, 1H, one of C2-H2), 1.85 (m, 1H, one of C2-H2), 1.92 (dt, J = 4.3, 13.2 Hz, 1H, one of C1-H2), 2.26 (d, J = 13.4 Hz, 1H, one of C3-H2), 2.30 (dd, J = 4.3, 13.0 Hz, 1H, one of C7-H2), 2.51 (t, J = 13.0 Hz, 1H, one of C7-H2), 2.72 (ddq, J = 4.3, 13.0, 6.4 Hz, 1H, C8-H), 3.13 (s, 1H, C5-H), 3.75 (s, 3H, CO2CH3), 12.62 (br s, 1H, CO2H); 13C-NMR (125.7 MHz, CDCl3) δ 2.8 (CH3), 17.3 (CH3), 17.5 (CH3), 18.4 (CH2), 28.0 (CH3), 32.7 (CH2), 37.0 (CH), 38.7 (CH2), 43.6 (C), 46.4 (CH2), 49.6 (C), 52.0 (CH3), 60.3 (CH), 86.1 (C), 171.9 (C), 176.0 (C), 218.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C19H32O6SiNa 407.1860; found 407.1847.
Methyl (1S,4R,6R,7R,8S,12R)-1,6,8-trimethyl-2-oxo-7-(trimethylsilyl)oxy-3-oxatricyclo[6.3.1.04,12]dodecane-7-carboxylate (23). A solution of γ-ketocarboxylic acid 27 (38.6 mg, 0.10 mmol) in EtOH (0.25 mL plus 2 × 0.25 mL rinse) was added to an ice-cooled (0 °C) solution of NaBH4 (4.3 mg, 0.11 mmol) in EtOH (0.5 mL). After 30 min of stirring at 0 °C, NaBH4 (3.7 mg, 0.098 mmol) was added, and the mixture was stirred at 0 °C for another 30 min. The reaction was quenched with saturated aqueous NH4Cl (5 mL), and the resulting mixture was partitioned between AcOEt (10 mL) and H2O (3 mL). The aqueous layer was extracted with AcOEt (5 × 10 mL), and the combined organic extracts were washed with brine (10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (42.7 mg), which was used without further purification.
Ethyl chloroformate (50 μL, 0.523 mmol) was added to an ice-cooled (0 °C) mixture of crude γ-hydroxycarboxylic acid (42.7 mg) and Et3N (0.10 mL, 0.717 mmol) in CH2Cl2 (1.7 mL), and the mixture was stirred for 30 min. The reaction was quenched with saturated aqueous NaHCO3 (5 mL), and the mixture was partitioned between n-hexane/AcOEt (10:1, 22 mL) and H2O (5 mL). The aqueous layer was extracted with n-hexane/AcOEt (10:1, 22 mL), and the combined organic extracts were washed with brine (20 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo followed by column chromatography (silica gel 2.5 g, 9:1 n-hexane/AcOEt) afforded lactone 23 (32.6 mg, 88% for two steps) as a colorless oil. Rf 0.44 (4:1 n-hexane/AcOEt); [α + 24.6 (c 1.07, CHCl3); IR (neat) 2953, 1771, 1732, 1456, 1250, 1180, 1142, 1105, 1053, 841 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.15 (s, 9H, Si(CH3)3), 0.79 (d, J = 6.4 Hz, 3H, C8-CH3), 0.85 (dt, J = 13.0, 8.5 Hz, 1H, one of C1-H2), 1.10 (s, 3H, C10-CH3), 1.30 (s, 3H, C4-CH3), 1.42−1.56 (m, 3H, one of C2-H2, one of C3-H2, one of C7-H2), 1.69 (m, 1H, one of C2-H2), 1.77 (dd, J = 11.5, 13.0 Hz, 1H, one of C1-H2), 2.10−2.16 (m, 2H, one of C3-H2, C5-H), 2.22 (dd, J = 6.0, 16.2 Hz, 1H, one of C7-H2), 2.56 (m, 1H, C8-H), 3.73 (s, 3H, CO2CH3), 4.68 (br t, J = 5.3 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, acetone-d6) δ 2.6 (CH3), 18.38 (CH3), 18.40 (CH2), 22.3 (CH3), 23.4 (CH3), 29.0 (CH2), 29.4 (CH2), 30.6 (CH), 31.9 (CH2), 40.4 (C), 44.1 (C), 45.6 (CH), 52.0 (CH3), 76.0 (CH), 86.1 (C), 173.6 (C), 183.2 (C); HRMS (ESI) m/z [M + Na]+ calcd for C19H32O5SiNa 391.1911; found 391.1917.
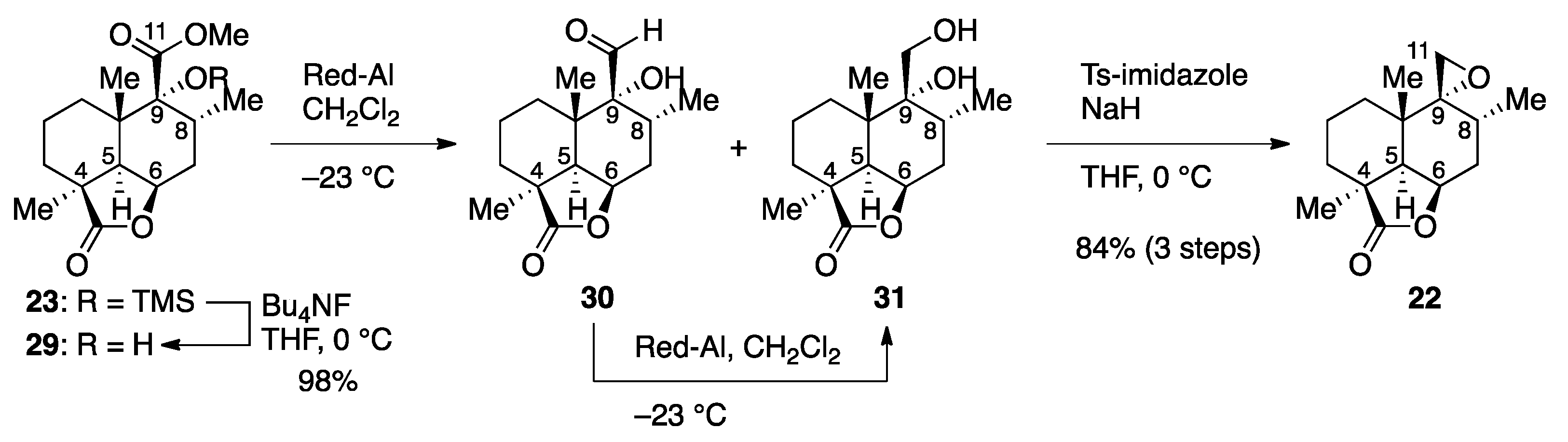
Methyl (1S,4R,6R,7R,8S,12R)-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane-7-carboxylate (29). Bu4NF in THF (1.0 M, 2.0 mL, 2.0 mmol) was added to an ice-cooled (0 °C) solution of TMS ether 23 (488 mg, 1.32 mmol) in THF (14 mL). After 1 h of stirring at 0 °C, the mixture was partitioned between AcOEt (40 mL) and H2O (15 mL), and the aqueous layer was extracted with AcOEt (40 mL). The combined organic extracts were washed with brine (2 × 20 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (634 mg), which was purified by column chromatography (silica gel 10 g, 10:1 → 3:1 n-hexane/AcOEt) to give α-hydroxyester 29 (383 mg, 98%) as a white solid. Rf 0.45 (2:1 n-hexane/AcOEt); mp 142−143 °C (colorless needles from n-hexane); [α + 25.0 (c 1.04, CHCl3); IR (KBr) 3455, 2959, 1761, 1732, 1466, 1375, 1236, 1144, 1098, 1045, 932 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.78 (d, J = 6.4 Hz, 3H, C8-CH3), 0.87 (dt, J = 13.0, 8.5 Hz, 1H, one of C1-H2), 1.22 (s, 3H, C10-CH3), 1.30 (s, 3H, C4-CH3), 1.44−1.71 (m, 5H, one of C1-H2, C2-H2, one of C3-H2, one of C7-H2), 2.15 (dt, J = 4.5, 13.4 Hz, 1H, one of C3-H2), 2.28 (dd, J = 6.3, 16.4 Hz, 1H, one of C7-H2), 2.32 (d, J = 4.6 Hz, 1H, C5-H), 2.53 (ddq, J = 6.3, 11.7, 6.4 Hz, 1H, C8-H), 3.33 (s, 1H, C9-OH), 3.82 (s, 3H, CO2CH3), 4.74 (dd, J = 4.6, 6.8 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.4 (CH3), 17.6 (CH2), 21.6 (CH3), 23.0 (CH3), 27.9 (CH2), 28.1 (CH2), 29.0 (CH), 30.7 (CH2), 38.9 (C), 43.4 (C), 44.7 (CH), 52.8 (CH3), 75.9 (CH), 80.8 (C), 175.9 (C), 183.6 (C); HRMS (ESI) m/z [M + Na]+ calcd for C16H24O5Na 319.1516; found 319.1519; Anal. Calcd for C16H24O5: C, 64.84; H, 8.16. Found: C, 64.82; H, 8.06.
(2′R,2aS,5aS,6R,7R,8aR)-2a,5a,7-Trimethyloctahydrospiro[6H-naphtho[1,8-bc]furan-6,2′-oxiran]-2(2aH)-one (22). Sodium bis(2-methoxyethoxy)aluminum hydride in toluene (3.3 M, 1.1 mL, 3.6 mmol) was diluted with CH2Cl2 (5.6 mL), and the solution was cooled to −78 °C. A solution of α-hydroxyester 29 (255 mg, 0.86 mmol) in CH2Cl2 (2 mL plus 2 × 0.5 mL rinse) was added, and the mixture was stirred at −23 °C for 22 h. The reaction was quenched by addition of MeOH (3 mL) followed by 10% aqueous potassium sodium tartrate (12 mL). After 3 h of stirring, the resulting mixture was extracted with AcOEt (2 × 40 mL), and the combined organic extracts were washed with brine (2 × 20 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (235 mg), which was used without further purification.
This sequence was repeated employing bis(2-methoxyethoxy)aluminum hydride in toluene (3.3 M, 0.16 mL, 0.53 mmol) and CH2Cl2 (4 mL) with the reaction time of 16 h at −23 °C. The crude product (216 mg) was used without further purification.
