
Synthesis, Antiproliferative Activity and Molecular Docking Studies of Novel Doubly Modified Colchicine Amides and Sulfonamides as Anticancer Agents

 ,
,  , ,
, , 
Abstract
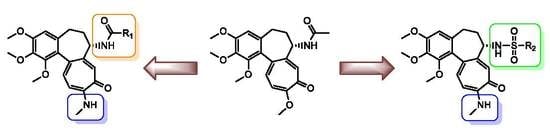
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. X-ray Crystal Analysis
2.3. Molecular Electrostatic Potential Map Analysis
2.4. In Vitro Determination of Drug-Induced Inhibition of Human Cancer Cell Line Growth
2.5. In Silico Determination of the Molecular Mode of Action
3. Materials and Methods
3.1. General
3.2. Spectroscopic Measurements
3.3. Synthesis
3.3.1. Synthesis of 2
3.3.2. Synthesis of 3
3.3.3. General Procedure for the Synthesis of Colchicine Derivatives 4, 8, 13, 15–16, 18 and 21
Compound 4
Compound 8
Compound 13
Compound 15
Compound 16
Compound 18
Compound 21
3.3.4. General Procedure for the Synthesis of Colchicine Derivatives 5–7, 9–12 and 14
Compound 5
Compound 6
Compound 7
Compound 9
Compound 10
Compound 11
Compound 12
Compound 14
3.3.5. Synthesis of 17
3.3.6. Synthesis of 19
3.3.7. Synthesis of 20
3.4. Single Crystal X-ray Measurement
3.5. DFT Molecular Modeling
3.6. In Vitro Antiproliferative Activity
3.6.1. Cell Lines and Culturing Conditions
3.6.2. Cell Viability Assays
SRB
3.7. Molecular Docking Studies
3.7.1. Ligand Preparation
3.7.2. Tubulin Model
3.7.3. Molecular Dynamics Simulations
3.7.4. Docking Simulations
3.7.5. Binding Energy and Pairwise Per-Residue Free Energy Decomposition Calculations Using MM/GBSA Method
3.8. Calculation clogP
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Amos, L.A. Microtubule structure and its stabilisation. Org. Biomol. Chem. 2004, 2, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Wade, R.H. On and around microtubules: An overview. Mol. Biotechnol. 2009, 43, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, T.; Mirigian, M.; Selcuk Yasar, M.; Ross, J.L. Mechanics of microtubules. J. Biomech. 2010, 43, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Hyman, A.; Desai, A. The spindle: A dynamic assembly of microtubules and motors. Nat. Cell Biol. 2001, 3. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules and actin filaments: Dynamic targets for cancer chemotherapy. Curr. Opin. Cell Biol. 1998, 10, 123–130. [Google Scholar] [CrossRef]
- Pellegrini, F.; Budman, D.R. Review: Tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest. 2005, 23, 264–273. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, R.; Skvortsova, I.; Kumar, V. Mechanisms of Tubulin Binding Ligands to Target Cancer Cells: Updates on their Therapeutic Potential and Clinical Trials. Curr. Cancer Drug Targets 2016, 17, 357–375. [Google Scholar] [CrossRef]
- Capraro, H.G.; Brossi, A. Chapter 1 Tropolonic Colchicum Alkaloids. Alkaloids Chem. Pharmacol. 1984, 23, 1–70. [Google Scholar]
- Boyé, O.; Brossi, A. Tropolonic Colchicum Alkaloids and Allo Congeners. Alkaloids Chem. Pharmacol. 1992, 41, 125–176. [Google Scholar]
- Hastie, S.B. Interactions of colchicine with tubulin. Pharmacol. Ther. 1991, 51, 377–401. [Google Scholar] [CrossRef]
- Sapra, S.; Bhalla, Y.; Nandani Sharma, S.; Singh, G.; Nepali, K.; Budhiraja, A.; Dhar, K.L. Colchicine and its various physicochemical and biological aspects. Med. Chem. Res. 2013, 22, 531–547. [Google Scholar] [CrossRef]
