Abstract
Two novel series of compounds based on the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine and 4,5,6,7-tetrahydrobenzo[b]thiophene molecular skeleton, characterized by the presence of a 3′,4′,5′-trimethoxyanilino moiety and a cyano or an alkoxycarbonyl group at its 2- or 3-position, respectively, were designed, synthesized, and evaluated for antiproliferative activity on a panel of cancer cell lines and for selected highly active compounds, inhibition of tubulin polymerization, and cell cycle effects. We have identified the 2-(3′,4′,5′-trimethoxyanilino)-3-cyano-6-methoxycarbonyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivative 3a and its 6-ethoxycarbonyl homologue 3b as new antiproliferative agents that inhibit cancer cell growth with IC50 values ranging from 1.1 to 4.7 μM against a panel of three cancer cell lines. Their interaction with tubulin at micromolar levels leads to the accumulation of cells in the G2/M phase of the cell cycle and to an apoptotic cell death. The cell apoptosis study found that compounds 3a and 3b were very effective in the induction of apoptosis in a dose-dependent manner. These two derivatives did not induce cell death in normal human peripheral blood mononuclear cells, suggesting that they may be selective against cancer cells. Molecular docking studies confirmed that the inhibitory activity of these molecules on tubulin polymerization derived from binding to the colchicine site.
1. Introduction
Cancer, which is the result of deviation of cell growth control mechanisms, has become a major worldwide disease and the second leading cause of mortality in developed countries, with almost 20 million deaths annually [1,2]. Many of the current treatments, involving an integrated treatment of surgery, radiation, and chemotherapy, have problems with toxicity and drug resistance [3,4]. Therefore, there is a strong demand for the discovery and development of effective chemotherapeutic agents with high selectivity and low toxicity [5,6,7].
The central role of microtubules in cell division and mitosis makes them an important target for the development of potential new anticancer agents [8,9,10]. A large number of small molecules displaying wide structural diversity and derived from natural sources or obtained by chemical synthesis have been identified within the last several decades [11,12,13] and have been shown to interfere with microtubules, polymeric structures composed of α- and β-tubulin heterodimers [14,15,16].
Prominent examples of tubulin-binding compounds used clinically for cancer treatment include microtubule-stabilizers that inhibit microtubule function by promoting abnormally high levels of tubulin polymerization. Examples of such drugs include paclitaxel (Taxol) and its semisynthetic analogue docetaxel (Taxotere) [17,18]. In contrast, the vinca alkaloids vincristine, vinorelbine, and vinblastine destabilize microtubules and act as microtubule depolymerizers by inhibiting tubulin polymerization [19]. Both taxoids and vinca alkaloids interact with β-tubulin, but at different sites, named the taxol and vinca sites, respectively [20]. The colchicine-site binders that inhibit tubulin assembly [21] are another class of compounds in discovery and development, including combretastatin A-4 phosphate (CA-4P) [22], that bind to the colchicine site located between the α- and β-tubulin subunits in the heterodimer, distinct from the vinca site located between adjacent α- and β-tubulin heterodimers [23].
Although many chemically diverse synthetic tubulin inhibitors have been synthesized, there is still a need to identify novel small molecules that target microtubules [24,25,26,27,28]. Among such compounds, thiophene [29,30,31,32,33,34] and fused thiophenes such as benzo[b]thiophenes [35,36,37,38,39,40], thieno[3,2-d]pyrimidines [41,42,43], and thieno[2,3-b]pyridines [44] were present as molecular skeletons in a significant number of synthetic inhibitors of microtubule polymerization, but there are limited examples of small molecules showing interesting activities in inhibiting both microtubule assembly and cell proliferation based on the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine molecular skeleton as the core structure.
In an earlier published study, a series of molecules with general structure 1 (Figure 1), based on the 2-amino-3-(3′,4′,5′-trimethoxybenzoyl)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine skeleton, yielded the promising derivatives 1a and 1b. These compounds inhibit cancer cell growth with IC50 values ranging from 25 to 440 nM against a panel of four cancer cell lines and exert strong inhibition of tubulin polymerization by binding to the colchicine site of tubulin [45]. For this series of molecules, the presence of a N-methoxy/ethoxycarbonyl moiety at the 6-position of the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine system was needed for potent antiproliferative activity.
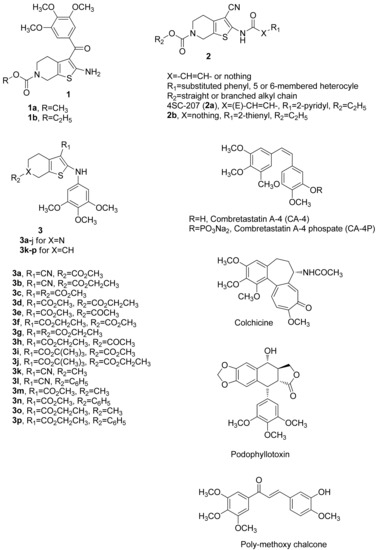
Figure 1.
Structures of tetrahydrothieno[2,3-c]pyridines 1–3. Compounds with a 3,4,5-trimethoxyphenyl moiety, such as colchicine, CA-4, podophyllotoxin, and poly-methoxychalcone.
A second series of compounds with general structure 2, reported by Beckers et al. [46,47], incorporated the 2-carboxamido-3-cyano-4,5,6,7-tetrahydrothieno[2,3-c]pyridine system, and these agents were active at micromolar concentrations (IC50 = 0.2–5 μM) as antiproliferative agents against human colon adenocarcinoma (RKOp27) cells. Bausch et al. [48] reported a molecule named 4SC-207 (2a), characterized by the presence of the 2-[(pyridin-3′-yl)acrylamido]-3-cyano-4,5,6,7-tetrahydrothieno[2,3-c]pyridine skeleton, able to inhibit microtubule polymerization at high concentrations and showing strong antiproliferative activity at low nanomolar concentrations in a large panel of cancer cell lines. These studies [46,47,48] showed that the presence of a cyano group at the 3-position of the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine nucleus was required for antiproliferaive activity. These findings prompted us to analyze this class of compounds in more detail.
Building upon these observations, since the 6-N-alkoxycarbonyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine structure was demonstrated to be essential for bioactivity, we retained this pharmacophoric unit throughout the present investigation and kept the 3-cyano group in compounds we synthesized. We were intrigued by the idea of studying the biological effects of replacing the carboxamido group at the 2-position of the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine ring of compounds with general structure 2 with a 3′,4′,5′-trimethoxyanilino moiety to yield the 2-(3′,4′,5′-trimethoxyanilino)-3-cyano-6-methoxy/ethoxycarbonyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridines 3a and 3b. Previous studies have shown that the 3,4,5-trimethoxyphenyl moiety is a pharmacophoric group that is an essential structural requirement for the anticancer effect of numerous inhibitors of tubulin polymerization, such as colchicine, combretastatin A-4 (CA-4), podophyllotoxin, and poly-methoxychalcone [49,50]. Starting from compounds 3a and 3b, additional analogues were also generated by modification of the 3-cyano function of the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine skeleton. The cyano group was replaced with a 3-alkoxycarbonyl moiety, such as 3-methoxycarbonyl (3c–3e), 3-ethoxycarbonyl (3f–3h), or 3-tert-butoxycarbonyl (3i–3j). Moreover, the influence on antiproliferative activity of the substituent (R2) at the 6-position was evaluated with R2 as acetyl or methoxy/ethoxycarbonyl.
Additional structural modifications involved the nitrogen at the 6-position of 4,5,6,7-tetrahydrothieno[2,3-c]pyridine scaffold, by the classical bioisosteric replacement with a CH group to furnish a novel series of 2-(3′,4′,5′-trimethoxyanilino)-3,6-substituted-4,5,6,7-tetrahydrobenzo[b]thiophene derivatives 3k–3p. For this latter series of compounds, besides the C-3 cyan and methoxy/ethoxycarbonyl moieties, the substituents examined at the C-6 position were methyl (3k, 3m, and 3o) and phenyl (3l, 3n, and 3p) groups.
2. Chemistry
The 2-(3′,4′,5′-trimethoxyanilino)-3-cyano/alkoxycarbonyl-6-substituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridines 3a–3j and the corresponding 4,5,6,7-tetrahydrobenzo[b]thiophenes 3k–3p were prepared through the three-step procedure shown in Scheme 1.
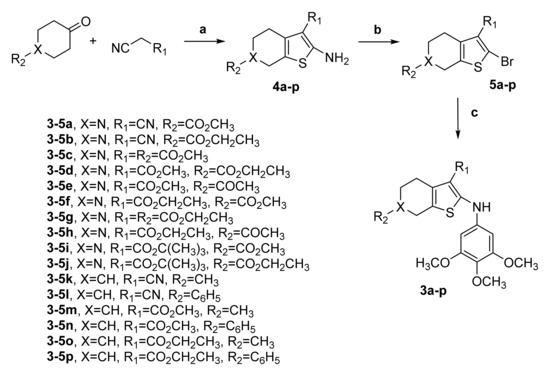
Scheme 1.
Synthesis of compounds 3a-p. Reagents. a: appropriate ketone (4-methyl/phenylcyclohexanone, N-acetyl-4-piperidone, or methoxy/ethoxycarbonyl-4-piperidone), malonitrile or methyl/ethyl/t-butylcyanoacetate, S8, TEA, MeOH or EtOH, reflux; b: t-BuONO, CuBr2, CH3CN, 65 °C; c: 3,4,5-trimethoxyaniline, Pd(OAc)2, BINAP, Cs2CO3, PhMe, 100 °C, 18 h.
The one-pot Gewald reaction [51] applied to commercially available 4-methyl/phenylcyclohexanone, N-acetyl-4-piperidone or methoxy/ethoxycarbonyl-4-piperidone with an activated nitrile (malonitrile or a cyanoacetate, such as methyl/ethyl/t-butylcyanoacetate) and elemental sulfur in refluxing methanol or ethanol in the presence of triethylamine (TEA) as base furnished 2-amino-3-cyano/alkoxycarbonyl-6-substituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridines 4a–4j and the related 4,5,6,7-tetrahydrobenzo[b]thiophene analogues 4k–4p. These latter compounds were transformed by substitutive deamination with tert-butyl nitrite (t-BuONO) and copper (II) bromide (CuBr2) in acetonitrile into the 2-bromo-4,5,6,7-tetrahydrothieno[2,3-c]pyridines 5a–5j and 4,5,6,7-tetrahydrobenzo[b]thiophene analogues 5k–5p, respectively [52]. Finally, the novel 2-(3′,4′,5′-trimethoxyanilino)-4,5,6,7-tetrahydrothieno[2,3-c]pyridines 3a–3j and 2-(3′,4′,5′-trimethoxyanilino)-4,5,6,7-tetrahydrobenzo[b]thiophenes 3k–3p were prepared in good yields by palladium-catalyzed arylamination conditions of the appropriate 2-bromo-4,5,6,7-tetrahydrothieno[2,3-c]pyridine 5a-j and 2-bromo-4,5,6,7-tetrahydrobenzo[b]thiophene analogues 5k–5p, respectively, with 3,4,5-trimethoxyaniline in the presence of palladium (II) acetate (Pd(OAc)2), rac-2,2′-bis-(diphenylphosphane)-1,1′-binaphtyl (BINAP), and cesium carbonate (Cs2CO3) (as catalyst, ligand, and base, respectively) in toluene [53].