A solution of crude diol 31 (216 mg) in THF (2 mL plus 1 mL and 2 × 0.5 mL rinse) was added dropwise to an ice-cooled (0 °C) suspension of NaH (60% in oil, 240 mg, 6.01 mmol) in THF (4.6 mL). After 30 min of stirring at 0 °C, p-toluenesulfonyl imidazole (577 mg, 2.59 mmol) was added, and the mixture was stirred at 0 °C for 11 h. The reaction was quenched with saturated aqueous NH4Cl (10 mL), and the resulting mixture was extracted with AcOEt (2 × 40 mL). The combined organic extracts were washed with brine (2 × 20 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (563 mg), which was purified by column chromatography (silica gel 15 g, 40:1 → 10:1 n-hexane/AcOEt) to give epoxide 22 (182 mg, 84% for three steps) as a white solid. Rf 0.53 (2:1 n-hexane/AcOEt); mp 83−84 °C (colorless needles from 8:1 n-hexane/Et2O); [α +13.7 (c 1.21, CHCl3); IR (KBr) 2941, 1761, 1464, 1393, 1354, 1209, 1105, 997 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.76 (d, J = 6.6 Hz, 3H, C8-CH3), 0.97 (dt, J = 12.4, 8.5 Hz, 1H, one of C1-H2), 1.24 (s, 3H, C10-CH3), 1.30 (s, 3H, C4-CH3), 1.35 (dd, J = 10.1, 12.4 Hz, 1H, one of C1-H2), 1.45−1.51 (m, 2H, one of C2-H2, one of C3-H2), 1.66 (ddd, J = 4.7, 11.8, 16.0 Hz, 1H, one of C7-H2), 1.72 (m, 1H, one of C2-H2), 1.90 (d, J = 4.7 Hz, 1H, C5-H), 2.14 (dt, J = 5.4, 15.5 Hz, 1H, one of C3-H2), 2.37 (dd, J = 6.1, 16.0 Hz, 1H, one of C7-H2), 2.55 (ddq, J = 6.1, 11.8, 6.6 Hz, 1H, C8-H), 2.63 (d, J = 3.9 Hz, 1H, one of C11-H2), 2.71 (d, J = 3.9 Hz, 1H, one of C11-H2), 4.79 (br t, J = 4.7 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 15.0 (CH3), 17.3 (CH2), 22.4 (CH3), 24.9 (CH), 25.0 (CH3), 26.8 (CH2), 28.1 (C), 32.5 (CH2), 34.6 (C), 44.1 (C), 45.9 (CH2), 47.5 (CH), 63.7 (C), 76.0 (CH), 183.3 (C); HRMS (ESI) m/z [M + Na]+ calcd for C15H22O3Na 273.1461; found 273.1469.
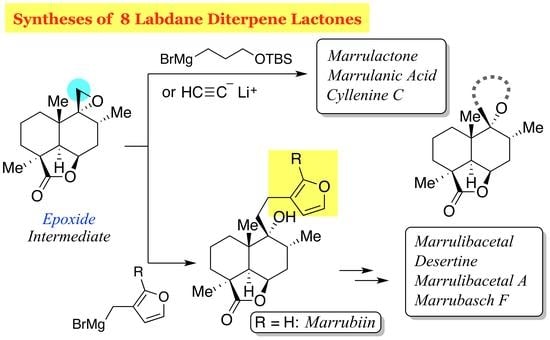
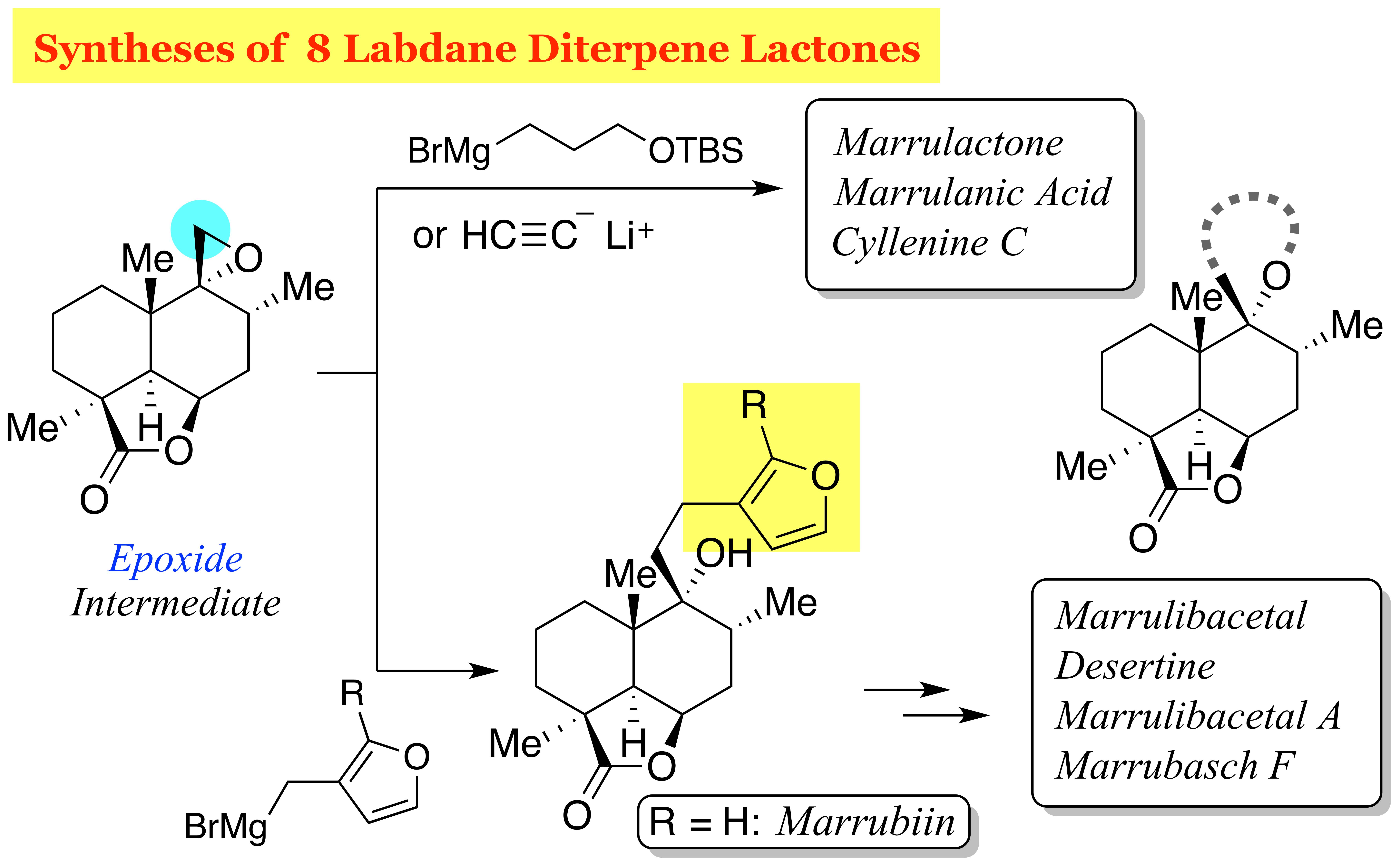
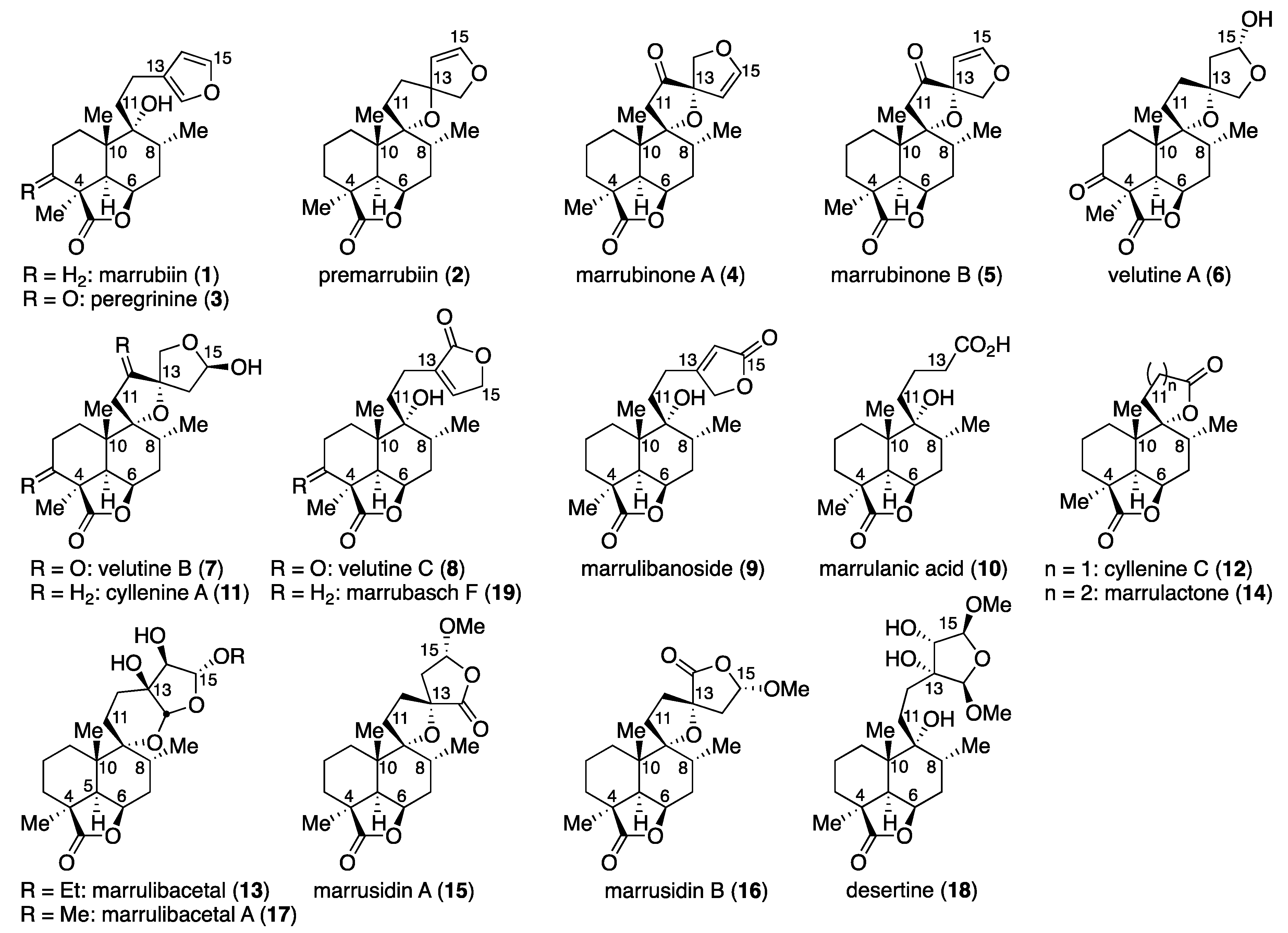
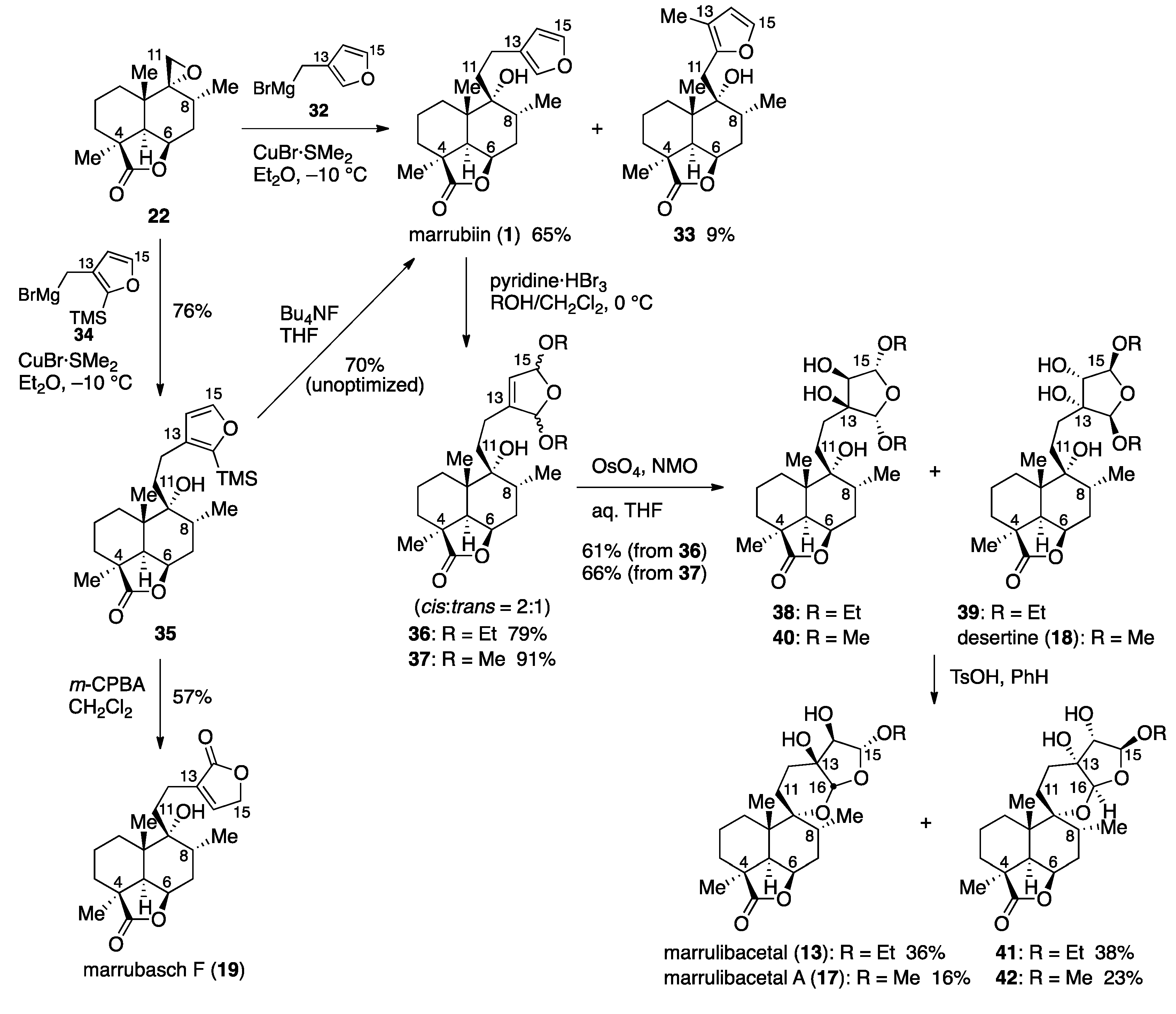
Marrubiin (
1). To a cooled (−10 °C) suspension of CuBr·SMe
2 (65.1 mg, 0.316 mmol) in Et
2O (1.5 mL) was added a 0.14 M solution of (3-furylmethyl)magnesium bromide (
32) in Et
2O (3.35 mL, 0.69 mmol) [prepared from 3-(bromomethyl)furan (563 mg, 3.37 mmol) and magnesium (103 mg, 4.22 mmol) in Et
2O (4 mL) at 0 °C], followed by addition of a solution of epoxide
22 (40.2 mg, 0.161 mmol) in Et
2O (0.5 mL plus 2 × 0.5 mL rinse). After 2 h, the reaction was quenched with saturated aqueous NH
4Cl (5 mL), and the resulting mixture was extracted with AcOEt (3 × 10 mL). The combined organic extracts were washed with brine (20 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the crude product (200 mg), which was chromatographed twice (silica gel 5 g, 20:1 → 10:1
n-hexane/AcOEt) to give marrubiin (
1, 34.7 mg, 65%) and isomer
33 (4.7 mg, 9%) as white solids.
Rf 0.59 (1:1
n-hexane/AcOEt); mp 160−161 °C (colorless needles from 4:1
n-hexane/AcOEt) (lit. [
38], mp 160 °C); [α
+34.4 (
c 1.04, CHCl
3) [lit. [
