
Phenacylation of 6-Methyl-Beta-Nitropyridin-2-Ones and Further Heterocyclization of Products



Abstract
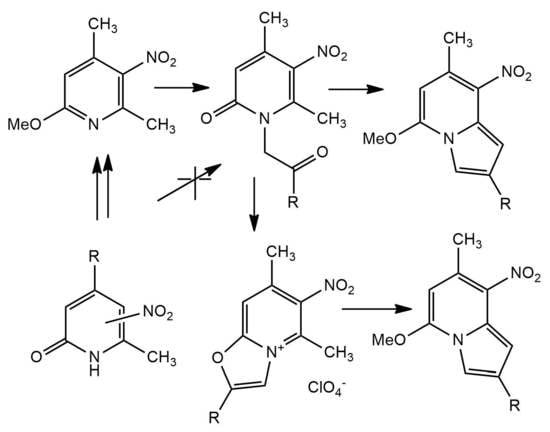
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Isomers and Homologues of Beta-Nitro-6-Methylpyridin-2-One (1a–d)
2.2. Attempts of Direct Phenacylation of Homologues of Beta-Nitropyridin-2-Ones
2.3. Synthesis of Beta-Nitro-2-Methoxypyridine Homologues and Their Phenacylation
2.4. Cyclocondensation of N-Phenacylpyridones under Basic Condition
2.5. Synthesis of 5-Substituted Indolizine via Oxazolopyridinium Salt
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.3. X-ray Diffraction Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flitsch, W. Pyrroles with fused six-membered heterocyclic rings: A-fused. In Comprehensive Heterocyclic Chemistry; Katritzky, A., Rees, C.W., Eds.; Pergamon Press: Oxford, UK, 1984; Volume 4, pp. 443–496. [Google Scholar]
- Renard, M.; Gubin, J. Metallation of 2-Phenylindolizine. Tetrahedron Lett. 1992, 33, 4433–4434. [Google Scholar] [CrossRef]
- Kuznetsov, A.G.; Bush, A.A.; Rybakov, V.B.; Babaev, E.V. An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation. Molecules 2005, 10, 1074–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzhevskii, S.A.; Rybakov, V.B.; Khrustalev, V.N.; Babaev, E.V. Reactions of 5-Indolizyl Lithium Compounds with Some Bielectrophiles. Molecules 2017, 22, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kost, A.N.; Sagitullin, R.S.; Gromov, S.P. Nucleophilic amination and recyclization of the indolizine nucleus. Heterocycles 1977, 7, 997–1001. [Google Scholar]
- Gevald, K.; Jansch, H.J. 3-Amino-furo[2.3-b]pyridine. J. Prakt. Chem. 1976, 318, 313–320. [Google Scholar]
- Babaev, E.V.; Vasilevich, N.I.; Ivushkina, A.S. Efficient synthesis of 5-substituted 2-aryl-6-cyanoindolizines via nucleophilic substitution reactions. Beilstein J. Org. Chem. 2005, 1, 9–11. [Google Scholar] [CrossRef]
- Babaev, E.V. Fused Munchnones in Recyclization Tandems. J. Heterocycl. Chem. 2000, 37, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Babaev, E.V.; Alifanov, V.L.; Efimov, A.V. Oxazolo[3,2-a]pyridinium and Oxazolo[3,2-a]pyrimidinium Salts in Organic Synthesis. Russ. Chemical Bull. 2008, 57, 845–862. [Google Scholar] [CrossRef]
- Bradsher, C.K.; Zinn, M.F. Oxazolo[3,2-a] pyridinium salts. J. Heterocycl. Chem. 1967, 4, 66–70. [Google Scholar] [CrossRef]
- Krasnaya, Z.A.; Stytsenko, T.S.; Prokof’ev, E.P.; Yakovlev, I.P.; Kucherov, V.F. Reaction of enaminocarbonyl compounds with nitroacetic ester. Bull. Acad. Sci. USSR 1974, 23, 809–816. [Google Scholar] [CrossRef]
- Goerlitzer, K.; Wilpert, C.; Ruebsamen-Waigmann, H.; Suhartono, H.; Wang, L.; Immelmann, A. Pyrido[3,2-e][1,4] diazepine - Synthese und Prüfung auf Anti-HIV-1-Wirkung. Arch. Pharm. 1995, 328, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Kislyi, V.P.; Shestopalov, A.M.; Kagramanov, N.D.; Semenov, V.V. Synthesis of 3-nitropyrid-2(1H)-ones from C-nitroacetamide and 1,3-dicarbonyl compounds. Russ. Chem. Bull. 1997, 46, 539–542. [Google Scholar] [CrossRef]
- Mariella, R.P.; Callahan, J.J.; Jibril, A.O. Some novel color reactions of some pyridine derivatives. J. Org. Chem. 1955, 20, 1721–1728. [Google Scholar] [CrossRef]