3. Results and Discussion
3.1. In Vitro Antiproliferative Activities Against A Panel of Three Different Cancer Cell Lines (L1210, CEM, and HeLa)
In Table 1 we report the in vitro antiproliferative activity of 2-(3′,4′,5′-trimethoxyanilino)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivatives 3a–3j and the related 4,5,6,7-tetrahydrobenzo[b]thiophenes 3k–3p against a panel of three cell lines, which were derived from different cancer types, including murine leukemia (L1210), human T-lymphoblastoid leukemia (CEM), and human cervix carcinoma (HeLa) cells. CA-4 and 2-carboxamido-3-cyano-6-ethoxycarbonyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine 2b used in Beckers’ study [46,47] were also used as positive controls. All compounds that had an IC50 > 20 μM are considered inactive for purposes of this discussion.

Table 1.
In vitro growth inhibition activity of compounds 3a–3p and reference compounds 2b and CA-4 against the proliferation of murine leukemia (L1210), human T-lymphocyte (CEM), and human cervix carcinoma (HeLa) cells.
Compound 3b with an ethoxycarbonyl substituent at the C-6 position of the 2-(3′,4′,5′-trimethoxyanilino)-3-cyano-4,5,6,7-tetrahydrothieno[2,3-c]pyridine system, along with its 6-methoxycarbonyl analogue 3a, exhibited the greatest antiproliferative activity among the tested compounds, with IC50 values of 1.1–2.8 and 1.9–4.7 μM, respectively, showing improved activity against HeLa cells as compared with the other two cell lines. The 3-cyano-6-ethoxycarbonyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivative 3b possessed the highest potency, inhibiting the growth of L1210, CEM, and HeLa cells with IC50 values of 2.8, 2.3, and 1.1 μM, respectively. These values are 1.5-fold lower than those obtained with the 6-methoxycarbonyl derivative 3a against L1210 and HeLa cells, while both compounds were equipotent against CEM cells. The activities of 2-(3′,4′.5′-trimethoxyanilino) derivatives 3a and 3b were quite similar to that of 2-(thiophene-2′-carboxamido) analogue 2b against L1210 and CEM cells, while there was greater difference with the HeLa cell line, with compounds 3a and 3b 40- and 20-fold more active than 2b (IC50: 41 μM). However, CA-4 was more potent than any of the new compounds in all cell lines examined. The activity of the most effective compounds (3a and 3b) was also evaluated on the human chronic myelogenous leukemia K562 cell line. The data obtained demonstrated that both 3a and 3b were highly efficient in inhibiting K562 cells, displaying IC50 values of 0.75 and 0.70 μM, respectively.
The C-3-position of the 2-(3′,4′,5′-trimethoxyanilino)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine nucleus plays an essential role in the antiproliferative activity of derivatives 3a–3j. The activity of compounds 3a and 3b demonstrated that the 3-cyano substitution was crucial for potent cell growth inhibition. Its substitution with an alkoxycarbonyl moiety led to a reduction of growth inhibition activity (compare 3a with 3c, 3f, and 3i as well as 3b with 3d, 3g, and 3j). For the most active compound of the series (3b), replacement of the 3-cyano group with alkoxycarbonyl moieties, such as methoxycarbonyl, ethoxycarbonyl or tert-butoxycarbonyl (compounds 3d, 3g, and 3j, respectively) reduced antiproliferative activity from 5- to 18-fold against the three cancer cell lines.
Finally, although only a few compounds were synthesized thus far in the 4,5,6,7-tetrahydrobenzo[b]thophene series, all compounds of this series proved inactive (IC50 >50 μM), significantly less potent than the corresponding 4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivatives. The reduced antiproliferative activity of the 4,5,6,7-tetrahydrobenzo[b]thiophene derivatives as compared with the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine compounds demonstrates that the presence of a piperidine ring fused with the thiophene scaffold is critical for activity.
In order to investigate the selective antiproliferative activities of derivatives 3a and 3b in normal human cells, both these molecules were tested in vitro against human peripheral blood mononuclear cells (PBMC) isolated from healthy donors. After treatment of both resting and phytohemagglutin (PHA)-stimulated PBMC for 72 h, the IC50 values were over 20 µM, indicating that 3a and 3b did not substantially affect the viability of these cells, suggesting that these two compounds have cancer cell selective killing properties.
3.2. Inhibition of Tubulin Polymerization and Colchicine Binding
To investigate whether the antiproliferative activities of these compounds were related to an interaction with the microtubule system, the two most active compounds of the series (3a and 3b) as antiproliferative agents were evaluated for their inhibitory effects on tubulin polymerization and on the binding of [3H]colchicine to tubulin (in the latter assay, tubulin was examined at a concentration of 1 μM, while compounds and colchicine were at 5 μM). For comparison, CA-4 was examined in contemporaneous experiments as a reference compound (Table 2). Derivatives 3a and 3b inhibited the assembly reaction with IC50 values of 3.6 and 3.8 μM, respectively, considerably higher than the value obtained with CA-4 (IC50: 0.54 μM), which is consistent with the reduced antiproliferative activity of 3a and 3b with respect to CA-4. These results indicated that tubulin may be the intracellular target of compounds 3a and 3b. In competition experiments, both these compounds inhibited the binding of [3H]colchicine to its binding site on tubulin, with 31% and 29% of inhibition, respectively. These derivatives were less potent than CA-4, which in these experiments inhibited colchicine binding by 98%.
Table 2.
Inhibition of tubulin polymerization and colchicine binding by compounds 3a, 3b, and CA-4.
The results are consistent with the conclusion that the antiproliferative activity of these compounds derives from an interaction with the colchicine site of tubulin and interference with microtubule assembly. In conclusion, manipulation of the scaffold of compounds with general structures 1 and 2 led to the successful identification of a new series of inhibitors of tubulin assembly, characterized by a 3,4,5-trimethoxy aniline and a cyano function at the 2- and 3-positions, respectively, of the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine nucleus.
3.3. Analysis of Effects on the Cell Cycle
Since molecules exhibiting effects on tubulin assembly should cause alteration of cell cycle parameters, leading to a preferential G2/M blockade, the effects of compounds 3a and 3b on cell cycle distribution were investigated on K562 cells using flow cytometry. The cells were cultured for 72 h in the presence of two different concentrations (IC50 and IC75) for each tested derivative and the obtained results were compared with non-treated K562 cells as control. Both compounds caused a significant (p < 0.01) and dose-dependent accumulation of the cells in the G2/M phase of the cell cycle, with a concomitant decrease of cells in the other phases of the cell cycle (especially the G0/G1 phase). As shown in Figure 2 (panels I and L), treatment of K562 cells at different concentrations of 3b (IC50: 0.70 μM and IC75: 0.90 μM) increased the percentage of G2/M-phase cells from 22.9% ± 0.7% (control group) to 30.3% ± 1.3% and 33.5% ± 0.6% respectively, indicating that compound 3b impact the cell cycle distribution in a dose-dependent manner. Similarly, for derivative 3a (IC50: 0.75 μM and IC75: 1.0 μM), an increase of the percentage of G2/M-phase cells from 22.2% ± 0.7% (control group) to 33.6% ± 1.1%, and 34.9% ± 1.3% was observed at the values, respectively. These data confirm that compounds 3a and 3b act selectively on the G2-M phase of the cell cycle, as expected for inhibitors of tubulin assembly.
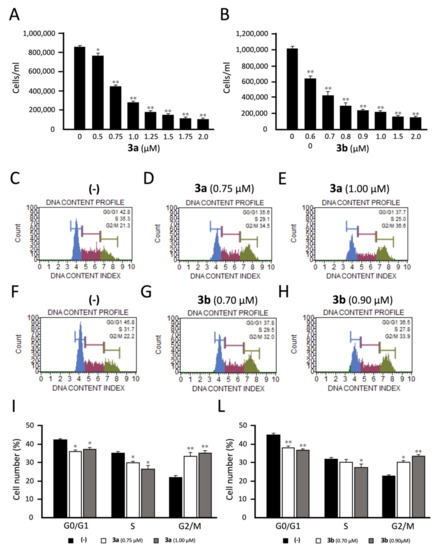
Figure 2.
Effects of the tetrahydrothieno[2,3-c]pyridines 3a and 3b on cell growth and cell cycle. K562 cells were treated with compounds 3a (A,C–E,I) and 3b (B,F–H,L) and the following biological parameters were evaluated: cell growth (A,B) and cell cycle distribution (C–H). Analyses were performed after 3 days of cell culture at the indicated concentrations of the compounds. In panels C-H, representative cell cycle analyses are reported. In panels I and L, a summary of three independent experiments is reported. The data represent the average ± S.D. *: significant (p < 0.05); **: highly significant (p < 0.01).
3.4. Effects on Apoptosis
In order to characterize the mode of cell death induced by compounds 3a and 3b, a biparametric flow cytometry analysis was performed using propidium iodide (PI), which stains DNA and is permeable only to dead cells, and fluorescent immunolabeling of the protein annexin-V, which binds to the phospholipid phosphatidylserine (PS) in a highly selective manner. This phospholipid flips from the inner to the outer leaflet of the plasma membrane during apoptosis. Positive staining with annexin-V correlates with the loss of plasma membrane polarity, but this staining precedes the complete loss of membrane integrity that accompanies the later stages of cell death, resulting from either apoptosis or necrosis. In contrast, PI can only enter cells after complete loss of membrane integrity. Thus, dual staining for annexin-V and with PI permits discrimination between unaffected cells (annexin-V−/PI−), early apoptotic cells (annexin-V+/PI−), late apoptotic cells (annexin-V+/PI+), and necrotic cells (annexin-V−/PI+). The results obtained are shown in Figure 3.