- Skoufias, D.A.; Wilson, L. Mechanism of Inhibition of Microtubule Polymerization by Colchicine: Inhibitory Potencies of Unliganded Colchicine and Tubulin-Colchicine Complexes. Biochemistry 1992, 31, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.; Panda, D.; Gupta, S.; Banerjee, M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008, 28, 155–183. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Wiesenfeld, P.L.; Garthoff, L.H.; Sobotka, T.J.; Suagee, J.K.; Barton, C.N. Acute oral toxicity of colchicine in rats: Effects of gender, vehicle matrix and pre-exposure to lipopolysaccharide. J. Appl. Toxicol. 2007, 27, 421–433. [Google Scholar] [CrossRef]
- Spiller, H.A. Colchicine. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Elsevier Inc.: London, UK, 2014; pp. 1007–1008. [Google Scholar]
- Roubille, F.; Kritikou, E.; Busseuil, D.; Barrere-Lemaire, S.; Tardif, J.-C. Colchicine: An Old Wine in a New Bottle? Antiinflamm. Antiallergy. Agents Med. Chem. 2013, 12, 14–23. [Google Scholar] [CrossRef]
- Mendis, S. Colchicine cardiotoxicity following ingestion of Gloriosa superba tubers. Postgrad. Med. J. 1989, 65, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.L.; Wilson, L. Addition of colchicine tubulin complex to microtubule ends: The mechanism of substoichiometric colchicine poisoning. Proc. Natl. Acad. Sci. USA 1977, 74, 3466–3470. [Google Scholar] [CrossRef]
- Kuncl, R.W.; Duncan, G.; Watson, D.; Alderson, K.; Rogawski, M.A.; Peper, M. Colchicine Myopathy and Neuropathy. N. Engl. J. Med. 1987, 316, 1562–1568. [Google Scholar] [CrossRef]
- Finkelstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine poisoning: The dark side of an ancient drug. Clin. Toxicol. 2010, 48, 407–414. [Google Scholar] [CrossRef]
- Cocco, G.; Chu, D.C.C.; Pandolfi, S. Colchicine in clinical medicine. A guide for internists. Eur. J. Intern. Med. 2010, 21, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Zemer, D.; Revach, M.; Pras, M.; Modan, B.; Schor, S.; Sohar, E.; Gafni, J. A Controlled Trial of Colchicine in Preventing Attacks of Familial Mediterranean Fever. N. Engl. J. Med. 1974, 291, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Cerquaglia, C.; Diaco, M.; Nucera, G.; La Regina, M.; Montalto, M.; Manna, R. Pharmacological and clinical basis of treatment of Familial Mediterranean Fever (FMF) with colchicine or analogues: An update. Curr. Drug Targets Inflamm. Allergy 2005, 4, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Urayama, A.; Kogure, M.; Nakajima, A.; Nakae, K.; Inaba, G. Double-masked trial of cyclosporin versus colchicine and long-term open study of cyclosporin in Behçet’s disease. Lancet 1989, 333, 1093–1096. [Google Scholar] [CrossRef]
- Keith, M.P.; Gilliland, W.R. Updates in the Management of Gout. Am. J. Med. 2007, 120, 221–224. [Google Scholar] [CrossRef]
- Hitzeman, N.; Stephens, R. Colchicine for acute gout. Am. Fam. Physician 2015, 91, 759–760. [Google Scholar] [PubMed]
- Kerekes, P.; Sharma, P.N.; Brossi, A.; Chignell, C.F.; Quinn, F.R. Synthesis and Biological Effects of Novel Thiocolchicines. 3. Evaluation of N-Acyldeacetylthiocolchicines, N-(Alkoxycarbonyl)deacetylthiocolchicines, and O-Ethyldemethylthiocolchicines. New Synthesis of Thiodemecolcine and Antileukemic Effects of 2-Demeth. J. Med. Chem. 1985, 28, 1204–1208. [Google Scholar] [CrossRef]
- Sun, L.; Hamel, E.; Lin, C.M.; Hastie, S.B.; Pyluck, A.; Lee, K.H. Antitumor Agents. 141. Synthesis and Biological Evaluation of Novel Thiocolchicine Analogs: N-Acyl-, N-Aroyl-, and N-(Substituted benzyl)deacetylthiocolchicines as Potent Cytotoxic and Antimitotic Compounds. J. Med. Chem. 1993, 36, 1474–1479. [Google Scholar] [CrossRef]