- Rybakov, V.B.; Babaev, E.V.; Paronikyan, E.G. X-Ray Mapping in Heterocyclic Design: 18. X-Ray Diffraction Study of a Series of Derivatives of 3-Cyanopyridine-2-one with Annelated Heptane and Octane Cycles. Crystallogr. Rep. 2017, 62, 219–231. [Google Scholar] [CrossRef]
- Rybakov, V.B.; Babaev, E.V. Transformations of Substituted Oxazolo[3,2-a]Pyridines to 5,6-Disubstituted Indolizines: Synthesis And X-ray Structural Mapping. Chem. Heterocycl. Comp. 2014, 50, 225–236. [Google Scholar] [CrossRef]
- Okul’, E.M.; Rybakov, V.B.; Babaev, E.V. The structure of products of phenacylation and subsequent (re)cyclizations of 3-acetyl-4,6-dimethylpyridin-2(1H)-one according to X-ray structural analysis. Chem. Heterocycl. Comp. 2017, 53, 997–1002. [Google Scholar] [CrossRef]
- Feklicheva, E.M.; Rybakov, V.B.; Babaev, E.V.; Oficerov, E.N. Physical-chemical investigations of transformations in the series of 2,4-dimethyl-6-oxo-1,6-dihydropyridine-3-carboxanmide. Part 1. Synthesis and X-ray study of derivatives of 2,4-dimethyl-6-oxo-1,6-dihydropyridine-3-carboxanmide. Butlerov Communications 2019, 60, 1–23. [Google Scholar]
- Bush, A.A.; Babaev, E.V. Synthesis of 6-Nitroderivatives of Oxazolo[3,2-a]pyridines and their Reactions with Nucleophiles. Molecules 2003, 8, 460–466. [Google Scholar] [CrossRef]
- Babaev, E.V.; Efimov, A.V.; Maiboroda, D.A. Hetarenes with a nitrogen bridging atom. 1. Phenacylation of 2-substituted 6-methylpyridines. Chem. Heterocycl. Comp. 1995, 31, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Babaev, E.V.; Tsisevich, A.A.; Al’bov, D.V.; Rybakov, V.B.; Aslanov, L.A. Heterocycles with a bridgehead nitrogen atom. Part 16. Assembly of a peri-fused system from an angular tricycle by recyclization of an oxazole ring into pyrrole one. Russ. Chem. Bull. 2005, 54, 259–261. [Google Scholar] [CrossRef]
- Parker, E.D.; Shive, W. Substituted 2-Picolines Derived from 6-Amino-2-picoline. J. Amer. Chem. Soc. 1947, 69, 63–67. [Google Scholar] [CrossRef]
- Remennikov, G.Y.; Kurilenko, L.K.; Boldyrev, I.V.; Cherkasov, V.M. The recyclization of 5-nitropyrimidine and its methoxy derivatives upon reaction with the acetylacetone carbanion. Chem. Heterocycl. Comp. 1987, 23, 422–425. [Google Scholar] [CrossRef]
- Frydman, B.; Reil, S.J.; Boned, J.; Rapoport, H. Synthesis of substituted 4- and 6-azaindoles. J. Org. Chem. 1968, 33, 3762–3766. [Google Scholar] [CrossRef]
- Baumgarten, H.E.; Su, H.C.-F. Synthesis of 3- and 5-nitro-2-picoline and derivatives. J. Amer. Chem. Soc. 1952, 74, 3828–3830. [Google Scholar] [CrossRef]
- Sawanishi, H.; Tajima, K.; Tsuchiya, T. Studies on Diazepines. XXVIII. Syntheses of 5H-1,3-Diazepines and 2H-1,4-Diazepines from 3-Azidopyridines. Chem. Pharm. Bull. 1987, 35, 4101–4109. [Google Scholar] [CrossRef] [Green Version]
- Kislyi, V.P.; Semenov, V.V. Investigation of the regioselectivity of alkylation of 3-nitropyridine-2(1H)-ones. Russ. Chem. Bull. 2001, 50, 460–463. [Google Scholar] [CrossRef]
- Babaev, E.V.; Rybakov, V.B. CCDC 1986879. Experimental Crystal Structure Determination. 2020. Available online: https://doi.org/10.5517/ccdc.csd.cc24phw7 (accessed on 27 February 2020).