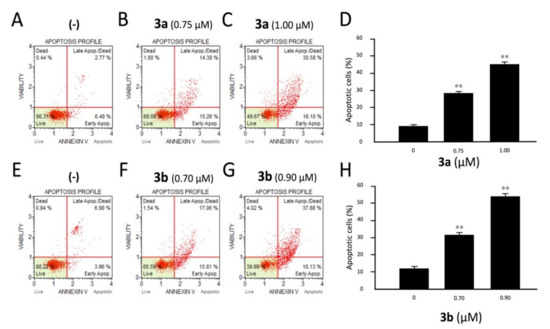
Figure 3.
Effects of the tetrahydrothieno[2,3-c]pyridines 3a and 3b on apoptosis. K562 cells were treated with compounds 3a (A–D) and 3b (E–H), and apoptosis was analyzed by cytofluometry. Analyses were performed after 3 days of cell culture at the indicated concentrations of 3a and 3b compounds. In panels A–C and E–G, representative analyses of apoptosis are reported for compound 3a (A–C) and compound 3b (E–G). In panels D and H, a summary of three independent experiments is presented. The data represent the average ± S.D. **: highly significant (p < 0.01).
The obtained two parameter histograms demonstrate the effects of different concentrations of 3a (IC50: 0.75 μM and IC75: 1.00 μM) and 3b (IC50: 0.70 μM and IC75: 0.90 μM) on K562 cells after 72 h of treatment. Both compounds induced an accumulation of annexin-V positive cells in comparison with the control, and this accumulation was dose dependent. In the representative experiment shown in Figure 3, the amount of total apoptotic cells did not exceed 11% in the negative controls (not treated samples). On the contrary, compound 3b at the IC50 (0.70 μM) and IC75 (0.90 μM) values after 72 h of treatment showed 32.87% and 56.01% cells undergoing apoptosis, respectively. Similarly, 3a is also very effective in the induction of apoptosis in a dose-dependent manner, showing 29.64% and 46.68% cells in apoptotic phase at its IC50 (0.75 μM) and IC75 (1.00 μM) values, respectively. The results indicated that most of K562 cells treated with 3a and 3b undergo apoptosis.
3.5. Molecular Modeling Studies
The potential interaction between compounds 3a and 3b and the colchicine site was investigated through molecular docking studies, using Glide. [54] The colchicine-tubulin complex (PDB ID: 4O2B) crystal structure was selected as the protein for the docking simulation. [55]
Both compounds seem to occupy the binding site partially overlapping the co-crystallized colchicine, with the trimethoxyphenyl ring orientated towards the nearby α-tubulin subunit, with interactions αSer178 and αThr179 (Figure 4A,B). The N-methoxy/ethoxycarbonyl moiety at the 6-position of the tetrahydrothieno[2,3-c]pyridine ring is placed in proximity and interacts with either the polypeptide backbone of β-tubulin or with the sidechain of βCys241, a key interaction point for tubulin polymerization inhibition. Interestingly, the two molecules adopt a different orientation than the canonical binding previously reported for several tubulin inhibitors, including combretastatin A-4, in which the trimethoxyphenyl ring is placed in the β-tubulin subunit in proximity to βCys241 [56,57]. This occupation of the colchicine site seems to be fundamental for strong inhibition of tubulin polymerization, and therefore, the different orientation found for compounds 3a and 3b could explain their apparently weaker binding to tubulin and causing their reduced inhibitory effects on tubulin assembly/colchicine binding described above.
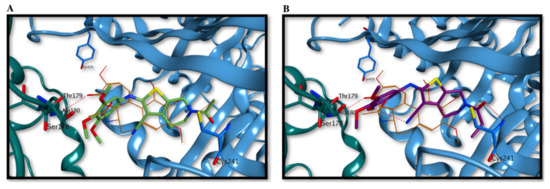
Figure 4.
Proposed binding modes for compounds 3a (A) and 3b (B) in the colchicine site. The trimethoxyphenyl ring is oriented towards the α-tubulin subunit, while the N-methoxy/ethoxycarbonyl moiety at the 6-position of the tetrahydrothieno[2,3-c]pyridine ring is placed in proximity of βCys241. Co-crystallized colchicine is shown in orange. The tubulin α subunit is shown as a dark green ribbon, while the β subunit is represented as a cornflower blue ribbon.
4. Materials and Methods
4.1. Chemistry
4.1.1. Materials and Methods
1H experiments were recorded on either a Bruker AC 200 or a Varian 400 Mercury Plus spectrometer, while 13C-NMR spectra were recorded on a Varian 400 Mercury Plus spectrometer. Chemical shifts (δ) are given in ppm upfield, and the spectra were recorded in appropriate deuterated solvents, as indicated. Positive-ion electrospray ionization (ESI) mass spectra were recorded on a double-focusing Finnigan MAT 95 instrument with BE geometry. Melting points (mp) were determined on a Büchi-Tottoli apparatus and are uncorrected. All products reported showed 1H and 13C-NMR spectra in agreement with the assigned structures. The purity of tested compounds was determined by combustion elemental analyses conducted by the Microanalytical Laboratory of the Chemistry Department of the University of Ferrara with a Yanagimoto MT-5 CHN recorder elemental analyzer. All tested compounds yielded data consistent with a purity of at least 95% as compared with the theoretical values. All reactions were carried out under an inert atmosphere of dry nitrogen, unless otherwise indicated. Standard syringe techniques were used for transferring dry solvents. Reaction courses and product mixtures were routinely monitored by Thin Layer Chromatography (TLC) on silica gel (precoated F254 Merck plates), and compounds were visualized with aqueous KMnO4. Flash chromatography was performed using 230–400 mesh silica gel and the indicated solvent system. Organic solutions were dried over anhydrous Na2SO4.
4.1.2. General Procedure A for the Synthesis of Compounds (4a–4p)
To a suspension of malonitrile or cyanoacetate (methyl/ethyl/t-butylcyanoacetate) (5 mmol), the appropriate ketone corresponding to 4-methyl/phenylcyclohexanone, N-acetyl-4-piperidone or methoxy/ethoxycarbonyl-4-piperidone (5 mmol), TEA (0.44 mL, 5 mmol) and sulfur (164 mg, 5 mmol) in the appropriate solvent (methanol or ethanol, 10 mL) was stirred for 2 h at 70 °C. The solvent was evaporated, and the residue was diluted with dichloromethane (15 mL). After washing with water (2 × 5 mL) and brine (5 mL), the organic layer was dried over anhydrous Na2SO4 and evaporated. The crude product was purified by column chromatography on silica gel or by crystallization from ethyl ether.
Methyl 2-amino-3-cyano-4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate (4a). Following general procedure A, the crude residue obtained by the condensation between malononitrile and methyl 4-oxopiperidine-1-carboxylate in ethanol as solvent was purified by crystallization with ethyl ether to furnish 4a as an orange solid. Yield: 87%, m.p. 131–133 °C. 1H-NMR (d6-DMSO) δ: 2.38–2.44 (m, 2H), 3.56–3.59 (m, 2H), 3.62 (s, 3H), 4.29 (s, 2H), 7.14 (bs, 2H). MS (ESI): [M + 1]+ = 238.3.
Ethyl 2-amino-3-cyano-4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate (4b). Following general procedure A, the crude residue obtained by the condensation between malononitrile and ethyl 4-oxopiperidine-1-carboxylate in ethanol as solvent was purified by crystallization with ethyl ether to furnish 4b as an orange solid. Yield: 87%, m.p. 171–174 °C. 1H–NMR (CDCl3) δ: 1.29 (t, J = 7.2 Hz, 3H), 2.77 (t, J = 5.8 Hz, 2H), 3.74 (t, J = 5.8 Hz, 2H), 4.21 (q, J = 7.2 Hz, 2H), 4.56 (s, 2H), 4.67 (bs, 2H). MS (ESI): [M + 1]+ = 252.3.
Dimethyl 2–amino-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (4c). Following general procedure A, the crude residue obtained by the condensation between methyl 2-cyanoacetate and methyl 4-oxopiperidine-1-carboxylate in methanol as solvent was purified by crystallization with ethyl ether to furnish 4c as a brown solid. Yield: 67%, m.p. 135–137 °C. 1H-NMR (CDCl3) δ: 2.80–2.82 (m, 2H), 3.66 (t, J = 6.0 Hz, 2H), 3.74 (s, 3H), 3.86 (s, 3H), 4.42 (s, 2H), 4.52 (bs, 2H). MS (ESI): [M + 1]+ = 271.3.
6-Ethyl 3-methyl 2-amino-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (4d). Following general procedure A, the crude residue obtained by the condensation between methyl 2-cyanoacetate and ethyl 4-oxopiperidine-1-carboxylate in methanol as solvent was purified by crystallization with ethyl ether to furnish 4d as a yellow solid. Yield: 84%, m.p. 152–154 °C. 1H-NMR (CDCl3) δ: 1.27 (t, J = 7.0 Hz, 3H), 2.81 (t, J = 6.0 Hz, 2H), 3.66 (t, J = 6.0 Hz, 2H), 3.90 (s, 3H), 4.11 (bs, 2H), 4.15 (q, J = 7.0 Hz, 2H), 4.42 (s, 2H). MS (ESI): [M + 1]+ = 285.4.
Methyl 6-acetyl-2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (4e). Following general procedure A, the crude residue obtained by the condensation between methyl 2-cyanoacetate and 1-acetylpiperidin-4-one in methanol as solvent was purified by crystallization with ethyl ether to furnish 4e as a yellow solid. Yield: 72%, m.p. 144–146 °C. 1H-NMR (CDCl3) δ: 2.24 (s, 3H), 2.87 (t, J = 6.0 Hz, 2H), 3.72 (t, J = 6.0 Hz, 2H), 3.80 (s, 3H), 4.51 (s, 2H), 5.14 (bs, 2H). MS (ESI): [M + 1]+ = 255.3.
3-Ethyl 6-methyl 2-amino-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (4f). Following general procedure A, the crude residue obtained by the condensation between ethyl 2-cyanoacetate and methyl 4-oxopiperidine-1-carboxylate in ethanol as solvent was purified by crystallization with ethyl ether to furnish 4f as a yellow solid. Yield: 64%, m.p. 151–153 °C. 1H-NMR (CDCl3) δ: 1.33 (t, J = 7.0 Hz, 3H), 2.80-2.82 (m, 2H), 3.66 (t, J = 6.0 Hz, 2H), 3.74 (s, 3H), 3.78 (bs, 2H), 4.24 (q, J = 7.0 Hz, 2H), 4.41 (s, 2H). MS (ESI): [M + 1]+ = 285.4.