38], [α
+35.8 (
c 3.1, CHCl
3)]; IR (KBr) 3466, 2940, 2870, 1740, 1468, 1395, 1356, 1304, 1269, 1200, 1153, 1101, 1024, 984 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 0.97 (d,
J = 6.4 Hz, 3H, C8-C
H3), 1.07 (s, 3H, C10-C
H3), 1.26 (s, 1H, C9-O
H), 1.29 (s, 3H, C4-C
H3), 1.32 (dt,
J = 12.8, 8.5 Hz, 1H, one of C1-
H2), 1.43−1.55 (m, 2H, one of C2-
H2, one of C3-
H2), 1.66−1.79 (m, 4H, one of C1-
H2, one of C2-
H2, one of C7-
H2, one of C11-
H2), 1.90 (ddd,
J = 7.2, 10.1, 14.4 Hz, 1H, one of C11-
H2), 2.09−2.18 (m, 3H, one of C3-
H2, one of C7-
H2, C8-
H), 2.23 (d,
J = 4.6 Hz, 1H, C5-
H), 2.48−2.58 (m, 2H, C12-
H2), 4.74 (br dd,
J = 4.6, 6.5 Hz, 1H, C6-
H), 6.27 (s, 1H, C14-
H), 7.24 (s, 1H, C16-
H), 7.37 (s, 1H, C15-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 16.6 (CH
3), 18.2 (CH
2), 21.0 (CH
2), 22.3 (CH
3), 22.9 (CH
3), 28.3 (CH
2), 28.6 (CH
2), 31.5 (CH
2), 32.4 (CH), 35.1 (CH
2), 39.7 (C), 43.8 (C), 44.8 (CH), 75.8 (C), 76.2 (CH), 110.7 (CH), 125.0 (C), 138.6 (CH), 143.1 (CH), 183.8 (C); HRMS (ESI)
m/
z [M + Na]
+ calcd for C
20H
28O
4Na 355.1880; found 355.1867.
Data for (1S,4R,6R,7R,8S,12R)-7-[(3-methylfuran-2-yl)methyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (33): Rf 0.27 (3:1 n-hexane/AcOEt); mp 173−174 °C (colorless needles from 3:1 n-hexane/AcOEt); [α +12.0 (c 0.38, CHCl3); IR (KBr) 3453, 2955, 2928, 2874, 1738, 1456, 1393, 1352, 1300, 1279, 1198, 1152, 1138, 999, 982 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.85 (d, J = 6.4 Hz, 3H, C8-CH3), 1.07 (s, 3H, C10-CH3), 1.15 (m, 1H, one of C1-H2), 1.28 (s, 3H, C4-CH3), 1.41−1.52 (m, 2H, one of C1-H2, one of C2-H2), 1.66−1.74 (m, 3H, one of C2-H2, one of C3-H2, one of C7-H2), 1.98 (s, 3H, C13-CH3), 2.06−2.21 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.28 (d, J = 4.7 Hz, 1H, C5-H), 2.33 (s, 1H, C9-OH), 2.78 (d, J = 15.5 Hz, 1H, one of C11-H2), 2.88 (d, J = 15.5 Hz, 1H, one of C11-H2), 4.73 (ddd, J = 1.4, 4.7, 6.1 Hz, 1H, C6-H), 6.19 (d, J = 1.8 Hz, 1H, C14-H), 7.28 (d, J = 1.8 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 10.3 (CH3), 16.3 (CH3), 18.3 (CH2), 22.2 (CH3), 22.9 (CH3), 28.23 (CH2), 28.24 (CH2), 30.6 (CH2), 31.93 (CH), 31.94 (CH2), 39.6 (C), 43.8 (C), 44.6 (CH), 76.2 (CH), 76.9 (C), 113.3 (CH), 116.4 (C), 140.5 (CH), 148.0 (C), 183.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C20H28O4Na 355.1880; found 355.1871.
(1S,4R,6R,7R,8S,12R)-7-[2-[2-(Trimethylsilyl)furan-3-yl]ethyl]]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (35). Phosphorus tribromide (0.28 mL, 2.98 mmol) was added to an ice-cooled (0 °C) solution of [2-(trimethylsilyl)furan-3-yl]methanol (998 mg, 5.86 mmol) in Et2O (30 mL). After 30 min of stirring, the reaction was quenched with brine (30 mL), and the resulting mixture was extracted with Et2O (30 mL). The organic extract was dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale brown oil, which was purified by distillation to give 3-(bromomethyl)-2-(trimethylsilyl)furan (618 mg, 45%) as a pale brown oil.