- Majcher, U.; Urbaniak, A.; Maj, E.; Moshari, M.; Delgado, M.; Wietrzyk, J.; Bartl, F.; Chambers, T.C.; Tuszynski, J.A.; Huczyński, A. Synthesis, antiproliferative activity and molecular docking of thiocolchicine urethanes. Bioorg. Chem. 2018, 81, 553–566. [Google Scholar] [CrossRef]
- Marzo-Mas, A.; Falomir, E.; Murga, J.; Carda, M.; Marco, J.A. Effects on tubulin polymerization and down-regulation of c-Myc, hTERT and VEGF genes by colchicine haloacetyl and haloaroyl derivatives. Eur. J. Med. Chem. 2018, 150, 591–600. [Google Scholar] [CrossRef]
- Shen, L.H.; Li, H.Y.; Shang, H.X.; Tian, S.T.; Lai, Y.S.; Liu, L.J. Synthesis and cytotoxic evaluation of new colchicine derivatives bearing 1,3,4-thiadiazole moieties. Chinese Chem. Lett. 2013, 24, 299–302. [Google Scholar] [CrossRef]
- Shen, L.H.; Li, Y.; Zhang, D.H.; Lai, Y.S.; Liu, L.J. Synthesis and evaluation of nitrate derivatives of colchicine as anticancer agents. Chinese Chem. Lett. 2011, 22, 768–770. [Google Scholar] [CrossRef]
- Shen, L.H.; Wang, S.L.; Li, H.Y.; Lai, Y.S.; Liu, L.J. Synthesis and bioactivity of furoxan-based nitric oxide-releasing colchicine derivatives as anticancer agents. Asian, J. Chem. 2013, 25, 3294–3296. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.M.; Kim, H.; Seok, H.; Kim, S.O.; Kwon, T.K.; Chang, J.S. The colchicine derivative CT20126 shows a novel microtubule-modulating activity with apoptosis. Exp. Mol. Med. 2013, 45, e19-e7. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.K.; Kim, J.M.; Kim, M.H.; Kim, K.H.; Chun, K.W.; Cho, K.H.; Youn, J.Y.; Namgoong, S.K. New synthetic thiocolchicine derivatives as low-toxic anticancer agents. Arch. Pharm. (Weinheim) 2005, 338, 582–589. [Google Scholar] [CrossRef]
- Saeedi, M.; Goli, F.; Mahdavi, M.; Dehghan, G.; Faramarzi, M.A.; Foroumadi, A.; Shafiee, A. Synthesis and biological investigation of some novel sulfonamide and amide derivatives containing coumarin moieties. Iran. J. Pharm. Res. 2014, 13, 881–892. [Google Scholar]
- Ashraf, Z.; Mahmood, T.; Hassan, M.; Afzal, S.; Rafique, H.; Afzal, K.; Latip, J. Dexibuprofen amide derivatives as potential anticancer agents: Synthesis, in silico docking, bioevaluation, and molecular dynamic simulation. Drug Des. Devel. Ther. 2019, 13, 1643–1657. [Google Scholar] [CrossRef]
- Ragha Suma, V.; Sreenivasulu, R.; Subramanyam, M.; Rao, K.R.M. Design, Synthesis, and Anticancer Activity of Amide Derivatives of Structurally Modified Combretastatin-A4. Russ. J. Gen. Chem. 2019, 89, 499–504. [Google Scholar] [CrossRef]
- Kachaeva, M.V.; Hodyna, D.M.; Semenyuta, I.V.; Pilyo, S.G.; Prokopenko, V.M.; Kovalishyn, V.V.; Metelytsia, L.O.; Brovarets, V.S. Design, synthesis and evaluation of novel sulfonamides as potential anticancer agents. Comput. Biol. Chem. 2018, 74, 294–303. [Google Scholar] [CrossRef]
- Gul, H.I.; Yamali, C.; Sakagami, H.; Angeli, A.; Leitans, J.; Kazaks, A.; Tars, K.; Ozgun, D.O.; Supuran, C.T. New anticancer drug candidates sulfonamides as selective hCA IX or hCA XII inhibitors. Bioorg. Chem. 2018, 77, 411–419. [Google Scholar] [CrossRef]
- Kiyoshi, A. N-methyldeacetylcolchiceinamide. Patent EP0607647(A1), 27 July 1994. [Google Scholar]
- Winum, J.Y.; Toupet, L.; Barragan, V.; Dewynter, G.; Montero, J.L. N-(tert-Butoxycarbonyl)-N-[4-(dimethylazaniumylidene)-1,4-dihydropyridin-1-ylsulfonyl]azanide: A new sulfamoylating agent. Structure and reactivity toward amines. Org. Lett. 2001, 3, 2241–2243. [Google Scholar] [CrossRef] [PubMed]