Diethyl 2-amino-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (4g). Following general procedure A, the crude residue obtained by the condensation between ethyl 2-cyanoacetate and ethyl 4-oxopiperidine-1-carboxylate in ethanol as solvent was purified by crystallization with ethyl ether to furnish 4g as a yellow solid. Yield: 78%, m.p. 145–147 °C. 1H-NMR (d6-DMSO) δ: 1.15-1.28 (m, 6H), 2.64-2.67 (m, 2H), 3.55 (t, J = 6.0 Hz, 2H), 4.04 (q, J = 7.2 Hz, 2H), 4.16 (q, J = 7.2 Hz, 2H), 4.29 (s, 2H), 7.32 (bs, 2H). MS (ESI): [M + 1]+ = 299.1.
Ethyl 6-acetyl-2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (4h). Following general procedure (A), the crude residue obtained by the condensation between ethyl 2-cyanoacetate and 1-acetylpiperidin-4-one in ethanol as solvent was purified by crystallization with ethyl ether to furnish 4h as a yellow solid. Yield: 69%, m.p. 135–137 °C. 1H-NMR (CDCl3) δ: 1.36 (t, J = 7.4 Hz, 3H), 2.26 (s, 3H), 2.89 (t, J = 6.0 Hz, 2H), 3.73 (t, J = 6.0 Hz, 2H), 4.29 (q, J = 7.2 Hz, 2H), 4.38 (s, 2H), 4.96 (bs, 2H). MS (ESI): [M + 1]+ = 269.3.
3-tert-butyl 6-methyl 2-amino-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (4i). Following general procedure A, the crude residue obtained by the condensation between methyl 2-cyanoacetate and tert-butyl 4-oxopiperidine-1-carboxylate in methanol as solvent was purified by crystallization with ethyl ether to furnish 4i as a yellow solid. Yield: 78%, m.p. 134–136 °C. 1H-NMR (CDCl3) δ: 1.57 (s, 9H), 2.76–2.81 (m, 2H), 3.66–3.70 (m, 2H), 3.73 (s, 3H), 4.45 (s, 2H), 4.54 (bs, 2H). MS (ESI): [M + 1]+ = 313.4.
3-tert-butyl 6-ethyl 2-amino-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (4j). Following general procedure A, the crude residue obtained by the condensation between ethyl 2-cyanoacetate and tert-butyl 4-oxopiperidine-1-carboxylate in ethanol as solvent was purified by crystallization with ethyl ether to furnish 4j as a yellow solid. Yield: 84%, m.p. 152–154 °C. 1H-NMR (CDCl3) δ: 1.29 (t, J = 7.2 Hz, 3H), 1.55 (s, 9H), 2.78–2.81 (m, 2H), 3.66-3.69 (m, 2H), 4.19 (q, J = 7.2 Hz, 2H), 4.46 (s, 2H), 4.84 (bs, 2H). MS (ESI): [M + 1]+ = 327.4.
2. -Amino-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (4k). Following general procedure A, the crude residue obtained by the condensation between malononitrile and 4-methylcyclohexanone in methanol as solvent was purified by trituration of the crude product with ethyl ether to furnish 4k as a yellow solid. Yield: 58%, m.p. 124–126 °C. 1H-NMR (CDCl3) δ: 1.03 (d, J = 6.6 Hz, 3H), 1.39–1.42 (m, 1H), 1.84–1.88 (m, 2H), 2.08–2.12 (m, 1H), 2.56–2.60 (m, 3H), 4.29 (bs, 2H). MS (ESI): [M + 1]+ = 193.3.
2. -Amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (4l). Following general procedure (A), the crude residue obtained by the condensation between malononitrile and 4-phenylcyclohexanone in ethanol as solvent was purified by trituration of the crude product with ethyl ether to furnish 4l as a yellow solid. Yield: 71%, m.p. 180–182 °C. 1H-NMR (CDCl3) δ: 1.19–1.22 (m, 1H), 2.07–2.10 (m, 1H), 2.66–2.71 (m, 3H), 2.72–2.76 (m, 2H), 2.97–3.00 (m, 1H), 3.39 (bs, 2H), 7.32 (m, 5H). MS (ESI): [M + 1]+ = 255.2.
Methyl 2-amino-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (4m). Following general procedure A, the crude residue obtained by the condensation between methyl 2-cyanoacetate and 4-methylcyclohexanone in methanol as solvent was purified by flash chromatography on silica gel, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 4m as a yellow solid. Yield: 65%, m.p. 109–110 °C. 1H-NMR (CDCl3) δ: 1.03 (d, J = 6.2 Hz, 3H), 1.34–1.38 (m, 1H), 1.84–1.88 (m, 2H), 2.15–2.19 (m, 1H), 2.63–2.66 (m, 2H), 2.84–2.87 (m, 1H), 3.88 (s, 3H), 4.64 (bs, 2H). MS (ESI): [M + 1]+ = 226.3.
Methyl 2-amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (4n). Following general procedure A, the crude residue obtained by the condensation between methyl 2-cyanoacetate and 4-phenylcyclohexanone in methanol as solvent was purified by flash chromatography on silica gel, using ethyl acetate:petroleum ether 4:6 (v:v) as eluent, to furnish 4n as a white solid. Yield: 62%, m.p. 132–134. °C 1H-NMR (CDCl3) δ: 1.80–1.84 (m, 1H), 2.02–2.06 (m, 1H), 2.75–2.79 (m, 3H), 2.99–3.02 (m, 2H), 3.86 (s, 3H), 4.82 (bs, 2H), 7.23–7.36 (m, 5H). MS (ESI): [M + 1]+ = 288.2.
Ethyl 2-amino-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (4o). Following general procedure A, the crude residue obtained by the condensation between ethyl 2-cyanoacetate and 4-methylcyclohexanone in ethanol as solvent was purified by flash chromatography on silica gel, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 4o as a yellow solid. Yield: 63%, m.p. 112–114. °C 1H-NMR (CDCl3) δ: 1.03 (d, J = 6.2 Hz, 3H), 1.29–1.32 (m, 1H), 1.35 (t, J = 7.0 Hz, 3H), 1.84–1.87 (m, 2H), 2.14–2.17 (m, 1H), 2.63–2.68 (m, 2H), 2.84–2.88 (m, 1H), 4.32 (q, J = 7.0 Hz, 2H), 5.82 (bs, 2H). MS (ESI): [M + 1]+ = 240.3.
Ethyl 2-amino-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (4p). Following general procedure A, the crude residue obtained by the condensation between ethyl 2-cyanoacetate and 4-phenylcyclohexanone in ethanol as solvent was purified by flash chromatography on silica gel, using ethyl acetate:petroleum ether 4:6 (v:v) as eluent, to furnish 4p as a yellow oil. Yield: 59%. 1H-NMR (CDCl3) δ: 1.34 (t, J = 7.0 Hz, 3H), 1.90–1.95 (m, 1H), 2.10–2.14 (m, 1H), 2.77–2.82 (m, 3H), 3.02–3.06 (m, 2H), 4.34 (q, J = 7.0 Hz, 2H), 4.76 (bs, 2H), 7.24–7.33 (m, 5H). MS (ESI): [M + 1]+ = 302.3.
4.1.3. General Procedure B for the Synthesis of Compounds (5a–p)
In a dry three-necked round-bottom flask, anhydrous CuBr2 (536 mg, 2.4 mmol) and tert-butyl nitrite (360 μL, 3 mmol) were dissolved in anhydrous acetonitrile (10 mL) under an argon atmosphere. The resulting mixture was warmed at 65 °C and 2-amino derivative 4a–p (2 mmol) in acetonitrile (5 mL) was slowly added. The reaction was complete after 2 h, as monitored by TLC. The dark mixture was allowed to reach room temperature, poured in a saturated aqueous NH4Cl solution (10 mL), and extracted with CH2Cl2 (30 mL). The organic phase was washed twice with a saturated aqueous NH4Cl solution (10 mL) and brine (10 mL), dried over Na2SO4, and concentrated at reduced pressure to furnish a residue that was purified by flash chromatography on silica gel.
Methyl 2-bromo-3-cyano-4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate (5a). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 5a as a yellow solid. Yield: 64%, m.p. 153–155 °C. 1H-NMR (CDCl3) δ: 2.77 (t, J = 5.8 Hz, 2H), 3.71 (t, J = 5.8 Hz, 2H), 3.75 (s, 3H), 4.56 (s, 2H). MS (ESI): [M] + = 301.2, [M + 2]+ = 303.2.
Ethyl 2-bromo-3-cyano-4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate (5b). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 5b as a white solid. Yield: 64%, m.p. 171–173 °C. 1H-NMR (CDCl3) δ: 1.28 (t, J = 7.0 Hz, 3H), 2.81 (t, J = 5.8 Hz, 2H), 3.73 (t, J = 5.8 Hz, 2H), 4.17 (q, J = 7.0 Hz, 2H), 4.66 (s, 2H). MS (ESI): [M]+ = 315.2, [M + 2]+ = 317.2.
Dimethyl 2-bromo-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (5c). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 5c as a colorless oil. Yield: 64%. 1H-NMR (CDCl3) δ: 2.89 (t, J = 5.8 Hz, 2H), 3.68 (t, J = 5.8 Hz, 2H), 3.73 (s, 3H), 3.83 (s, 3H), 4.54 (s, 2H). MS (ESI): [M]+ = 334.1, [M + 2]+ = 336.3.
6-Ethyl 3-methyl 2-bromo-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (5d). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 5d as a yellow oil. Yield: 73%. 1H-NMR (CDCl3) δ: 1.28 (t, J = 7.2 Hz, 3H), 2.89 (t, J = 6.0 Hz, 2H), 3.68 (t, J = 5.8 Hz, 2H), 3.87 (s, 3H), 4.16 (q, J = 7.2 Hz, 2H), 4.54 (s, 2H). MS (ESI): [M]+ = 348.3, [M + 2]+ = 350.3.
Methyl 6-acetyl-2-bromo-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5e). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as eluent, to furnish 5e as a brown oil. Yield: 68%. 1H-NMR (CDCl3) δ: 2.21 (s, 3H), 3.05 (t, J = 6.0 Hz, 2H), 3.68 (t, J = 5.8 Hz, 2H), 3.83 (s, 3H), 4.76 (s, 2H). MS (ESI): [M]+ = 318.3, [M + 2]+ = 320.3.
3-Ethyl 6-methyl 2-bromo-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (5f). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 5f as a yellow solid. Yield: 65%, m.p. 110–112 °C. 1H-NMR (CDCl3) δ: 1.32 (t, J = 7.2 Hz, 3H), 2.90 (t, J = 6.0 Hz, 2H), 3.68 (t, J = 5.8 Hz, 2H), 3.74 (s, 3H), 4.29 (q, J = 7.2 Hz, 2H), 4.55 (s, 2H). MS (ESI): [M]+ = 348.3, [M + 2]+ = 350.3.