A solution of 3-(bromomethyl)-2-(trimethylsilyl)furan (584 mg, 2.50 mmol) in Et2O (1 mL) was added to a cooled (−10 °C) suspension of magnesium tuning (102 mg, 4.20 mmol) in Et2O (1 mL), and the reaction mixture was stirred for 1.5 h. The 0.23 M solution of [2-(trimethylsilyl)furan-3-yl]methylmagnesium bromide (34) in Et2O (1.5 mL, 0.345 mmol) thus obtained was added to a cooled (−10 °C) suspension of CuBr·SMe2 (69.6 mg, 0.339 mmol) in Et2O (0.6 mL), followed by addition of a solution of epoxide 22 (42.6 mg, 0.170 mmol) in Et2O (1.0 mL). After 4 h of stirring, the reaction was quenched with saturated aqueous NH4Cl (3 mL), and the resulting mixture was extracted with AcOEt (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale brown oil (206 mg), which was purified by flash column chromatography (silica gel 4 g, n-hexane → 40:1 → 20:1 → 5:1 n-hexane/AcOEt) to give silylated marrubiin 35 (52.0 mg, 76%) as a colorless solid. Rf 0.32 (3:1 n-hexane/AcOEt); [α +29.3 (c 2.05, acetone); IR (neat) 3479, 3417, 2954, 2870, 1749, 1633, 1568, 1464, 1387, 1248, 1197, 1149, 1089, 1045, 1020, 991, 914, 840 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.29 (s, 9H, Si(CH3)3), 0.99 (d, J = 6.5 Hz, 3H, C8-CH3), 1.06 (s, 3H, C10-CH3), 1.29 (s, 3H, C4-CH3), 1.30 (m, 1H, one of C1-H2), 1.45 (m, 1H, one of C3-H2), 1.52 (m, 1H, one of C2-H2), 1.68−1.75 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C11-H2), 1.87 (ddd, J = 7.1, 10.3, 14.5 Hz, 1H, one of C11-H2), 2.09−2.19 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.23 (d, J = 4.6 Hz, 1H, C5-H), 2.56−2.75 (m, 2H, C12-H2), 4.74 (dd, J = 4.6, 6.3 Hz, 1H, C6-H), 6.27 (d, J = 1.2 Hz, 1H, C14-H), 7.55 (d, J = 1.2 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ −0.9 (CH3), 16.6 (CH3), 18.2 (CH2), 21.7 (CH2), 22.3 (CH3), 22.9 (CH3), 28.3 (CH2), 28.7 (CH2), 31.5 (CH2), 32.4 (CH), 36.2 (CH2), 39.7 (C), 43.8 (C), 44.9 (CH), 75.8 (C), 76.2 (CH), 110.7 (CH), 135.0 (C), 146.4 (CH), 154.3 (C), 183.9 (C); HRMS (ESI) m/z [M + Na]+ calcd for C23H36O4SiNa 427.2275; found 427.2259.
Marrubiin (1). Bu4NF in THF (1.0 M, 20 μL, 20 μmol) was added to a solution of silylated marrubiin 35 (0.7 mg, 1.7 μmol) in THF (0.2 mL). After 4 h of stirring, the mixture was partitioned between AcOEt (8 mL) and H2O (8 mL), and the aqueous layer was extracted with AcOEt (8 mL). The combined organic extracts were washed with brine (8 mL) and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (0.5 mg), which was purified by column chromatography (silica gel 1 g, 4:1 → 1:1 n-hexane/AcOEt) to give marrubiin (1, 0.4 mg, 70%) as a white solid.
(1S,4R,6R,7R,8S,12R)-7-[2-(2,5-Diethoxy-2,5-dihydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (36). Pyridinium tribromide (11.0 mg, 34 μmol) was added to an ice-cooled (0 °C) solution of marrubiin (1, 10.4 mg, 31 μmol) in CH2Cl2/EtOH (1:1, 0.6 mL). After 10 min of stirring, the reaction was quenched with a mixture of saturated aqueous NaHCO3 (3 mL) and 1 M aqueous Na2S2O3 (2 mL), and the resulting mixture was extracted with AcOEt (2 × 20 mL). The combined organic extracts were washed with brine (30 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (14.7 mg), which was purified by column chromatography (silica gel 2.5 g, 3:1 → 1:1 n-hexane/AcOEt) to give bisacetals 36 (10.4 mg, 79%, dr = 2:2:1:1) as a colorless oil. Rf 0.58 (2:3 n-hexane/AcOEt); [α +33.9 (c 1.19, CHCl3); IR (neat) 3516, 2972, 2930, 1767, 1755, 1458, 1373, 1346, 1198, 1101, 1045, 1018, 984 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.92 (d, J = 6.3 Hz, 3H, C8-CH3), 1.05 (s, 3H, C10-CH3), 1.21−1.34 (m, 11H), 1.43−1.56 (m, 2H), 1.66−1.77 (m, 4H), 1.86 (m, 1H, one of CH2), 2.05−2.32 (m, 6H), 3.53−3.82 (m, 4H, 2 × OCH2CH3), 4.74 (m, 1H, C6-H), 5.48 (s, 0.65H, C15-H), 5.60 (s, 0.65H, C16-H), 5.67 (s, 1H, C14-H), 5.75 (br d, J = 3.6 Hz, 0.35H, C16-H), 5.85 (br d, J = 3.6 Hz, 0.35H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 15.3 (CH3), 15.4 (CH3), 16.50 (CH3), 16.51 (CH3), 16.54 (CH3), 18.09 (CH2), 18.12 (CH2), 22.22 (CH3), 22.23 (CH3), 22.26 (CH3), 22.28 (CH3), 22.31 (CH2), 22.4 (CH2), 22.49 (CH2), 22.52 (CH2), 22.87 (CH3), 22.89 (CH3), 22.96 (CH3), 22.97 (CH3), 28.27 (CH2), 28.28 (CH2), 28.59 (CH2), 28.61 (CH2), 28.63 (CH2), 31.4 (CH2), 31.46 (CH2), 31.47 (CH2), 32.0 (CH2), 32.11 (CH2), 32.15 (CH), 32.22 (CH), 32.29 (CH2), 32.33 (CH2), 32.5 (CH), 39.69 (C), 39.70 (C), 39.71 (C), 39.73 (C), 43.71 (C), 43.72 (C), 43.76 (C), 44.7 (CH), 44.8 (CH), 62.3 (CH2), 62.61 (CH2), 62.62 (CH2), 62.8 (CH2), 63.1 (CH2), 63.41 (CH2), 63.42 (CH2), 63.5 (CH2), 75.37 (C), 75.388 (C), 75.394 (C), 76.06 (CH), 76.12 (CH), 76.13 (CH), 105.95 (CH), 105.97 (CH), 106.937 (CH), 106.943 (CH), 107.5 (CH), 107.6 (CH), 108.2 (CH), 108.3 (CH), 123.8 (CH), 124.0 (CH), 124.1 (CH), 124.3 (CH), 145.3 (C), 145.78 (C), 145.81 (C), 183.7 (C), 183.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C24H38O6Na 445.2561; found 445.2550.
[1S,4R,6R,7R,7(2S,3S,4R,5R),8S,12R]-7-[2-(2,5-Diethoxy-3,4-dihydroxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (38) and [1S,4R,6R,7R,7(2R,3R,4S,5S),8S, 12R]-7-[2-(2,5-diethoxy-3,4-dihydroxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatri cyclo[6.3.1.04,12]dodecane (39). A 0.157 M solution of OsO4 in t-BuOH (0.06 mL, 9.4 μmol) was added to a solution of bisacetals 36 (21.7 mg, 51 μmol) and NMO (4.8 M in H2O, 0.04 mL, 0.19 mmol) in THF/H2O (10:1, 0.55 mL). After 1 h of stirring, the reaction was quenched with 1 M aqueous Na2S2O3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (15 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the crude product (29.1 mg), which was purified by flash column chromatography (silica gel 2.3 g, 1:1 n-hexane/AcOEt) to give a 1:1 mixture of triols 38 and 39 (14.3 mg, 61%) as a colorless oil, along with recovered bisacetals 36 (8.2 mg, 38%) as a colorless oil. Rf 0.49 (1:4 n-hexane/AcOEt); [α +29.1 (c 0.56, CHCl3); IR (neat) 3462, 2930, 1751, 1458, 1389, 1375, 1101, 978 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.5 Hz, 1.5H, C8-CH3), 0.92 (d, J = 6.4 Hz, 1.5H, C8-CH3), 1.04 (s, 3H, C10-CH3), 1.207 (t, J = 7.1 Hz, 1.5H, OCH2CH3), 1.214 (t, J = 7.1 Hz, 1.5H, OCH2CH3), 1.23 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.29 (s, 3H, C4-CH3), 1.42−1.52 (m, 2H, CH2), 1.60−1.97 (m, 8H, 4 × CH2), 2.03−2.15 (m, 3.5H, CH2, C8-H, OH), 2.24 (d, J = 4.7 Hz, 1H, C5-H), 2.33 (br s, 0.5H, OH), 2.99 (d, J = 6.9 Hz, 0.5H, OH), 3.05 (m, 1H, OH), 3.44−3.57 (m, 2.5H, OCH2CH3, OH), 3.76−3.84 (m, 2H, OCH2CH3), 3.96 (m, 1H, C14-H), 4.73 (br s, 1H, C6-H), 4.85 (s, 0.5H, C16-H), 4.86 (s, 0.5H, C16-H), 4.97 (d, J = 4.3 Hz, 0.5H, C15-H), 4.98 (d, J = 4.3 Hz, 0.5H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 14.88 (CH3), 14.93 (CH3), 15.2 (CH3), 16.5 (CH3), 16.8 (CH3), 18.1 (CH2), 22.2 (CH3), 22.3 (CH3), 22.85 (CH3), 22.93 (CH3), 27.4 (CH2), 28.1 (CH2), 28.27 (CH2), 28.28 (CH2), 28.31 (CH2), 28.4 (CH2), 28.5 (CH2), 28.6 (CH2), 31.5 (CH2), 31.6 (CH2), 32.47 (CH), 32.49 (CH), 39.97 (C), 40.03 (C), 43.88 (C), 43.91 (C), 44.90 (CH), 44.93 (CH), 63.07 (CH2), 63.10 (CH2), 64.55 (CH2), 64.57 (CH2), 75.30 (C), 75.33 (C), 76.3 (CH), 76.4 (CH), 80.3 (CH), 80.4 (CH), 81.2 (C), 81.3 (C), 106.5 (CH), 106.7 (CH), 109.17 (CH), 109.19 (CH), 184.10 (C), 184.12 (C); HRMS (ESI) m/z [M + Na]+ calcd for C24H40O8Na 479.2615; found 479.2631.
Marrulibacetal (
13). TsOH (2.9 mg, 17 μmol) was added to a mixture of triols
38 and
39 (12.5 mg, 27 μmol) in benzene (1 mL), and the mixture was stirred for 1.5 h. The reaction was quenched with saturated aqueous NaHCO
3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (15 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the crude product (19.2 mg), which was purified by flash column chromatography (silica gel 2 g, 2:1
n-hexane/AcOEt) to give a mixture of marrulibacetal (
13) and its diastereomers. The mixture was flash chromatographed (silica gel 4.5 g, 30:1 CHCl
3/ acetone) to provide a mixture of marrulibacetal (
13) and C13,14,15,16-epimer
41, along with a 1:1 mixture of C15-epimer and C13,14,16-epimer (2.0 mg, 18%). Separation of marrulibacetal (
13) and
41 by flash column chromatography (silica gel 4.5 g, 50:1 CHCl
3/acetone) yielded marrulibacetal (
13, 4.1 mg, 36%) and C13,14,15,16-epimer
41 (4.3 mg, 38%) as white solids.