- Al-Riyami, L.; Pineda, M.A.; Rzepecka, J.; Huggan, J.K.; Khalaf, A.I.; Suckling, C.J.; Scott, F.J.; Rodgers, D.T.; Harnett, M.M.; Harnett, W. Designing anti-inflammatory drugs from parasitic worms: A synthetic small molecule analogue of the acanthocheilonema viteae product ES-62 prevents development of collagen-induced arthritis. J. Med. Chem. 2013, 56, 9982–10002. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Ortega-Muñoz, M.; López-Jaramillo, F.J.; Hernández-Mateo, F.; Blanco, V.; Santoyo-González, F. Vinyl Sulfonates: A Click Function for Coupling-and-Decoupling Chemistry and their Applications. Adv. Synth. Catal. 2016, 358, 3394–3413. [Google Scholar] [CrossRef]
- Lessinger, L.; Margulis, T.N. The crystal structure of colchicine. A new application of magic integers to multiple-solution direct methods. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1978, 34, 578–584. [Google Scholar] [CrossRef]
- McClure, W.O.; Paulson, J.C. The interaction of colchicine and some related alkaloids with rat brain tubulin. Mol. Pharmacol. 1977, 13, 560–575. [Google Scholar] [PubMed]
- Hastie, S.B.; Williams, R.C.; Puett, D.; Macdonald, T.L. The binding of isocolchicine to tubulin. Mechanisms of ligand association with tubulin. J. Biol. Chem. 1989, 264, 6682–6688. [Google Scholar]
- Politzer, P.; Laurence, P.R.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef]
- Chemical Applications of Atomic and Molecular Electrostatic Potentials. Available online: https://www.springer.com/gp/book/9780306406577 (accessed on 13 April 2020).
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01.; Gaussian, Inc: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Patterson, J.D. Density-functional theory of atoms and molecules. Ann. Nucl. Energy 1989, 16, 611. [Google Scholar] [CrossRef]
- Bai, R.; Pei, X.F.; Boyé, O.; Getahun, Z.; Grover, S.; Bekisz, J.; Nguyen, N.Y.; Brossi, A.; Hamel, E. Identification of cysteine 354 of β-tubulin as part of the binding site for the a ring of colchicine. J. Biol. Chem. 1996, 271, 12639–12645. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Seetharamalu, P.; Schwarz, P.M.; Hausheer, F.H.; Ludueña, R.F. The interaction of the B-ring of colchicine with α-Tubulin: A novel footprinting approach. J. Mol. Biol. 2000, 303, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.H.A.; Wu, S.Y.; Lee, T.R.; Chang, C.Y.; Wu, J.S.; Hsieh, H.P.; Chang, J.Y. Cancer cells acquire mitotic drug resistance properties through beta i-tubulin mutations and alterations in the expression of beta-tubulin isotypes. PLoS ONE 2010, 5, 1–11. [Google Scholar] [CrossRef]
- Ravanbakhsh, S.; Gajewski, M.; Greiner, R.; Tuszynski, J.A. Determination of the optimal tubulin isotype target as a method for the development of individualized cancer chemotherapy. Theor. Biol. Med. Model. 2013. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis CCD and CrysAlis RED, version 1171.32.15; Oxford Diffraction Ltd: Abingdon, Oxford, UK, 2009.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; Mcmahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Nevozhay, D. Cheburator software for automatically calculating drug inhibitory concentrations from in vitroscreening assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef]
- Schrödinger Schrödinger Release 2019–4: LigPrep. 2019. Available online: https://www.schrodinger.com/ligprep (accessed on 13 April 2020).