Diethyl 2-bromo-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (5g). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 5g as a yellow oil. Yield: 58%. 1H-NMR (CDCl3) δ: 1.28 (t, J = 7.0 Hz, 3H), 1.35 (t, J = 7.0 Hz, 3H), 2.86 (t, J = 5.6 Hz, 2H), 3.68 (t, J = 5.8 Hz, 2H), 4.16 (q, J = 7.2 Hz, 2H), 4.34 (q, J = 7.2 Hz, 2H), 4.54 (s, 2H). MS (ESI): [M]+ = 362.1, [M + 2]+ = 364.3.
Ethyl 6-acetyl-2-bromo-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (5h). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as eluent, to furnish 5h as a brown oil. Yield: 69%. 1H-NMR (CDCl3) δ: 1.35 (t, J = 7.2 Hz, 3H), 2.22 (s, 3H), 3.05 (t, J = 6.0 Hz, 2H), 3.72 (t, J = 5.8 Hz, 2H), 4,27 (q, J = 7.2 Hz, 2H), 4.76 (s, 2H). MS (ESI): [M]+ = 332.3, [M + 2]+ = 334.3.
3-tert-butyl 6-methyl 2-bromo-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (5i). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 5i as a colorless oil. Yield: 63%. 1H-NMR (CDCl3) δ: 1.57 (s, 9H), 2.87 (t, J = 6.0 Hz, 2H), 3.67 (t, J = 5.8 Hz, 2H), 3.74 (s, 3H), 4.54 (s, 2H). MS (ESI): [M]+ = 376.3, [M + 2]+ = 378.3.
3-tert-butyl 6-ethyl 2-bromo-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (5j). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 5j as a colorless oil. Yield: 71%. 1H-NMR (CDCl3) δ: 1.26 (t, J = 7.2 Hz, 3H), 1.55 (s, 9H), 2.87 (t, J = 6.0 Hz, 2H), 3.71 (t, J = 5.8 Hz, 2H), 4.16 (q, J = 7.2 Hz, 2H), 4.53 (s, 2H). MS (ESI): [M]+ = 390.3, [M + 2]+ = 392.3.
2. -Bromo-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (5k). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as eluent, to furnish 5k as a green oil. Yield: 72%. 1H-NMR (CDCl3) δ: 1.06 (d, J = 6.2 Hz, 3H), 1.26–1.28 (m, 1H), 1.89–1.92 (m, 2H), 2.20–2.24 (m, 1H), 2.69–2.72 (m, 3H). MS (ESI): [M]+ = 256.2, [M + 2]+ = 258.2.
2. -Bromo-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (5l). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as eluent, to furnish 5l as a brown oil. Yield: 63%. 1H-NMR (CDCl3) δ: 1.80–1.83 (m, 1H), 2.17–2.20 (m, 1H), 2.75–2.79 (m, 3H), 2.98–3.01 (m, 2H), 7.26–7.31 (m, 5H). MS (ESI): [M]+ = 318.2, [M + 2]+ = 320.3.
Methyl 2-bromo-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (5m). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as eluent, to furnish 5m as a colorless oil. Yield: 63%. 1H-NMR (CDCl3) δ: 1.06 (d, J = 6.6 Hz, 3H), 1.30–1.32 (m, 1H), 1.89-1.92 (m, 2H), 2.24–2.28 (m, 1H), 2.74-2.78 (m, 2H), 3.03-3.06 (m, 1H), 3.84 (s, 3H). MS (ESI): [M]+ = 289.2, [M + 2]+ = 291.2.
Methyl 2-bromo-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (5n). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as eluent, to furnish 5n as a colorless oil. Yield: 70%. 1H-NMR (CDCl3) δ: 1.77–1.82 (m, 1H), 2.08–2.10 (m, 1H), 2.78–2.82 (m, 3H), 3.00–3.04 (m, 2H), 3.84 (s, 3H), 7.24–7.30 (m, 5H). MS (ESI): [M]+ = 351.3, [M + 2]+ = 353.4.
Ethyl 2-bromo-6-methyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (5o). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as eluent, to furnish 5o as a colorless oil. Yield: 72%. 1H-NMR (CDCl3) δ: 1.07 (d, J = 6.6 Hz, 3H), 1.31–1.34 (m, 1H), 1.37 (t, J = 7.0 Hz, 3H), 1.78-1.82 (m, 2H), 2.17–2.21 (m, 1H), 2.68–2.75 (m, 2H), 2.86–2.88 (m, 1H), 4.26 (q, J = 7.0 Hz, 2H). MS (ESI): [M]+ = 303.2, [M + 2]+ = 305.2.
Ethyl 2-bromo-6-phenyl-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (5p). Following general procedure B, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 0.5:9.5 (v:v) as eluent, to furnish 5p as a colorless oil. Yield: 68%. 1H-NMR (CDCl3) δ: 1.36 (t, J = 7.0 Hz, 3H), 1.78–1.82 (m, 1H), 2.00–2.05 (m, 1H), 2.76–2.80 (m, 3H), 2.97–3.02 (m, 2H), 4.33 (q, J = 7.0 Hz, 2H), 7.24–7.33 (m, 5H). MS (ESI): [M]+ = 365.4, [M + 2]+ = 367.4.
4.1.4. General Procedure C for the Preparation of Compounds (3a–3p)
A dry Schlenk tube was charged with dry toluene (5 mL), 2-bromo derivative 3a–p (0.5 mmol), Pd(OAc)2 (3 mol%, 15 mg), rac-BINAP (4 mol%, 15 mg), Cs2CO3 (230 mg, 0.7 mmol, 1.4 equiv.), and 3,4,5-trimethoxyaniline (137 mg, 0.75 mmol, 1.5 equiv.) under argon, and the mixture was heated at 100 °C for 18 h. After cooling, the mixture was filtered on a pad of celite and the filtrate diluted with EtOAc (10 mL) and water (5 mL). The organic phase was washed with brine (5 mL), dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel.
Methyl 3-cyano-2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate (3a). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 3a as a yellow solid. Yield: 58%, m.p. 180–181 °C. 1H-NMR (d6-DMSO) δ: 2.50–2.53 (m, 2H), 3.59 (s, 3H), 3.62 (t, J = 5.8 Hz, 2H), 3.69 (s, 3H), 3.72 (s, 6H), 4.39 (s, 2H), 6.53 (s, 2H), 9.55 (s, 1H). 13C-NMR (d6-DMSO) δ: 23.92, 30.58, 42.31, 52.54, 55.66 (2C), 60.02, 90.03, 95.76 (2C), 114.79, 117.84, 130.87, 132.83, 137.99, 153.22 (2C), 155.41, 157.49. MS (ESI): [M + 1]+ = 404.3. Anal. calcd for C19H21N3O5S: C, 56.56; H, 5.25; N, 10.42; found: 56.38; H, 5.11; N, 10.21.
Ethyl 3-cyano-2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-6(7H)-carboxylate (3b). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 4:6 (v:v) as eluent, to furnish 3b as a yellow solid. Yield: 51%, m.p. 134–136 °C. 1H-NMR (d6-DMSO) δ: 1.15 (t, J = 7.2 Hz, 3H), 2.50-2.53 (m, 2H), 3.59 (s, 3H), 3.63 (t, J = 5.8 Hz, 2H), 3.72 (s, 6H), 4.05 (q, J = 7.2 Hz, 2H), 4.39 (s, 2H), 6.53 (s, 2H), 9.55 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.46, 23.92, 42.22, 55.67 (2C), 60.02, 61.02, 61.18, 95.74 (2C), 105.76, 114.80, 117.35, 130.89, 132.82, 137.99, 153.22 (2C), 154.62, 157.47. MS (ESI): [M + 1]+ = 418.4. Anal. calcd for C20H23N3O5S: C, 57.54; H, 5.55; N, 10.07; found: C, 57.32; H, 5.38; N, 9.98.
Dimethyl 2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (3c). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 4:6 (v:v) as eluent, to furnish 3c as a white solid. Yield: 58%, m.p. 140–141 °C. 1H-NMR (d6-DMSO) δ: 2.75 (t, J = 5.6 Hz, 2H), 3.59 (t, J = 5.6 Hz, 2H), 3.63 (s, 6H), 3.78 (s, 9H), 4.39 (s, 2H), 6.68 (s, 2H), 9.62 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.52, 26.24, 42.82, 51.51, 56.38 (2C), 60.20, 60.57, 97.92 (2C), 105.92, 114.33, 131.23, 134.09, 137.31, 153.86 (2C), 155.65, 158.70, 165.79. MS (ESI): [M + 1]+ = 437.5. Anal. calcd for C20H24N2O7S: C, 55.03; H, 5.54; N, 6.42; found: C, 54.89; H, 5.23; N, 6.30.
6-Ethyl 3-methyl 2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (3d). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 4:6 (v:v) as eluent, to furnish 3d as a yellow solid. Yield: 55%, m.p. 76–78 °C. 1H-NMR (d6-DMSO) δ: 1.19 (t, J = 6.8 Hz, 3H), 2.82 (t, J = 5.8 Hz, 2H), 3.62 (t, J = 5.8 Hz, 2H), 3.64 (s, 3H), 3.77 (s, 3H), 3.78 (s, 6H), 4.06 (q, J = 6.8 Hz, 2H), 4.39 (s, 2H), 6.68 (s, 2H), 9.64 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.58, 26.51, 42.30, 51.08, 55.94 (2C), 59.77, 60.14, 61.00, 97.43 (2C), 105.48, 114.32, 130.80, 133.64, 136.87, 153.42 (2C), 155.02, 158.24, 165.37. MS (ESI): [M + 1]+ = 451.5. Anal. (C21H26N2O7S) C, H, N. Anal. calcd for C21H26N2O7S. C, 55.99; H, 5.82; N, 6.22; found: C, 55.78; H, 5.64; N, 6.10.
Methyl 6-acetyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (3e). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as eluent, to furnish 3e as a yellow solid. Yield: 54%, m.p. 90–91 °C. 1H-NMR (d6-DMSO) δ: 2.06 (s, 3H), 2.79-2.82 (m, 2H), 3.61-3.64 (m, 5H), 3.76 (s, 9H), 4.42 (s, 2H), 6.66 (s, 2H), 9.61 (s, 1H). 13C-NMR (d6-DMSO) δ: 20.98, 27.09, 42.94, 43.98, 50.97, 55.83 (2C), 60.02, 97.31 (2C), 105.38, 114.08, 130.65, 133.52, 136.76, 153.31 (2C), 158.02, 165.22, 168.33. MS (ESI): [M + 1]+ = 421.3. Anal. calcd for C20H24N2O6S: C, 57.13; H, 5.75; N, 6.66; found: 57.01; H, 5.58; N, 6.48.