Rf 0.44 (4:1 CH
2Cl
2/acetone); mp 177−179 °C (colorless needles from
n-hexane/benzene); [α
−21.7 (
c 1.16, CHCl
3) [lit. [
12], [α
−13.1 (
c 0.29, CHCl
3)]; IR (neat) 3435, 2961, 2928, 1773, 1740, 1458, 1389, 1244, 1200, 1111, 1053, 935 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 1.03 (s, 3H, C10-C
H3), 1.11 (d,
J = 6.8 Hz, 3H, C8-C
H3), 1.19 (m, 1H, one of C1-
H2), 1.22 (t,
J = 7.1 Hz, 3H, OCH
2C
H3), 1.28 (s, 3H, C4-C
H3), 1.42 (m, 1H, one of C3-
H2), 1.49 (m, 1H, one of C2-
H2), 1.71−1.78 (m, 2H, one of C2-
H2, one of C11-
H2), 1.80−1.90 (m, 3H, one of C7-
H2, one of C11-
H2, one of C12-
H2), 1.98 (m, 1H, one of C1-
H2), 2.05−2.20 (m, 4H, one of C3-
H2, one of C7-
H2, C8-
H, one of C12-
H2), 2.36 (d,
J = 4.7 Hz, 1H, C5-
H), 2.60 (s, 1H, C13-O
H), 2.61 (d,
J = 6.2 Hz, 1H, C14-O
H), 3.51 (dq,
J = 9.5, 7.1 Hz, 1H, one of OC
H2CH
3), 3.82 (dq,
J = 9.5, 7.1 Hz, 1H, one of OC
H2CH
3), 3.95 (dd,
J = 2.0, 6.2 Hz, 1H, C14-
H), 4.77 (br dd,
J = 4.7, 5.9 Hz, 1H, C6-
H), 5.04 (d,
J = 2.0 Hz, 1H, C15-
H), 5.46 (s, 1H, C16-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 15.0 (CH
3), 17.9 (CH
2), 19.5 (CH
3), 21.1 (CH
2), 22.1 (CH
3), 23.1 (CH
3), 27.8 (CH
2), 28.2 (CH
2), 29.6 (CH
2), 32.3 (CH
2), 33.6 (CH), 40.9 (C), 43.9 (C), 44.6 (CH), 63.9 (CH
2), 75.6 (C), 76.5 (CH), 78.5 (CH), 80.4 (C), 105.3 (CH), 108.7 (CH), 184.0 (C); HRMS (EI)
m/
z [M
+] calcd for C
22H
34O
7 410.2305; found 410.2300.
Data for (2S,2′aS,3S,3aR,5′aS,6R,7′R,7aR,8′aR,8′bR)-2-ethoxy-3,3a-dihydroxy-2′a,5′a,7′-trimethyltetradecahydrospiro[6H-furo[2,3-b]pyran-6,6′-[6H]naphtho[1,8-bc]furan]-2′(2′aH)-one (41). Rf 0.56 (4:1 CH2Cl2/acetone); [α +57.2 (c 1.26, CHCl3); IR (KBr) 3458, 2930, 1771, 1749, 1466, 1389, 1260, 1198, 1153, 1094, 1063, 1040, 989, 941 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.96 (d, J = 6.1 Hz, 3H, C8-CH3), 1.01 (s, 3H, C10-CH3), 1.21 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.30 (s, 3H, C4-CH3), 1.39−1.54 (m, 4H, one of C1-H2, one of C2-H2, one of C3-H2, one of C11-H2), 1.70−1.83 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C12-H2), 1.95−2.11 (m, 4H, one of C3-H2, one of C7-H2, C8-H, one of C11-H2), 2.20 (m, 1H, one of C12-H2), 2.34 (d, J = 4.5 Hz, 1H, C5-H), 2.75 (d, J = 4.8 Hz, 1H, C14-OH), 2.79 (s, 1H, C13-OH), 3.46 (dq, J = 9.5, 7.1 Hz, 1H, one of OCH2CH3), 3.74 (d, J = 4.8 Hz, 1H, C14-H), 3.86 (dq, J = 9.5, 7.1 Hz, 1H, one of OCH2CH3), 4.78 (br dd, J = 4.5, 7.7 Hz, 1H, C6-H), 5.01 (s, 1H, C15-H), 5.50 (s, 1H, C16-H); 13C-NMR (125.7 MHz, CDCl3) δ 14.8 (CH3), 16.1 (CH3), 18.1 (CH2), 21.5 (CH2), 23.2 (CH3), 23.9 (CH3), 28.3 (CH2), 30.4 (CH2), 30.9 (CH2), 31.3 (CH2), 35.0 (CH), 40.8 (C), 43.9 (C), 45.5 (CH), 63.7 (CH2), 75.9 (C), 76.7 (CH), 80.0 (CH), 80.5 (C), 106.1 (CH), 108.2 (CH), 183.9 (C); HRMS (EI) m/z [M+] calcd for C22H34O7 410.2305; found 410.2297.
[1S,4R,6R,7R,7(2S,3S,4R,5R),8S,12R]-7-[2-(3,4-Dihydroxy-2,5-dimethoxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (40) and desertine (18). Pyridinium tribromide (32.9 mg, 0.102 mmol) was added to an ice-cooled (0 °C) solution of marrubiin (1, 30.5 mg, 91.7 μmol) in CH2Cl2/MeOH (1:1, 1.8 mL). After 20 min of stirring, the reaction was quenched with saturated aqueous NaHCO3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (2 × 10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the yellow oil (53.1 mg), which was purified by column chromatography (silica gel 3 g, 4:1 → 2:1 → 1:1 n-hexane/AcOEt) to give bisacetals 37 (33.1 mg, 91%, dr = 2:2:1:1) as a yellow oil.
A 0.157 M solution of OsO4 in t-BuOH (0.08 mL, 12.5 μmol) was added to a solution of bisacetals 37 (28.3 mg, 71.7 μmol) and NMO (4.8 M in H2O, 0.06 mL, 0.29 mmol) in THF/H2O (1:1, 0.8 mL). After 1 h of stirring, the reaction was quenched with 1 M aqueous Na2S2O3 (5 mL), and the resulting mixture was extracted with AcOEt (2 × 15 mL). The combined organic extracts were washed with brine (2 × 10 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the brown oil (40.7 mg), which was purified by column chromatography (silica gel 3.1 g, 2:1 → 1:1 n-hexane/AcOEt) to give a 1:1 mixture of desertine (18) and its diastereomer 40 (20.2 mg, 66%) as a brown oil, along with recovered bisacetals 37 (8.8 mg, 29%) as a colorless oil. Rf 0.30 (1:1 n-hexane/AcOEt); [α +12.9 (c 1.02, CHCl3); IR (neat) 3464, 2951, 1748, 1454, 1391, 1258, 1198, 1148, 1101, 1043, 989 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.5 Hz, 3H, C8-CH3), 1.04 (s, 1.5H, C10-CH3), 1.05 (s, 1.5H, C10-CH3), 1.28 (m, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.45 (m, 1H, one of C3-H2), 1.51 (m, 1H, one of C2-H2), 1.65 (m, 1H, one of C7-H2), 1.60−1.98 (m, 6H, one of C1-H2, one of C2-H2, C11-H2, C12-H2), 2.07 (m, 1H, C8-H), 2.11 (m, 1H, one of C3-H2), 2.14 (m, 1H, one of C7-H2), 2.22 (d, J = 4.2 Hz, 0.5H, C5-H), 2.23 (d, J = 4.2 Hz, 0.5H, C5-H), 3.40 (s, 3H, C16-OCH3), 3.47 (s, 3H, C15-OCH3), 3.92 (d, J = 3.5 Hz, 0.5H, C14-H), 3.95 (d, J = 3.5 Hz, 0.5H, C14-H), 4.73 (m, 1H, C6-H), 4.76 (s, 0.5H, C16-H), 4.77 (s, 0.5H, C16-H), 4.89 (d, J = 3.5 Hz, 0.5H, C15-H), 4.90 (d, J = 3.5 Hz, 0.5H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.4 (CH3), 16.8 (CH3), 18.1 (CH2), 22.2 (CH3), 22.3 (CH3), 22.86 (CH3), 22.94 (CH3), 27.4 (CH2), 28.0 (CH2), 28.2 (CH2), 28.26 (CH2), 28.28 (CH2), 28.41 (CH2), 28.43 (CH2), 28.5 (CH2), 31.5 (CH2), 31.6 (CH2), 32.5 (CH), 32.6 (CH), 40.0 (C), 40.1 (C), 43.87 (C), 43.91 (C), 44.9 (CH), 45.0 (CH), 55.08 (CH3), 55.11 (CH3), 56.40 (CH3), 56.42 (CH3), 75.3 (C), 75.4 (C), 76.2 (CH), 76.3 (CH), 80.2 (CH), 80.3 (CH), 81.2 (C), 81.4 (C), 108.3 (CH), 108.6 (CH), 110.7 (CH), 110.8 (CH), 183.95 (C), 183.99 (C); HRMS (ESI) m/z [M + Na]+ calcd for C22H36O8Na 451.2302; found 451.2308.
Marrulibacetal A (
17). TsOH (8.6 mg, 50 μmol) was added to a mixture of desertine (
18) and its diastereomer
40 (29.6 mg, 69.0 μmol) in benzene (1.4 mL). After 5 h of stirring, an additional portion of TsOH (1.6 mg, 9.3 μmol) was added, and the reaction mixture was stirred for 3.5 h. The reaction was quenched with saturated aqueous NaHCO
3 (2 mL), and the resulting mixture was extracted with AcOEt (3 × 5 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the brown oil (68.1 mg), which was purified by flash column chromatography (silica gel 5 g, 1:1
n-hexane/AcOEt) to give marrulibacetal A (
17, 4.3 mg, 16%) and C13,14,15,16-isomer
42 (6.2 mg, 23%) as colorless amorphous solids.
Rf 0.73 (AcOEt); [α
−14.0 (
c 1.69, CHCl
3) (lit. [
14], [α
−10.77); IR (neat) 2953, 2928, 1769, 1748, 1456, 1259, 1198, 1117, 1053, 1015, 989, 935 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 1.03 (s, 3H, C10-C
H3), 1.11 (d,
J = 7.0 Hz, 3H, C8-C
H3), 1.19 (m, 1H, one of C1-
H2), 1.28 (s, 3H, C4-C
H3), 1.42 (m, 1H, one of C3-
H2), 1.49 (m, 1H, one of C2-
H2), 1.71−1.92 (m, 5H, one of C2-
H2, one of C7-
H2, C11-
H2, one of C12-
H2), 1.96 (t,
J = 10.6 Hz, 1H, one of C1-
H2), 2.05−2.15 (m, 3H, one of C3-
H2, C8-
H, one of C12-
H2), 2.20 (dd,
J = 5.5, 16.0 Hz, 1H, one of C7-
H2), 2.38 (d,
J = 4.5 Hz, 1H, C5-
H), 3.42 (s, 3H, OC
H3), 3.89 (d,
J = 1.2 Hz, 1H, C14-
H), 4.79 (dd,
J = 4.5, 6.3 Hz, 1H, C6-
H), 4.93 (d,
J = 1.2 Hz, 1H, C15-
H), 5.48 (s, 1H, C16-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 17.9 (CH
2), 19.4 (CH
3), 20.7 (CH
2), 22.0 (CH
3), 23.1 (CH
3), 27.8 (CH
2), 28.2 (CH
2), 30.0 (CH
2), 32.2 (CH
2), 33.5 (CH), 40.9 (C), 43.9 (C), 44.6 (CH), 55.5 (CH
3), 75.8 (C), 76.5 (CH), 78.6 (CH), 80.3 (C), 105.7 (CH), 109.8 (CH), 184.1 (C); HRMS (ESI)
m/
z [M + Na]
+ calcd for C
21H
32O
7Na 419.2045; found 419.2037.