- Babaev, E.V.; Rybakov, V.B. CCDC 1986629. Experimental Crystal Structure Determination. 2020. Available online: https://doi.org/10.5517/ccdc.csd.cc24p7tx (accessed on 27 February 2020).
- Babaev, E.V.; Rybakov, V.B. CCDC 1984804. Experimental Crystal Structure Determination. 2020. Available online: https://doi.org/10.5517/ccdc.csd.cc24mby2 (accessed on 18 February 2020).
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 6 and 7 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Yield, % | Mp | Mp, Lit | Reference |
|---|---|---|---|---|
| 2a | 45 | 68 | 70–70.8 67–69 | [22] [23] |
| 3a | 32 | 57 | 57–58 | [24] |
| 2b | 25 | 54–55 | 54 | [25] |
| 3b | 36 | 64 | 64–65 | [26] |
| 2c | 64 | 47 | 47–48 | [27] |
| 3c | 90 | 104–105 | 104 | [28] |
| 2d | 85 | 54 | 54–55 | [27] |
| 3d | 92 | 59–60 | 60 | [14] |
| Compound (No.) | Formula | Calculated | Found | ||||
|---|---|---|---|---|---|---|---|
| C | H | N | C | H | N | ||
| N-(p-chlorophenacyl)-4,6-dimethyl-5-nitropyridin-2-one (4a) | C15H13ClN2O4 | 56.17 | 4.09 | 8.73 | 55.82 | 4.13 | 8.68 |
| N-(p-bromophenacyl)-4,6-dimethyl-5-nitropyridin-2-one (4b) | C15H13BrN2O4 | 49.34 | 3.59 | 7.67 | 49.13 | 3.62 | 7.64 |
| 2-p-chlorophenyl-5-hydroxy-7-methyl-8-nitroindolizine (5a) | C15H11ClN2O3 | 59.52 | 3.66 | 9.25 | 59.13 | 3.71 | 9.19 |
| 2-p-Bromophenyl-7-methyl-8-nitro-5-hydroxyindolizine (5b) | C15H11BrN2O3 | 51.90 | 3.19 | 8.07 | 51.51 | 3.25 | 8.01 |
| 2-(p-Chlorophenyl)-5,7-dimethyl-6-nitrooxazolo [3,2-a]pyridinium perchlorate (6) | C15H12ClN2O3*ClO4 | 44.69 | 3.00 | 6.95 | 44.40 | 3.05 | 6.90 |
| 2-(p-Chlorophenyl)-5-methoxy-7-methyl-8-nitroindolizine (7) | C16H13ClN2O3 | 60.67 | 4.14 | 8.84 | 60.29 | 4.18 | 8.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babaev, E.V.; Rybakov, V.B. Phenacylation of 6-Methyl-Beta-Nitropyridin-2-Ones and Further Heterocyclization of Products. Molecules 2020, 25, 1682. https://doi.org/10.3390/molecules25071682
Babaev EV, Rybakov VB. Phenacylation of 6-Methyl-Beta-Nitropyridin-2-Ones and Further Heterocyclization of Products. Molecules. 2020; 25(7):1682. https://doi.org/10.3390/molecules25071682
Chicago/Turabian StyleBabaev, Eugene V., and Victor B. Rybakov. 2020. "Phenacylation of 6-Methyl-Beta-Nitropyridin-2-Ones and Further Heterocyclization of Products" Molecules 25, no. 7: 1682. https://doi.org/10.3390/molecules25071682