3-Ethyl 6-methyl 2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (3f). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 3f as a yellow solid. Yield: 59%, m.p. 128–130 °C. 1H-NMR (d6-DMSO) δ: 1.27 (t, J = 7.2 Hz, 3H), 2.73-2.75 (m, 2H), 3.52-3.54 (m, 2H), 3.58 (s, 6H), 3.79 (s, 6H), 4.21 (q, J = 7.2 Hz, 2H), 4.40 (s, 2H), 6.65 (s, 2H), 9.69 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.21, 26.63, 42.41, 52.57, 55.09, 55.94 (2C), 59.72, 60.13, 97.17 (2C), 105.63, 114.55, 130.71, 133.53, 136.82, 153.44 (2C), 155.12, 158.07, 165.10. MS (ESI): [M + 1]+ = 451.6. Anal. calcd for C21H26N2O7S: C, 55.99; H, 5.82; N, 6.22; found: 55.77; H, 5.63; N, 6.02.
Diethyl 2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (3g). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 3g as a yellow solid. Yield: 63%, m.p. 67–69 °C. 1H-NMR (d6-DMSO) δ: 1.18 (t, J = 7.2 Hz, 3H), 1.29 (t, J = 7.2 Hz, 3H), 2.77 (t, J = 5.6 Hz, 2H), 3.62 (t, J = 5.6 Hz, 2H), 3.64 (s, 3H), 3.78 (s, 6H), 4.06 (q, J = 7.2 Hz, 2H), 4.23 (q, J = 7.2 Hz, 2H), 4.39 (s, 2H), 6.67 (s, 2H), 9.73 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.20, 14.58, 26.33, 30.69, 42.31, 55.94, 59.72 (2C), 60.13, 61.00, 97.13 (2C), 105.62, 114.29, 130.74, 133.53, 136.81, 153.44 (2C), 154.67, 158.06, 165.12. MS (ESI): [M + 1]+ = 465.6. Anal. (C22H28N2O7S) C, H, N. Anal. calcd for C22H28N2O7S: C, 56.88; H, 6.08; N, 6.03; found: C, 56.65; H, 5.92; N, 5.88.
Ethyl 6-acetyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylate (3h). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 1:9 (v:v) as eluent, to furnish 3h as a white solid. Yield: 54%, m.p. 93-95 °C. 1H-NMR (d6-DMSO) δ: 1.28 (t, J = 7.2 Hz, 3H), 2.09 (s, 3H), 2.78-2.80 (m, 2H), 3.63 (s, 3H), 3.64-3.66 (m, 2H), 3.78 (s, 6H), 4.26 (q, J = 7.2 Hz, 2H), 4.45 (s, 2H), 6.67 (s, 2H), 9.72 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.14, 20.99, 27.14, 42.97, 44.05, 55.83 (2C), 59.60, 60.02, 97.08 (2C), 105.54, 114.56, 130.59, 133.41, 136.71, 153.33 (2C), 157.97, 164.99, 168.33. MS (ESI): [M + 1]+ = 435.4. Anal. calcd for C21H26N2O6S: C, 62.20; H, 6.71; N, 3.45; found: C, 62.04; H, 6.58; N, 3.23.
3-tert-butyl 6-methyl 2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (3i). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3i as a white solid. Yield: 64%, m.p. 50–52 °C. 1H-NMR (d6-DMSO) δ: 1.53 (s, 9H), 2.72–2.75 (m, 2H), 3.59 (t, J = 5.8 Hz, 2H), 3.63 (s, 3H), 3.76 (s, 3H), 3.78 (s, 6H), 4.40 (s, 2H), 6.67 (s, 2H), 9.74 (s, 1H). 13C-NMR (d6-DMSO) δ: 26.78, 27.98 (3C), 42.35, 52.47, 55.86 (2C), 59.67, 60.05, 80.50, 96.83 (2C), 106.89, 113.97, 130.67, 133.32, 136.74, 153.35 (2C), 155.02, 157.42, 164.75. MS (ESI): [M + 1]+ = 479.5. Anal. calcd for C23H30N2O7S: C, 57.72; H, 6.32; N, 5.85; found: C, 57.54; H, 6.21; N, 5.67.
3-tert-butyl 6-ethyl 2-((3,4,5-trimethoxyphenyl)amino)-4,5-dihydrothieno[2,3-c]pyridine-3,6(7H)-dicarboxylate (3j). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 3:7 (v:v) as eluent, to furnish 3j as a yellow solid. Yield: 70%, m.p. 60–62 °C. 1H-NMR (d6-DMSO) δ: 1.17 (t, J = 7.2 Hz, 3H), 1.53 (s, 9H), 2.74 (t, J = 5.8 Hz, 2H), 3.59 (t, J = 5.8 Hz, 2H), 3.63 (s, 3H), 3.78 (s, 6H), 4.06 (q, J = 7.2 Hz, 2H), 4.39 (s, 2H), 6.67 (s, 2H), 9.75 (s, 1H). 13C-NMR (d6-DMSO) δ: 15.04, 24.02, 28.50 (3C), 31.14, 42.77, 56.38 (2C), 60.57, 61.44, 81.03, 97.32 (2C), 107.39, 114.04, 131.42, 133.83, 137.26, 153.88 (2C), 155.31, 157.93, 165.30. MS (ESI): [M + 1]+ = 493.4. Anal. calcd for C24H32N2O7S: C, 58.52; H, 6.55; N, 5.69; found: C, 58.29; H, 6.29; N, 5.41.
6. -Methyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (3k). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3k as a white solid. Yield: 58%, m.p. 112–114 °C. 1H-NMR (d6-DMSO) δ: 1.01 (d, J = 6.4 Hz, 3H), 1.37–1.40 (m, 1H), 1.80–1.82 (m, 2H), 2.14–2.18 (m, 1H), 2.50–2.52 (m, 2H), 2.61–2.65 (m, 1H), 3.61 (s, 3H), 3.73 (s, 6H), 6.52 (s, 2H), 9.41 (s, 1H). 13C-NMR (d6-DMSO) δ: 21.61, 24.14, 29.62, 30.31, 32.19, 56.17 (2C), 60.57, 91.43, 95.78 (2C), 115.76, 121.73, 132.12, 133.00, 138.88, 153.74 (2C), 156.67. MS (ESI): [M + 1]+ = 359.4. Anal. calcd for C19H22N2O5S: C, 63.66; H, 6.19; N, 7.82; found: C, 63.47; H, 6.01; N, 7.64.
6. -Phenyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (3l). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3l as a yellow solid. Yield: 63%, m.p. 169–170 °C. 1H-NMR (d6-DMSO) δ: 1.96–1.99 (m, 2H), 2.60–2.63 (m, 3H), 2.74–2.78 (m, 1H), 2.98–3.01 (m, 1H), 3.61 (s, 3H), 3.74 (s, 6H), 6.54 (s, 2H), 7.20–7.23 (m, 1H), 7.30-7.32 (m, 4H), 9.48 (s, 1H). 13C-NMR (d6-DMSO) δ: 24.80, 29.43, 32.02, 40.02, 56.19 (2C), 60.58, 91.25, 95.92 (2C), 115.75, 121.70, 126.80, 126.32 (2C), 128.87 (2C), 132.22, 133.10, 138.82, 145.91, 153.75 (2C), 156.99. MS (ESI): [M + 1]+ = 421.5. Anal. (C24H24N2O5S) C, H, N. Anal. calcd for C24H24N2O5S: C, 68.55; H, 5.75; N, 6.66; found: C, 68.34; H, 5.54; N, 6.26.
Methyl 6-methyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (3m). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3m as a white solid. Yield: 55%, m.p. 127-129 °C. 1H-NMR (d6-DMSO) δ: 0.99 (d, J = 6.8 Hz, 3H), 1.26-1.30 (m, 1H), 1.76–1.80 (m, 2H), 2.13–2.16 (m, 1H), 2.50–2.54 (m, 2H), 2.82- 2.84 (m, 1H), 3.63 (s, 3H), 3.76 (s, 3H), 3.77 (s, 6H), 6.65 (s, 2H), 9.70 (s, 1H). 13C-NMR (d6-DMSO) δ: 21.67, 26.50, 29.30, 30.93, 32.36, 51.40, 56.35 (2C), 60.57, 97.35 (2C), 106.39, 117.77, 131.88, 133.74, 137.47, 153.83 (2C), 157.61, 166.18. MS (ESI): [M + 1]+ = 392.3. Anal. (C20H25NO5S) C, H, N. Anal. calcd for C24H24N2O5S: C, 61.36; H, 6.44; N, 3.58; found: C, 61.18; H, 6.23; N, 3.41.
Methyl 6-phenyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (3n). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3n as a white solid. Yield: 61%, m.p. 147-148 °C. 1H-NMR (d6-DMSO) δ: 1.79–1.82 (m, 1H), 1.96–2.00 (m, 1H), 2.68–2.72 (m, 3H), 2.83–2.86 (m, 2H), 3.62 (s, 3H), 3.78 (s, 9H), 6.68 (s, 2H), 7.22 (m, 1H), 7.31 (m, 4H), 9.71 (s, 1H). 13C-NMR (d6-DMSO) δ: 26.59, 29.47, 31.52, 40.02, 50.90, 55.82 (2C), 60.02, 97.00 (2C), 105.75, 117.27, 126.11, 126.73 (2C), 128.26 (2C), 131.49, 133.31, 136.90, 145.63, 153.29 (2C), 157.35, 165.60. MS (ESI): [M + 1]+ = 454.4. Anal. calcd for C25H27NO5S: C, 66.20; H, 6.00; N, 3.09; found: C, 66.01; H, 5.84; N, 2.92.
Ethyl 6-methyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (3o). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3o as a white solid. Yield: 56%, m.p. 90–92 °C. 1H-NMR (d6-DMSO) δ: 0.99 (d, J = 6.4 Hz, 3H), 1.26–1.28 (m, 1H), 1.32 (t, J = 7.0 Hz, 3H), 1.79–1.82 (m, 2H), 2.16–2.20 (m, 1H), 2.52–2.58 (m, 2H), 2.79-2.82 (m, 1H), 3.62 (s, 3H), 3.77 (s, 6H), 4.24 (q, J = 7.0 Hz, 2H), 6.64 (s, 2H), 9.79 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.15, 21.10, 26.01, 28.73, 30.43, 31.83, 55.80 (2C), 59.44, 60.02, 96.54 (2C), 105.98, 117.26, 131.29, 133.10, 136.88, 153.31 (2C), 156.89, 165.38. MS (ESI): [M + 1]+ = 406.3. Anal. calcd for C21H27NO5S: C, 62.20; H, 6.71; N, 3.45; found: C, 62.01; H, 6.48; N, 3.23.