Data for (2S,2′aS,3S,3aR,5′aS,6R,7′R,7aR,8′aR,8′bR)-3,3a-dihydroxy-2-methoxy-2′a,5′a,7′-trimethyl tetradecahydrospiro[6H-furo[2,3-b]pyran-6,6′-[6H]naphtho[1,8-bc]furan]-2′(2′aH)-one (42): Rf 0.78 (AcOEt); [α +49.1 (c 0.75, CHCl3); IR (neat) 2955, 2928, 1749, 1541, 1506, 1456, 1265 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.94 (d, J = 6.1 Hz, 3H, C8-CH3), 1.01 (s, 3H, C10-CH3), 1.31 (s, 3H, C4-CH3), 1.42−1.56 (m, 4H, one of C1-H2, one of C2-H2, one of C3-H2, one of C11-H2), 1.70−1.83 (m, 4H, one of C1-H2, one of C2-H2, one of C7-H2, one of C12-H2), 1.98 (dt, J = 3.8, 14.2 Hz, 1H, one of C11-H2), 1.99−2.12 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.17 (dt, J = 4.4, 14.2 Hz, 1H, one of C12-H2), 2.35 (d, J = 4.5 Hz, 1H, C5-H), 2.87 (br s, 1H, OH), 3.41 (s, 3H, OCH3), 3.73 (s, 1H, C14-H), 4.79 (dd, J = 4.5, 6.2 Hz, 1H, C6-H), 4.91 (s, 1H, C15-H), 5.52 (s, 1H, C16-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.0 (CH3), 18.1 (CH2), 21.4 (CH2), 23.1 (CH3), 24.0 (CH3), 28.3 (CH2), 30.6 (CH2), 30.9 (CH2), 31.3 (CH2), 35.0 (CH), 40.8 (C), 43.9 (C), 45.5 (CH), 55.2 (CH3), 76.1 (C), 76.7 (CH), 79.8 (CH), 80.5 (C), 106.3 (CH), 109.4 (CH), 184.1 (C); HRMS (ESI) m/z [M + Na]+ calcd for C21H32O7Na 419.2045; found 419.2059.
Marrubasch F (
19).
m-CPBA (ca. 70%, 10.7 mg, 43.4 μmol) was azeotropically dried with benzene, and dissolved in CH
2Cl
2 (0.2 mL). The
m-CPBA solution was added to an ice-cooled (0 °C) solution of silylated marrubiin
35 (8.1 mg, 20.0 μmol) in CH
2Cl
2 (0.8 mL). After 1.5 h of stirring, the reaction was quenched with a mixture of 1 M aqueous Na
2S
2O
3 (2 mL) and saturated aqueous NaHCO
3 (2 mL), and the resulting mixture was extracted with AcOEt (10 mL). The organic extract was washed with brine (4 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the colorless oil (28.4 mg), which was purified by preparative thin layer chromatography (200 mm × 100 mm × 0.25 mm preparative silica gel plate and elution with 1:1
n-hexane/AcOEt) to give marrubasch F (
19, 3.9 mg, 56%) as a white solid.
Rf 0.32 (1:1
n-hexane/AcOEt); mp 195−196 °C (pale yellow plates from 1:2
n-hexane/AcOEt) (lit. [
20], mp 191−193 °C); [α
+41.1 (
c 0.534, CHCl
3) [lit. [
20], [α]
D + 41.5 (
c 1.00, CHCl
3)]; IR (neat) 3536, 2926, 2873, 1747, 1456, 1199, 1139, 1095, 1076, 1041, 983 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 0.96 (d,
J = 6.4 Hz, 3H, C8-C
H3), 1.04 (s, 3H, C10-C
H3), 1.25 (dt,
J = 12.8, 8.5 Hz, 1H, one of C1-
H2), 1.30 (s, 3H, C4-C
H3), 1.45 (dt,
J = 14.4, 4.8 Hz, 1H, one of C3-
H2), 1.53 (m, 1H, one of C2-
H2), 1.69−1.84 (m, 5H, one of C1-
H2, one of C2-
H2, one of C7-
H2, C11-
H2), 2.05−2.17 (m, 3H, one of C3-
H2, one of C7-
H2, C8-
H), 2.28 (d,
J = 4.5 Hz, 1H, C5-
H), 2.41−2.52 (m, 2H, C12-
H2), 4.75 (dd,
J = 4.5, 6.5 Hz, 1H, C6-
H), 4.80 (br d,
J = 1.4 Hz, 2H, C15-
H2), 7.14 (t,
J = 1.4 Hz, 1H, C14-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 16.7 (CH
3), 18.1 (CH
2), 21.2 (CH
2), 22.3 (CH
3), 22.9 (CH
3), 28.3 (CH
2), 28.6 (CH
2), 31.6 (CH
2), 32.4 (CH
2), 32.6 (CH
2), 39.9 (C), 43.8 (C), 44.9 (CH), 70.4 (CH
2), 75.2 (C), 76.2 (CH), 134.7 (C), 144.3 (CH), 174.9 (C), 183.9 (C); HRMS (ESI)
m/z [M + Na]
+ calcd for C
20H
28O
5Na 371.1830; found 371.1834.
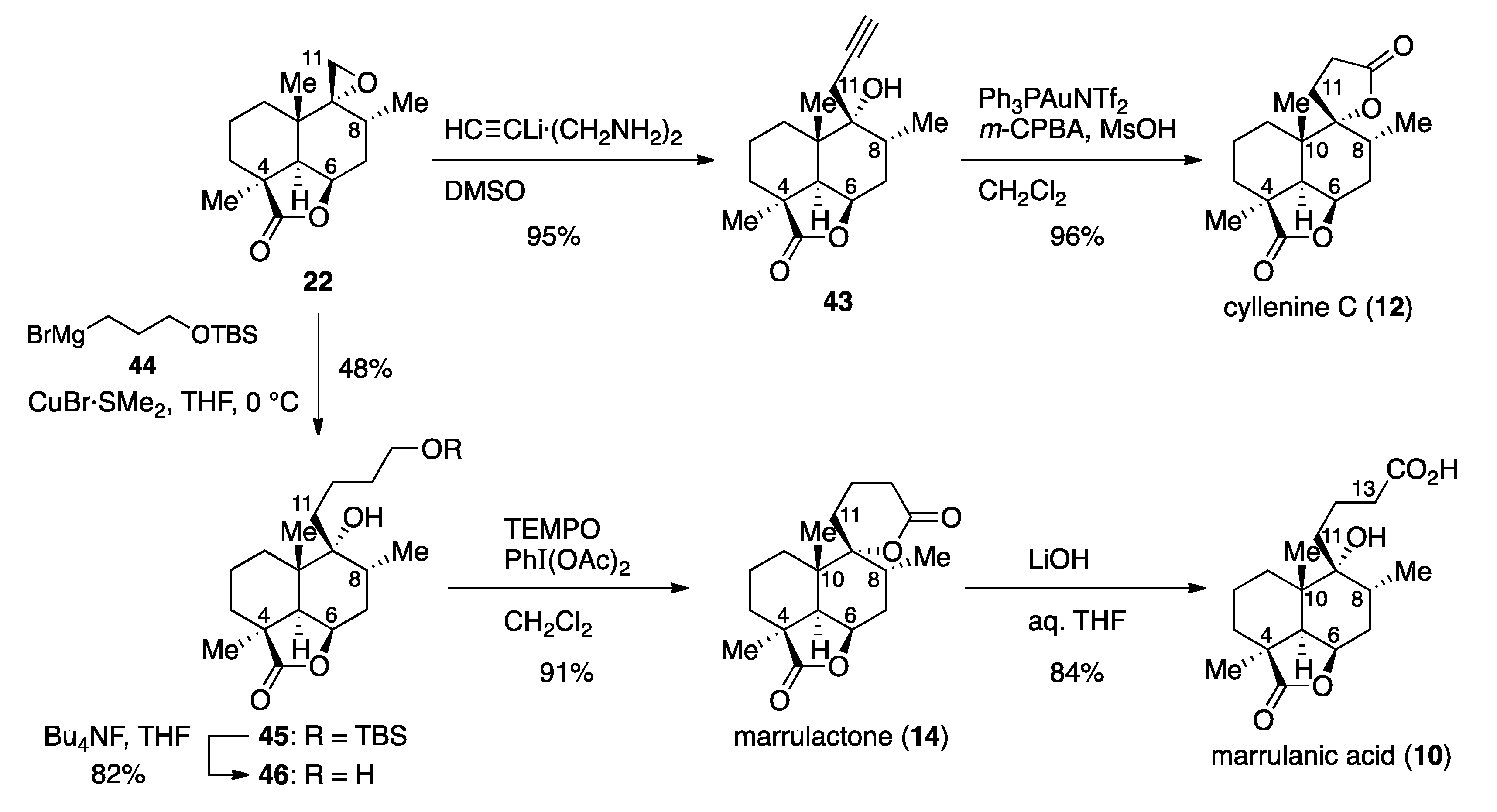
(1S,4R,6R,7R,8S,12R)-7-Hydroxy-1,6,8-trimethyl-2-oxo-7-propargyl-3-oxatricyclo[6.3.1.04,12]dodecane (43). Lithium acetylide ethylene diamine complex (36.5 mg, 0.397 mmol) was added to a solution of epoxide 22 (10.7 mg, 42.7 μmol) in DMSO (0.2 mL). After 30 min of stirring, the reaction was quenched with H2O (1 mL), and the resulting mixture was extracted with AcOEt (3 × 5 mL). The combined organic extracts were washed with brine (2 × 5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow solid (13.1 mg), which was purified by column chromatography (silica gel 3 g, 2:1 n-hexane/AcOEt) to give alcohol 43 (11.2 mg, 95%) as a pale yellow solid. Rf 0.57 (1:1 n-hexane/AcOEt); mp 195−196 °C (colorless plates from n-hexane/Et2O); [α +29.9 (c 0.249, CHCl3); IR (neat) 3306, 3019, 2955, 2934, 2872, 1757, 1458, 1427, 1393, 1136, 1096, 1045, 1003 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.99 (d, J = 6.3 Hz, 3H, C8-CH3), 1.05 (s, 3H, C10-CH3), 1.25 (dt, J = 13.0, 9.0 Hz, 1H, one of C1-H2), 1.30 (s, 3H, C4-CH3), 1.46 (dt, J = 14.7, 4.2 Hz, 1H, one of C3-H2), 1.53 (m, 1H, one of C2-H2), 1.69 (ddd, J = 6.3, 13.3, 17.7 Hz, 1H, one of C7-H2), 1.76 (m, 1H, one of C2-H2), 1.85 (dd, J = 9.6, 11.6 Hz, 1H, one of C1-H2), 2.09−2.18 (m, 3H, one of C3-H2, one of C7-H2, C8-H), 2.19 (t, J = 2.7 Hz, 1H, C13-H), 2.26 (d, J = 4.5 Hz, 1H, C5-H), 2.36 (dd, J = 2.7, 17.1 Hz, 1H, one of C11-H2), 2.59 (dd, J = 2.7, 17.1 Hz, 1H, one of C11-H2), 4.73 (dd, J = 4.5, 6.3 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.3 (CH3), 18.1 (CH2), 22.3 (CH3), 22.6 (CH3), 25.3 (CH2), 28.21 (CH2), 28.22 (CH2), 31.5 (CH2), 32.3 (CH), 39.2 (C), 43.9 (C), 44.8 (CH), 73.77 (C), 73.79 (CH), 76.1 (CH), 80.7 (C), 183.8 (C); HRMS (ESI) m/z [M + Na]+ calcd for C17H24O3Na 299.1618; found 299.1623.