- Löwe, J.; Li, H.; Downing, K.H.; Nogales, E. Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 2001, 313, 1045–1057. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc: Montreal, QC, Canada, 2012.
- Klejborowska, G.; Urbaniak, A.; Maj, E.; Preto, J.; Moshari, M.; Wietrzyk, J.; Tuszynski, J.A.; Chambers, T.C.; Huczyński, A. Synthesis, biological evaluation and molecular docking studies of new amides of 4-chlorothiocolchicine as anticancer agents. Bioorg. Chem. 2020, 97. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Preto, J.; Gentile, F. Assessing and improving the performance of consensus docking strategies using the DockBox package. J. Comput. Aided. Mol. Des. 2019, 33, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Molinspiration Property Calculation Service. Available online: http://www.molinspiration.com (accessed on 3 April 2020).
Sample Availability: Not available |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 | 11 | 12 | 14 | 15 | 16 | 18 | 19 | ||
|---|---|---|---|---|---|---|---|---|---|
| C1–C16–C13–C12 | 100 K 295 K DFT value | 53.1(4) 55.2(4) 53.1 | 55.5(6) 55.5(6) 52.3 | 53.7(3) 55.0(3) 53.4 | 54.0(5) 54.4(4) 53.5 | 53.6(4) 54.7(4) 52.9 | 54.1(3) 55.0(3) 53.3 | 56.1(4) 56.1(4) 54.5 | 55.3(3) 55.9(4) 56.0 |
| C17–O1–C1–C2 | 100 K 295 K DFT value | −79.0(4) −86.5(4) −73.0 | −86.4(6) −86.4(6) −70.7 | −89.3(3) −86.4(4) −80.3 | −90.6(5) −87.4(5) −80.0 | −87.3(4) −87.8(4) −72.9 | −86.8(4) −86.4(4) −79.4 | −108.6(3) −106.8(3) −74.8 | 66.2(3) 65.6(4) 59.2 |
| C18–O2–C2–C3 | 100 K 295 K DFT value | 108.0(3) 106.9(4) 70.1 | 105.2(6) 105.2(6) 68.8 | 96.1(2) 98.7(3) 82.1 | 95.5(4) 98.0(4) 82.3 | 99.7(4) 101.4(4) 71.1 | 94.5(3) 98.7(3) 81.1 | −77.5(4) −74.0(4) −59.6 | −102.7(3) −100.6(4) −77.2 |
| C19–O3–C3–C4 | 100 K 295 K DFT value | −14.1(5) −8.1(7) −4.0 | −10.5(9) −10.5(10) −3.7 | 8.6(4) 6.9(6) −2.0 | 14.4(6) 11.4(7) −1.9 | −6.0(6) −2.3(6)−3.8 | 7.4(6) 6.9(6) −1.9 | 1.2(5) 0.2(5) 0.7 | 7.7(4) 6.8(5) 3.1 |
| C20–N1–C10–C11 | 100 K 295 K DFT value | 7.7(5) 6.8(6) −0.9 | 8.8(8) 8.7(9) −0.9 | 4.6(3) 7.5(4) −1.4 | 4.4(6) 5.0(6) −1.0 | 5.0(5) 5.2(5) −1.2 | 6.6(4) 7.5(4) −1.3 | −0.2(5) −1.7(5) 0.2 | 4.7(4) 4.0(7) −1.6 |
| Compound | A549 | MCF-7 | LoVo | LoVo/DX | BALB/3T3 | |
|---|---|---|---|---|---|---|