Ethyl 6-phenyl-2-((3,4,5-trimethoxyphenyl)amino)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate (3p). Following general procedure C, the crude residue was purified by flash chromatography, using ethyl acetate:petroleum ether 2:8 (v:v) as eluent, to furnish 3p as a yellow solid. Yield: 73%, m.p. 98-100 °C. 1H-NMR (d6-DMSO) δ: 1.19 (t, J = 6.8 Hz, 3H), 1.84-1.86 (m, 1H), 1.98–2.02 (m, 1H), 2.79–2.84 (m, 3H), 2.97–3.00 (m, 2H), 3.60 (s, 3H), 3.82 (s, 6H), 4.28 (q, J = 7.0 Hz, 2H), 6.67 (s, 2H), 7.19–7.21 (m, 1H), 7.29–7.33 (m, 4H), 9.80 (s, 1H). 13C-NMR (d6-DMSO) δ: 14.17, 26.65, 29.57, 31.49, 55.82 (2C), 59.42, 59.65, 60.02, 96.72 (2C), 105.90, 117.32, 126.13, 126.73 (2C), 128,27 (2C), 131.44, 133.20, 136.86, 145.65, 153.31 (2C), 157.19, 165.35. MS (ESI): [M + 1]+ = 468.4. Anal. calcd for C26H29NO5S: C, 66.79; H, 6.25; N, 3.00; found: C, 66.58; H, 6.02; N, 2.78.
4.2. Biological Evaluation
4.2.1. Cell Growth Conditions and Antiproliferative Assay
Murine leukemia L1210, human T-lymphocyte leukemia CEM, human cervix carcinoma (HeLa), and human chronic myelogenous leukemia K562 cells (3–5 × 104 cells) and a serial (5-fold) dilution of the test compounds were added to a 96-well microtiter plate. The cells were incubated for 72 h at 37 °C in a humidified 5% CO2 atmosphere. At the end of the incubation period, the cells were counted in a Coulter Counter (Coulter Electronics Ltd., Harpenden Herts, United Kingdom). The IC50 (50% inhibitory concentration) was defined as the concentration of compound that inhibited cell proliferation by 50%. The IC50 values represent the average (± standard deviation) of three independent experiments. Human peripheral blood mononuclear cells (PBMC) were obtained from healthy donors by centrifugation with Ficoll-Paque Plus (GE Healthcare Bio-Sciences AB, Uppsala).
4.2.2. Effects on Tubulin Polymerization
Bovine brain tubulin was purified as described previously [58]. To evaluate the effect of the compounds on tubulin assembly in vitro [59], varying concentrations were preincubated with 10 μM tubulin in 0.8 M glutamate buffer at 30 °C and then cooled to 0 °C. After addition of GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C, and the assembly of tubulin was observed turbidimetrically. The IC50 was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The ability of the test compounds to inhibit colchicine binding to tubulin was measured as described earlier [60], except that the reaction mixtures contained 1 μM tubulin, 5 μM of [3H]colchicine, and test compounds 3a or 3b.
4.2.3. Effects on the Cell Cycle
Human leukemia K562 cells were treated with compounds 3a or 3b, and analysis of the distribution of the cells through the cell cycle was performed with the Muse Cell Analyzer instrument (Millipore Corporation, Billerica, MA, USA). Assay and cell counting were performed according to the instructions supplied by the manufacturer. Samples were collected after the treatment and washed in PBS. For analysis, 5 × 105 cells were resuspended in 200 μL of Muse Cell Cycle Reagent, incubated for 30 min at room temperature, and the cell suspension was transferred to a 1.5 mL microcentrifuge tube prior to analysis. Cells were analyzed by fluorescence-activated cell sorting analysis using a Muse Cell Analyzer.
4.2.4. Effects on Apoptosis
Apoptosis assays on K562 cells were performed with the Muse Cell Analyzer instrument, and assays were performed according to the instructions supplied by the manufacturer. The Muse Annexin V & Dead Cell Kit employs annexin V to detect phosphatidyl serine (PS) on the external membrane of apoptotic cells. A fluorescent DNA intercalator (7-ADD: 7-aminoactinomycin D) is also used as an indicator of cell membrane integrity. From live, healthy cells, as well as early apoptotic cells, 7-ADD is excluded while it is able to bind DNA in cells in late apoptosis and in dead cells. Cells were washed with sterile PBS 1X, suspended, and diluted (1:2) with the Muse Annexin V & Dead Cell reagent. Samples were gently mixed and incubated at room temperature, protected from the light for 15 min. Samples were analyzed using a Muse Cell Analyzer, and data from prepared samples were acquired and recorded utilizing the Annexin V and Dead Cell Software Module (Millipore) [61].
4.2.5. Molecular Modeling Methods
All molecular docking studies were performed on a Viglen Genie Intel®CoreTM i7-3770 vPro CPU@ 3.40 GHz x 8 running Ubuntu 18.04. Molecular Operating Environment (MOE) 2019.10 [62] and Maestro (Schrödinger Release 2019-3) [54] were used as molecular modeling software. The tubulin structure was downloaded from the PDB data bank (http://www.rcsb.org/; PDB code 4O2B). The protein was preprocessed using the Schrödinger Protein Preparation Wizard by assigning bond orders, adding hydrogens and performing a restrained energy minimization of the added hydrogens using the OPLS_2005 force field. Ligand structures were built with MOE and then prepared using the Maestro LigPrep tool by energy minimizing the structures (OPLS_2005 force field), generating possible ionization states at pH 7 ± 2, generating tautomers and low-energy ring conformers. After isolating a tubulin dimer structure, a 12 Å docking grid (inner-box 10 Å and outer-box 22 Å) was prepared using as centroid the co-crystallized colchicine. Molecular docking studies were performed using Glide SP precision, keeping the default parameters and setting 5 as the number of output poses per input ligand to include in the solution. The output database was saved as a mol2 file. The docking results were visually inspected for their ability to bind the active site.
5. Conclusions
In conclusion, we have described the synthesis and biological evaluation of two classes of compounds based on the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine and 4,5,6,7-tetrahydrobenzo[b]thiophene skeletons, both characterized by the presence of a common 3′,4′,5′-trimethoxyanilino moiety at the 2-position and a cyano or different alkoxycarbonyl groups at the 3-position. In the series of 2-(3′,4′,5′-trimethoxyanilino)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivatives, the results indicated that inhibition of cell growth was strongly dependent on the substituent at the 3-position, with the greatest activity occurring with the cyano group, the least with alkoxycarbonyl moieties such as methoxy/ethoxy/tert-butoxycarbonyl. We also demonstrated that replacement of the 4,5,6,7-tetrahydrothieno[2,3-c]pyridine moiety with a 4,5,6,7-tetrahydrobenzo[b]thiophene system was detrimental for antiproliferative activity. The 2-(3′,4′,5′-trimethoxyanilino)-3-cyano-6-methoxycarbonyl-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivative 3a and the corresponding 6-ethoxycarbonyl homologue 3b were the most promising compounds in this series, which inhibited cancer cell growth at low micromolar concentrations and interacted with tubulin by binding to the colchicine site. Comparing the two 4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carbonitrile derivatives 3a and 3b, the latter was about 1.5-fold more active than 3a against L1210 and HeLa cells, while both these derivatives were equipotent against CEM cells. We also showed by flow cytometry that derivatives 3a and 3b had cellular effects typical of agents that bind to tubulin, causing accumulation of cells in the G2-M phase of the cell cycle. Apoptotic cells also increased in a concentration-dependent manner, with an increase in the percentage of apoptotic K562 cells observed at the IC50 values examined for 3a and 3b (0.75 and 0.70 μM, respectively). Finally, the same compounds showed high sensitivity towards cancer over normal cells as they had no significant antiproliferative activity toward both quiescent and phytohemoagglutinin-stimulated cultures of PBMC (IC50 > 20 μM), suggesting that derivatives 3a and 3b may have selectivity against cancer cells.
Author Contributions
Conceptualization and supervision, R.R., R.G. and A.F.; methodology, B.C. and M.Z.; writing-review & editing, E.H., R.R., J.B. and R.G.; project administration and funding acquisition, S.M., A.B. and R.G.; software, A.B. and S.F.; investigation, F.P., P.O., E.H., A.F, M.Z. and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
R.R., B.C. and S.M. acknowledge the support of the PRIN 2017 by grant 2017E84AA4_002. R.G. was supported by an AIRC (IG 13575). A.B. and S.F. acknowledge the support of the Life Science Research Network Wales grant no. NRNPGSep14008, an initiative funded through the Welsh Government’s Sêr Cymru program. S.F. is supported by the Sêr Cymru programme, which is partially funded by the European Regional Development Fund through the Welsh Government. E.H. was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract No. HHSN261200800001E.