Cyllenine C (
12). To an ice-cooled (0 °C) mixture of alcohol
43 (6.8 mg, 24.6 μmol) and
m-CPBA (ca. 65%, 9.8 mg, 36.9 μmol) in CH
2Cl
2 (0.3 mL) was added a 0.123 M solution of methanesulfonic acid in CH
2Cl
2 (0.20 mL, 24.6 μmol) followed by Ph
3PAuNTf
2 (1.0 mg, 1.3 μmol). After 1 h of stirring at room temperature, the reaction was quenched with saturated aqueous NaHCO
3 (1 mL), and the resulting mixture was extracted with AcOEt (2 × 5 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the colorless oil (13.7 mg), which was purified by column chromatography (silica gel 2 g, 2:1
n-hexane/AcOEt) to give cyllenine C (
12, 6.9 mg, 96%) as a white solid.
Rf 0.32 (1:2
n-hexane/AcOEt); mp 164−165 °C (colorless needles from
n-hexane/Et
2O); [α
+22.1 (
c 0.69, CH
2Cl
2) [lit. [
11], [α
+11.82 (
c 0.33, CH
2Cl
2)]; IR (neat) 3019, 2934, 2870, 1763, 1474, 1458, 1420, 1391, 1273, 1120, 1067, 1042, 993 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 0.93 (d,
J = 6.5 Hz, 3H, C8-C
H3), 1.09 (s, 3H, C10-C
H3), 1.30 (dt,
J = 12.2, 9.8 Hz, 1H, one of C1-
H2), 1.31 (s, 3H, C4-C
H3), 1.46 (m, 1H, one of C1-
H2), 1.49 (m, 1H, one of C3-
H2), 1.52 (m, 1H, one of C2-
H2), 1.72 (ddd,
J = 6.3, 11.9, 16.4 Hz, 1H, one of C7-
H2), 1.78 (m, 1H, one of C2-
H2), 1.95 (ddd,
J = 4.4, 11.4, 13.8 Hz, 1H, one of C11-
H2), 2.12 (m, 1H, one of C3-
H2), 2.18 (ddq,
J = 6.3, 11.9, 6.5 Hz, 1H, C8-
H), 2.19 (d,
J = 4.5 Hz, 1H, C5-
H), 2.22 (ddd,
J = 8.9, 11.5, 13.8 Hz, 1H, one of C11-
H2), 2.28 (dd,
J = 6.3, 16.4 Hz, 1H, one of C7-
H2), 2.54 (ddd,
J = 4.4, 11.5, 18.8 Hz, 1H, one of C12-
H2), 2.62 (ddd,
J = 8.9, 11.4, 18.8 Hz, 1H, one of C12-
H2), 4.75 (dd,
J = 4.5, 6.3 Hz, 1H, C6-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 15.4 (CH
3), 17.7 (CH
2), 22.2 (CH
3), 22.4 (CH
3), 24.5 (CH
2), 27.7 (CH
2), 28.2 (CH
2), 29.3 (CH
2), 31.2 (CH
2), 32.3 (CH), 38.6 (C), 44.0 (C), 45.3 (CH), 75.6 (CH), 91.2 (C), 176.9 (C), 183.3 (C); HRMS (ESI)
m/
z [M + Na]
+ calcd for C
17H
24O
4Na 315.1567; found 315.1566.
(1S,4R,6R,7R,8S,12R)-7-[4-(tert-Butyldimethylsilyl)oxybutyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (45). To an ice-cooled (0 °C) solution of 3-(tert-butyldimethylsilyl)oxypropylmagnesium bromide (44) [prepared from (3-bromopropoxy)(tert-butyl)dimethylsilane (2.00 g, 7.90 mmol) and magnesium tuning (241 mg, 9.93 mmol)] in THF (14 mL) was added CuBr·SMe2 (129 mg, 0.627 mmol), followed by addition of a solution of epoxide 22 (39.1 mg, 0.156 mmol) in THF (0.20 mL plus 2 × 0.20 mL rinse). After 5 h of stirring at room temperature, the reaction was quenched with saturated aqueous NH4Cl (15 mL), and the resulting mixture was extracted with AcOEt (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow oil (869 mg), which was purified by column chromatography (silica gel 20 g, 10:1 → 6:1 → 3:1 n-hexane/AcOEt) to give TBS ether 45 (42.1 mg, 48%) as a pale yellow amorphous. Rf 0.46 (3:1 n-hexane/AcOEt); [α +23.2 (c 1.00, CHCl3); IR (neat) 3534, 2953, 2927, 2859, 1755, 1471, 1462, 1336, 1253, 1197, 1149, 1099, 994 cm−1; 1H-NMR (500 MHz, C6D6) δ 0.08 (s, 6H, Si(CH3)2), 0.65 (d, J = 6.6 Hz, 3H, C8-CH3), 0.78 (br s, 1H, OH), 0.96 (m, 1H, one of C1-H2), 0.97 (s, 6H, C4-CH3, C10-CH3), 1.00 (s, 9H, SiC(CH3)3), 1.14−1.30 (m, 6H, one of C2-H2, one of C3-H2, C11-H2, C12-H2), 1.35−1.45 (m, 4H, one of C2-H2, one of C7-H2, C13-H2), 1.47 (m, 1H, one of C1-H2), 1.77 (ddq, J = 6.2, 11.2, 6.6 Hz, 1H, C8-H), 1.92 (dd, J = 6.2, 15.8 Hz, 1H, one of C7-H2), 2.00 (d, J = 4.6 Hz, 1H, C5-H), 2.18 (dt, J = 4.3, 13.7 Hz, 1H, one of C3-H2), 3.52 (t, J = 6.1 Hz, 2H, C14-H2), 4.36 (dd, J = 4.6, 6.4 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, C6D6) δ −5.1 (CH3), 16.6 (CH3), 18.51 (C), 18.55 (CH2), 21.9 (CH2), 22.6 (CH3), 22.9 (CH3), 26.1 (CH3), 28.8 (CH2), 28.9 (CH2), 32.0 (CH2), 32.3 (CH), 34.0 (CH2), 34.8 (CH2), 39.8 (C), 43.7 (C), 44.9 (CH), 62.8 (CH2), 75.48 (C), 75.52 (C), 182.6 (C); HRMS (ESI) m/z [M + Na]+ calcd for C24H44O4SiNa 447.2907; found 447.2891.
(1S,4R,6R,7R,8S,12R)-7-Hydroxy-7-(4-hydroxybutyl)-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (46). Bu4NF in THF (1.0 M, 0.38 mL, 0.38 mmol) was added to an ice-cooled (0 °C) solution of TBS ether 45 (32.1 mg, 0.76 mmol) in THF (1.0 mL). After 1 h of stirring at room temperature, the mixture was partitioned between AcOEt (2 mL) and H2O (2 mL), and the aqueous layer was extracted with AcOEt (3 × 2 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the pale yellow oil (144 mg), which was purified by column chromatography (silica gel 2 g, 1:4 n-hexane/AcOEt) to give diol 46 (19.3 mg, 82%) as a colorless amorphous. Rf 0.33 (1:2 n-hexane/AcOEt); [α +24.4 (c 0.90, CHCl3); IR (neat) 3478, 2951, 2930, 2870, 1747, 1462, 1454, 1350, 1263, 1199, 1149, 1099, 1072, 1041, 991 cm−1; 1H-NMR (500 MHz, C6D6) δ 0.54 (s, 1H, OH), 0.61 (d, J = 6.7 Hz, 3H, C8-CH3), 0.86 (s, 1H, OH), 0.91 (m, 1H, one of C1-H2), 0.95 (s, 3H, C10-CH3), 0.97 (s, 3H, C4-CH3), 1.07−1.30 (m, 8H, one of C1-H2, one of C2-H2, C11-H2, C12-H2, C13-H2), 1.36−1.48 (m, 3H, one of C2-H2, one of C3-H2, one of C7-H2), 1.73 (ddq, J = 6.2, 11.3, 6.7 Hz, 1H, C8-H), 1.91 (ddd, J = 1.3, 6.2, 15.9 Hz, 1H, one of C7-H2), 2.00 (d, J = 4.8 Hz, 1H, C5-H), 2.18 (dt, J = 4.2, 13.8 Hz, 1H, one of C3-H2), 3.30 (t, J = 6.1 Hz, 2H, C14-H2), 4.35 (ddd, J = 1.3, 4.8, 6.2 Hz, 1H, C6-H); 13C-NMR (125.7 MHz, C6D6) δ 16.6 (CH3), 18.6 (CH2), 21.8 (CH2), 22.5 (CH3), 22.9 (CH3), 28.7 (CH2), 28.9 (CH2), 32.0 (CH2), 32.3 (CH), 33.7 (CH2), 34.7 (CH2), 39.8 (C), 43.7 (C), 44.9 (CH), 62.2 (CH2), 75.4 (C), 75.6 (CH), 182.7 (C); HRMS (ESI) m/z [M + Na]+ calcd for C18H30O4Na 333.2042; found 333.2052.
Marrulactone (
14). TEMPO (2.3 mg, 14.7 μmol) was added to an ice-cooled (0 °C) mixture of diol
46 (13.1 mg, 42.2 μmol) and PhI(OAc)
2 (40.5 mg, 0.125 mmol) in CH
2Cl
2 (0.3 mL). After 4 h of stirring at room temperature, the reaction was quenched with 1 M aqueous Na
2S
2O
3 (1 mL), and the resulting mixture was extracted with AcOEt (3 × 1 mL). The combined organic extracts were washed with brine (3 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the colorless oil (64.0 mg), which was purified by column chromatography (silica gel 1 g, 1:1
n-hexane/AcOEt) to give marrulactone (
14, 11.8 mg, 91%) as a colorless amorphous.
Rf 0.60 (1:2
n-hexane/AcOEt); [α
−11.6 (
c 0.61, CHCl
3) [lit. [
12], [α
−23.80 (
c 0.22, CHCl
3)]; IR (neat) 3017, 2961, 2928, 2872, 1769, 1719, 1458, 1391, 1329, 1261, 1244, 1215, 1188, 1117, 1070, 1030, 1003 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 0.98 (d,
J = 6.3 Hz, 3H, C8-C
H3), 1.11 (s, 3H, C10-C
H3), 1.31 (s, 3H, C4-C
H3), 1.34−1.42 (m, 2H, C1-
H2), 1.48 (m, 1H, one of C3-
H2), 1.55 (m, 1H, one of C2-
H2), 1.74−1.80 (m, 2H, one of C2-
H2, one of C11-
H2), 1.80−1.94 (m, 4H, one of C7-
H2, one of C11-
H2, C12-
H2), 2.09−2.20 (m, 3H, one of C3-
H2, one of C7-
H2, C8-
H), 2.25 (ddd,
J = 6.2, 11.2, 17.3 Hz, 1H, one of C13-
H2), 2.32 (d,
J = 4.4 Hz, 1H, C5-
H), 2.57 (ddt,
J = 2.0, 17.3, 4.1 Hz, 1H, one of C13-
H2), 4.78 (ddd,
J = 0.7, 4.4, 5.5 Hz, 1H, C6-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 16.5 (CH
3), 17.8 (CH
2), 18.9 (CH
2), 22.0 (CH
3), 22.8 (CH
3), 26.1 (CH
2), 28.1 (CH
2), 28.6 (CH
2), 30.4 (CH
2), 30.9 (CH
2), 34.3 (CH), 40.8 (C), 44.0 (C), 44.3 (CH), 75.8 (CH), 88.3 (C), 172.2 (C), 183.5 (C); HRMS (ESI)
m/
z [M + Na]
+ calcd for C
18H
26O
4Na 329.1723; found 329.1711.
Marrulanic acid (
10). LiOH (18.7 mg, 0.78 mmol) was added to a solution of marrulactone (
14, 11.8 mg, 38.5 μmol) in THF/H
2O (2:1, 1.2 mL). After 31 h of stirring, the mixture was acidified with 1 M aqueous HCl (3 mL), and the resulting mixture was extracted with AcOEt (3 × 3 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the colorless oil (11.7 mg), which was purified by column chromatography (silica gel 1 g, 1:1
n-hexane/AcOEt → AcOEt → 20:1 AcOEt/MeOH) to give marrulanic acid (
10, 10.5 mg, 84%) as a colorless amorphous.
Rf 0.19 (1:2
n-hexane/AcOEt); [α
+25.4 (
c 0.53, CHCl
3) [lit. [
12], [α
−10.8 (
c 1.2, CHCl
3)]; IR (neat) 3536, 3196, 2949, 2928, 2868, 1761, 1732, 1464, 1412, 1391, 1287, 1260, 1196, 1179, 1159, 1126, 1094, 1078, 1042, 1018 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 0.92 (d,
J = 6.5 Hz, 3H, C8-C
H3), 1.04 (s, 3H, C10-C
H3), 1.26 (m, 1H, one of C1-
H2), 1.29 (s, 3H, C4-C
H3), 1.44−1.80 (m, 9H, one of C1-
H2, C2-
H2, one of C3-
H2, one of C7-
H2, C11-
H2, C12-
H2), 2.04−2.17 (m, 3H, one of C3-
H2, one of C7-
H2, C8-
H), 2.24 (d,
J = 4.6 Hz, 1H, C5-
H), 2.38 (dt,
J = 1.8, 7.0 Hz, 2H, C13-
H2), 4.74 (ddd,
J = 1.2, 4.6, 5.8 Hz, 1H, C6-
H);
13C-NMR (125.0 MHz, CDCl
3) δ 16.5 (CH
3), 18.2 (CH
2), 20.3 (CH
2), 22.3 (CH
3), 22.9 (CH
3), 28.3 (CH
2), 28.6 (CH
2), 31.5 (CH
2), 32.2 (CH), 34.29 (CH
2), 34.31 (CH
2), 39.7 (C), 43.8 (C), 44.8 (CH), 75.6 (C), 76.2 (CH), 178.5 (C), 183.9 (C); HRMS (ESI)
m/
z [M + Na]
+ calcd for C
18H
28O
5Na 347.1834; found 347.1813.
[1S,4R,6R,7R,7(2S,3S,4R,5R),8S,12R]-7-[2-(3,4-Dihydroxy-2,5-dimethoxytetrahydrofuran-3-yl)ethyl]-7-hydroxy-1,6,8-trimethyl-2-oxo-3-oxatricyclo[6.3.1.04,12]dodecane (40). TsOH (0.4 mg, 2.3 μmol) was added to a solution of marrulibacetal A (17, 0.90 mg, 2.3 μmol) in MeOH (1.0 mL), and the mixture was heated at reflux for 5 h. After cooling to 0 °C, the reaction was quenched with half saturated aqueous NaHCO3 (4 mL), and the resulting mixture was extracted with AcOEt (2 × 5 mL). The combined organic extracts were washed with brine (5 mL), and dried over anhydrous Na2SO4. Filtration and evaporation in vacuo furnished the colorless oil (1.4 mg), which was purified by flash column chromatography (silica gel 0.8 g, 1:1 n-hexane/AcOEt → AcOEt) to give triol 40 (0.24 mg, 25%) as a colorless oil. Rf 0.30 (1:1 n-hexane/AcOEt); [α +16.5 (c 0.46, benzene); IR (neat) 3447, 2930, 1744, 1466, 1449, 1389, 1288, 1198, 1130, 1094, 1042, 991 cm−1; 1H-NMR (500 MHz, CDCl3) δ 0.91 (d, J = 6.4 Hz, 3H, C8-CH3), 1.05 (s, 3H, C10-CH3), 1.28 (m, 1H, one of C1-H2), 1.29 (s, 3H, C4-CH3), 1.45 (m, 1H, one of C3-H2), 1.51 (m, 1H, one of C2-H2), 1.65 (m, 1H, one of C7-H2), 1.74 (m, 1H, one of C2-H2), 1.60−1.98 (m, 5H, one of C1-H2, C11-H2, C12-H2), 2.07 (m, 1H, C8-H), 2.11 (m, 1H, one of C3-H2), 2.14 (m, 1H, one of C7-H2), 2.22 (d, J = 4.5 Hz, 1H, C5-H), 3.40 (s, 3H, C16-OCH3), 3.48 (s, 3H, C15-OCH3), 3.92 (d, J = 3.7 Hz, 1H, C14-H), 4.73 (dd, J = 4.5, 6.5 Hz, 1H, C6-H), 4.77 (s, 1H, C16-H), 4.89 (d, J = 3.7 Hz, 1H, C15-H); 13C-NMR (125.7 MHz, CDCl3) δ 16.4 (CH3), 18.1 (CH2), 22.3 (CH3), 22.9 (CH3), 28.0 (CH2), 28.26 (CH2), 28.28 (CH2), 28.4 (CH2), 31.5 (CH2), 32.6 (CH), 40.0 (C), 43.9 (C), 44.9 (CH), 55.1 (CH3), 56.4 (CH3), 75.4 (C), 76.2 (CH), 80.3 (CH), 81.4 (C), 108.3 (CH), 110.7 (CH), 184.0 (C); HRMS (ESI) m/z [M + Na]+ calcd for C22H36O8Na 451.2302; found 451.2308.
Desertine (
18). TsOH (1.6 mg, 9.3 μmol) was added to a solution of bisacetal
42 (2.0 mg, 4.7 μmol) in MeOH (0.5 mL), and the mixture was heated at reflux for 3.5 h. After cooling to 0 °C, the reaction was quenched with saturated aqueous NaHCO
3 (2 mL), and the resulting mixture was extracted with AcOEt (2 × 5 mL). The combined organic extracts were washed with brine (10 mL), and dried over anhydrous Na
2SO
4. Filtration and evaporation in vacuo furnished the pale yellow oil (3.6 mg), which was purified by flash column chromatography (silica gel 0.8 g, 1:1
n-hexane/AcOEt) to give desertine (
18, 0.34 mg, 16%) as a colorless oil.
Rf 0.30 (1:1
n-hexane/AcOEt); [α
+22.8 (
c 0.50, benzene) (lit. [
14], [α
−56.6); IR (neat) 3464, 2930, 1748, 1463, 1450, 1391, 1256, 1198, 1152, 1099, 1043, 989 cm
−1;
1H-NMR (500 MHz, CDCl
3) δ 0.91 (d,
J = 6.2 Hz, 3H, C8-C
H3), 1.04 (s, 3H, C10-C
H3), 1.28 (m, 1H, one of C1-
H2), 1.29 (s, 3H, C4-C
H3), 1.45 (m, 1H, one of C3-
H2), 1.51 (m, 1H, one of C2-
H2), 1.65 (m, 1H, one of C7-
H2), 1.74 (m, 1H, one of C2-
H2), 1.60−1.98 (m, 5H, one of C1-
H2, C11-
H2, C12-
H2), 2.07 (m, 1H, C8-
H), 2.11 (m, 1H, one of C3-
H2), 2.14 (m, 1H, one of C7-
H2), 2.23 (d,
J = 4.0 Hz, 1H, C5-
H), 3.41 (s, 3H, C16-OC
H3), 3.47 (s, 3H, C15-OC
H3), 3.95 (d,
J = 3.5 Hz, 1H, C14-
H), 4.73 (dd,
J = 4.0, 6.9 Hz, 1H, C6-
H), 4.76 (s, 1H, C16-
H), 4.90 (d,
J = 3.5 Hz, 1H, C15-
H);
13C-NMR (125.7 MHz, CDCl
3) δ 16.8 (CH
3), 18.2 (CH
2), 22.2 (CH
3), 23.0 (CH
3), 27.4 (CH
2), 28.3 (CH
2), 28.4 (CH
2), 28.5 (CH
2), 31.6 (CH
2), 32.5 (CH), 40.1 (C), 43.9 (C), 45.0 (CH), 55.1 (CH
3), 56.4 (CH
3), 75.3 (C), 76.2 (CH), 80.2 (CH), 81.2 (C), 108.6 (CH), 110.8 (CH), 183.8 (C); HRMS (ESI)
m/
z [M + Na]
+ calcd for C
22H
36O
8Na 451.2302; found 451.2313.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}