Acknowledgments
The authors would like to acknowledge Alberto Casolari for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- Jemal, A.; Center, M.M.; De Sanis, C.; Ward, E.M. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worlwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Akhdar, H.; Legendre, C.; Aninat, C.; Morel, F. Anticancer drug metabolism: Chemotherapy resistance and new therapeutic approaches. In Topics on Drug Metabolism; Paxton, J., Ed.; InTech: Rijeka, Croatia, 2012; pp. 137–170. [Google Scholar]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Narang, A.S.; Desai, D.S. Anticancer drug development: Unique aspects of pharmaceutical development. In Pharmaceutical Perspectives of Cancer Therapeutics; Lu, Y., Mahato, R.I., Eds.; Springer Science: New York, NY, USA, 2009; pp. 49–92. [Google Scholar]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, G.; Gill, R.K.; Soni, R.; Bariwal, J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. [Google Scholar] [CrossRef]
- van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef]
- Nitika, V.; Kapil, K. Microtubule targeting agents: A benchmark in cancer therapy. Curr. Drug Ther. 2014, 8, 189–196. [Google Scholar] [CrossRef]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- Seligmann, J.; Twelves, C. Tubulin: An example of targeted chemotherapy. Future Med. Chem. 2013, 5, 339–352. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Dráber, P. Tubulins as therapeutic targets in cancer: From bench to bedside. Curr. Pharm. Des. 2012, 18, 2778–2792. [Google Scholar] [CrossRef] [PubMed]
- Manka, S.W.; Moores, C.A. The role of tubulin–tubulin lattice contacts in the mechanism of microtubule dynamic instability. Nat. Struct. Mol. Biol. 2018, 25, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Dynamics of microtubules and their associated proteins: Recent insights and clinical implications. Neurology 2016, 86, 1911–1920. [Google Scholar] [CrossRef]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-tubulin agents of natural origin: Targeting taxol, vinca, and colchicine binding domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef]
- Manfredi, J.; Parness, J.; Horwitz, S. Taxol binds to cellular microtubules. J. Cell Biology 1982, 94, 688–696. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.; Nazre, M. Vinca alkaloids. Int. J. Prev. Medicine. 2013, 4, 1231–1235. [Google Scholar]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef]
- Skoufias, D.; Wilson, L. Mechanism of inhibition of microtubule polymerization by colchicine: Inhibitory potencies of unliganded colchicine and tubulin-colchicine complexes. Biochemistry 1992, 31, 738–746. [Google Scholar] [CrossRef]
- Garon, E.B.; Neidhart, J.D.; Gabrail, N.Y.; de Oliveira, M.R.; Balkissoon, J.; Kabbinavar, F. A randomized Phase II trial of the tumor vascular disrupting agent CA-4P (fosbretabulin tromethamine) with carboplatin, paclitaxel, and bevacizumab in advanced nonsquamous non-small-cell lung cancer. Onco Targets Ther. 2016, 9, 7275–7283. [Google Scholar] [CrossRef]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than just vascular targeting agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Vindya, N.G.; Sharma, N.; Yadav, M.; Ethiraj, K.R. Tubulins-the target for anticancer therapy. Curr. Top. Med. Chem. 2015, 15, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.N.; Zheng, L.L.; Wang, D.; Liang, X.X.; Gao, F.; Zhou, X.L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806–828. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmaol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Theeramunkong, S.; Caldarelli, A.; Massarotti, A.; Aprile, S.; Caprifoglio, D.; Zaninetti, R.; Teruggi, A.; Pirali, T.; Grosa, G.; Tron, G.C.; et al. Regioselective Suzuki coupling of dihaloheteroaromatic compounds as a rapid strategy to synthesize potent rigid combretastatin analogues. J. Med. Chem. 2011, 54, 4977–4986. [Google Scholar] [CrossRef]
- Flynn, B.L.; Flynn, G.P.; Hamel, E.; Jung, M.K. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin A–4. Bioorg. Med. Chem. Lett. 2001, 11, 2341–2343. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Q.; Bai, Z.; Sun, J.; Jiang, X.; Song, H.; Wub, Y.; Zhang, W. Synthesis and biological evaluation of 2,3–diarylthiophene analogues of combretastatin A-4. Med. Chem. Commun. 2015, 6, 971–976. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Remusat, V.; Carrion, M.D.; Lopez Cara, C.; Preti, D.; Fruttarolo, F.; Pavani, M.G.; Aghazadeh Tabrizi, M.; Tolomeo, M.; et al. Synthesis and biological evaluation of 2-(3′,4′,5′–trimethoxybenzoyl)-3-amino 5-aryl thiophenes as a new class of tubulin inhibitors. J. Med. Chem. 2006, 49, 6425–6428. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Pavani, M.G.; Tabrizi, M.A.; Preti, D.; Fruttarolo, F.; Piccagli, L.; Jung, M.K.; Hamel, E.; Borgatti, M.; et al. Synthesis and biological evaluation of 2-amino-3-(3′, 4′, 5′-trimethoxybenzoyl)-5-aryl thiophenes as a new class of potent antitubulin agents. J. Med. Chem. 2006, 49, 3906–3915. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Balzarini, J.; Nussbaumer, P.; Bassetto, M.; Brancale, A.; et al. Design, synthesis and biological evaluation of 3,5-disubstituted 2-amino thiophene derivatives as a novel class of antitumor agents. Bioorg. Med. Chem. 2014, 22, 5097–5109. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Somappa, S.B.; Patil, S.A.; Nagaraja, B.M. An overview of benzo[b]thiophene-based medicinal chemistry. Eur. J. Med. Chem. 2017, 138, 1002–1033. [Google Scholar] [CrossRef] [PubMed]
- Pinney, K.G.; Bounds, A.D.; Dingerman, K.M.; Mocharla, V.P.; Pettit, G.R.; Bai, R.; Hamel, E. A new anti-tubulin agent containing the benzo[b]thiophene ring system. Bioorg. Med. Chem. Lett. 1999, 9, 1081–1086. [Google Scholar] [CrossRef]
- Flynn, B.L.; Verdier-Pinard, P.; Hamel, E. A novel palladium-mediated coupling approach to 2,3-disubstituted benzo[b]thiophenes and its application to the synthesis of tubulin binding agents. Org. Lett. 2001, 3, 651–654. [Google Scholar] [CrossRef]
- Chen, Z.; Mocharla, V.P.; Farmer, J.M.; Pettit, G.R.; Hamel, E.; Pinney, K.G. Preparation of new anti-tubulin ligands through a dual-mode, addition-elimination reaction to a bromo-substituted α, β-unsaturated sulfoxide. J. Org. Chem. 2000, 65, 8811–8815. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Lopez Cara, C.; Hamel, E.; Basso, G.; Bortolozzi, R.; Viola, G. Synthesis and biological evaluation of 2-(3′,4′,5′-trimethoxybenzoyl)-3-aryl/arylaminobenzo[b]thiophene derivatives as a novel class of antiproliferative agents. Eur. J. Med. Chem. 2010, 45, 5781–5791. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Bassetto, M.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Bortolozzi, R.; et al. Synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilino benzo[b]thiophenes and thieno[2,3-c]pyridines as new potent anticancer agents. J. Med. Chem. 2013, 56, 2606–2618. [Google Scholar] [CrossRef]
- Kemnitzer, W.; Sirisoma, N.; May, C.; Tseng, B.; Drewe, J.; Xiong Cai, S. Discovery of 4-anilino-N-methylthieno[3,2-d]pyrimidines and 4-anilino-N-methylthieno[2,3-d]pyrimidines as potent apoptosis inducers. Bioorg. Med. Chem. Lett. 2009, 19, 3536–3540. [Google Scholar] [CrossRef]
- Tian, C.; Chen, X.; Zhang, Z.; Wang, X.; Liu, J. Design and synthesis of (2-(phenylamino)thieno[3,2-d]pyrimidin-4-yl)(3,4,5-trimethoxyphenyl)methanone analogues as potent antitubulin polymerization agents. Eur. J. Med. Chem. 2019, 183, 111679. [Google Scholar] [CrossRef]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Baraldi, S.; Baraldi, P.G.; Schiaffino Ortega, S.; Chayah, M.; Kimatrai Salvador, M.; Lopez-Cara, L.C.; Brancale, A.; et al. Design, synthesis and biological evaluation of 6-substituted thieno[3,2-d]pyrimidine analogues as dual epidermal growth factor receptor kinase and microtubule inhibitors. J. Med. Chem. 2019, 62, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- Eurtivong, C.; Semenov, V.; Semenova, M.; Konyushkin, L.; Atamanenko, O.; Reynisson, J.; Kiselyov, A. 3-Amino-thieno[2,3-b]pyridines as microtubule-destabilising agents: Molecular modelling and biological evaluation in the sea urchin embryo and human cancer cells. Bioorg. Med. Chem. 2017, 25, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Lopez Cara, C.; Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Pipitone, M.R.; Balzarini, J.; et al. Synthesis and biological evaluation of 2-amino-3-(3′,4′,5′-trimethoxybenzoyl)-6-substituted-4,5,6,7-tetrahydrothieno[2,3-c]pyridine derivatives as antimitotic agents and inhibitors of tubulin polymerization. Bioorg. Med. Chem. Lett. 2008, 18, 5041–5045. [Google Scholar] [CrossRef] [PubMed]
- Bartels, B.; Gimmnich, P.; Pekari, K.; Baer, T.; Schmidt, M.; Beckers, T. Tetrahydropyridothiophenes for use in the treatment of cancer. WO2005118592, 15 December 2005. [Google Scholar]
- Pekari, K.; Baer, T.; Bertels, B.; Schmidt, M.; Beckers, T. Novel tetrahydropyridothiophenes. WO2005118071, 15 December 2005. [Google Scholar]
- Bausch, E.; Kohlhof, H.; Hamm, S.; Krauss, R.; Baumgartner, R.; Sironi, L. A novel microtubule inhibitor 4SC-207 with anti-proliferative activity in taxane-resistant cells. PLoS ONE 2013, 8, e79594. [Google Scholar] [CrossRef]
- Li, L.; Jiang, S.; Li, X.; Liu, Y.; Su, J.; Chen, J. Recent advances in trimethoxyphenyl (TMP) based tubulin inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2018, 151, 482–494. [Google Scholar] [CrossRef]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef]
- Wang, K.; Kim, D.; Domling, A. Cyanoacetamide MCR (III): Three-component Gewald reactions revisited. J. Comb. Chem. 2010, 12, 111–118. [Google Scholar] [CrossRef]
- Doyle, M.P.; Siegfried, B.; Dellaria, J.F., Jr. Alkyl nitrite-metal halide deamination reactions. 2. Substitutive deamination of arylamines by alkyl nitrites and copper (II) halides. A direct and remarkably efficient conversion of arylamines to aryl halides. J. Org. Chem. 1977, 42, 2426–2431. [Google Scholar] [CrossRef]
- Queiroz, M.-J.R.P.; Begouin, A.; Ferreira, I.C.F.R.; Kirsch, G.; Calhelha, R.C.; Barbosa, S.; Estevinho, L.M. Palladium-catalysed amination of electron-deficient or relatively electron-rich benzo[b]thienyl bromides-Preliminary studies of antimicrobial activity and SARs. Eur. J. Org. Chem. 2004, 3679–3685. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-3: Maestro, Schrödinger, LLC: New York, NY, USA, 2019. Available online: https://www.chemcomp.com (accessed on 7 April 2020).
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargstem, K.; Cavalli, A.; Steinmetz, M.O. Structural basis of cis- and trans-Combretastatin binding to tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Kimatrai Salvador, M.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, synthesis and biological evaluation of 2-alkoxycarbonyl-3-anilinoindoles as a new class of potent inhibitors of tubulin polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E.; Lin, C.M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–21. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Lai, J.-Y.; Yoo, H.-D.; Yu, J.; Marquez, B.; Nagle, D.G.; Nambu, M.; White, J.D.; Falck, J.R.; Gerwick, W.H.; et al. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998, 53, 62–67. [Google Scholar] [CrossRef]
- Milani, R.; Brognara, E.; Fabbri, E.; Manicardi, A.; Corradini, R.; Finotti, A.; Gasparello, J.; Borgatti, M.; Cosenza, L.C.; Lampronti, I.; et al. Targeting miR-155-5p and miR-221-3p by peptide nucleic acids induces caspase-3 activation and apoptosis in temozolomide-resistant T98G glioma cells. Int. J. Oncol. 2019, 55, 59–68. [Google Scholar] [CrossRef]
- ULC, C.C.G. Molecular Operating Environment (MOE), 2019.10, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2019. Available online: https://www.schrodinger.com (accessed on 7 April 2020).
Sample Availability: Samples of the compounds are not available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).