| IC50 [nM] | IC50 [nM] | IC50 [nM] | IC50 [nM] | RI | IC50 [nM] | |
| 1 | 115.3 ± 23.6 | 22.6 ± 1.3 | 17.5 ± 2.5 | 1646.6 ± 314.0 | 93.9 | 115.3 ± 36.8 |
| 2 | 10.8 ± 1.3 | 8.6 ± 1.3 | 4.3 ± 1.3 | 271.3 ± 99.9 | 63.0 | 10.8 ± 1.3 |
| 3 | 16.9 ± 2.8 | 19.7 ± 1.7 | 14.0 ± 1.7 | 129.2 ± 11.8 | 9.2 | 19.7 ± 7.0 |
| 4 | 14.6 ± 2.4 | 14.6 ± 1.5 | 9.7 ± 1.5 | 271.8 ± 104.4 | 28.0 | 19.4 ± 4.1 |
| 5 | 14.1 ± 2.4 | 14.1 ± 1.4 | 9.6 ± 0.5 | 194.8 ± 51.9 | 20.2 | 16.4 ± 3.5 |
| 6 | 13.6 ± 1.4 | 15.9 ± 6.6 | 6.8 ± 3.9 | 102.3 ± 20.7 | 15.0 | 13.6 ± 2.3 |
| 7 | 613.8 ± 194.4 | 464.8 ± 186.7 | 62.4 ± 16.9 | 2435.7 ± 923.4 | 39.0 | 545.9 ± 104.4 |
| 8 | 11.7 ± 1.4 | 18.8 ± 9.9 | 7.0 ± 1.4 | 171.2 ± 41.3 | 24.3 | 28.1 ± 10.8 |
| 9 | 11.6 ± 2.8 | 9.2 ± 1.4 | 1.8 ± 0.4 | 62.4 ± 6.7 | 35.2 | 11.6 ± 2.3 |
| 10 | 8.6 ± 1.3 | 8.6 ± 1.2 | 1.5 ± 0.5 | 38.5 ± 21.6 | 25.8 | 10.7 ± 1.3 |
| 11 | 14.6 ± 2.1 | 12.7 ± 0.4 | 10.4 ± 1.3 | 289.6 ± 165.2 | 27.8 | 81.3 ± 20.4 |
| 12 | 13.0 ± 1.3 | 13.0 ± 1.3 | 8.5 ± 0.4 | 99.9 ± 10.0 | 11.8 | 13.0 ± 3.3 |
| 13 | 6.3 ± 3.2 | 9.2 ± 0.8 | 0.7 ± 0.1 | 9.6 ± 3.3 | 14.0 | 6.2 ± 1.6 |
| 14 | 10.7 ± 0.6 | 12.6 ± 1.3 | 8.6 ± 0.8 | 102.5 ± 24.9 | 12.0 | 12.6 ± 2.1 |
| 15 | 36.8 ± 12.1 | 13.0 ± 2.2 | 10.8 ± 1.3 | 832.1 ± 292.7 | 76.8 | 43.3 ± 29.7 |
| 16 | 17.3 ± 3.7 | 12.8 ± 0.9 | 10.8 ± 1.3 | 946.9 ± 260.5 | 87.4 | 52.0 ± 29.3 |
| 17 | 56.6 ± 14.7 | 10.3 ± 3.9 | 21.1 ± 17.5 | 6466.0 ± 264.2 | 306.0 | 100.1 ± 24.1 |
| 18 | 10.6 ± 0.6 | 15.5 ± 1.7 | 9.3 ± 1.1 | 540.2 ± 107.2 | 57.8 | 14.2 ± 13.2 |
| 19 | 11.4 ± 1.7 | 13.0 ± 4.3 | 8.4 ± 0.7 | 306.7 ± 144.9 | 36.7 | 8.3 ± 3.8 |
| 20 | 800.3 ± 130.0 | 150.0 ± 25.3 | 268.4 ± 94.0 | 44385.7 ± 23852.0 | 165.4 | 991.1 ± 280.5 |
| 21 | 85.5 ± 5.4 | 134.3 ± 41.5 | 73.5 ± 15.7 | 6122.0 ± 825.1 | 83.3 | 87.6 ± 13.5 |
| Doxorubicin | 141.7 ± 46.0 | 204.2 ± 47.8 | 99.4 ± 41.0 | 8732.0 ± 2540.7 | 87.9 | 149.0 ±126.8 |
| Cisplatin | 5741.0 ± 968.0 | 7139.8 ± 1218.7 | 7076.3 ± 1596.2 | 8336.5 ± 1119.2 | 1.2 | 5665.1 ± 31.8 |
| Compound | 3D Representation of the Interactions | 2D Representation of the Interactions | Binding Energy [kcal/mol] | clogP | Active Residues |
|---|---|---|---|---|---|
| 1 |  |  | −41.0 | 1.1 | Ala179 Val180 Cys674 Leu688 Asn691 Ala749 Lys785 |
| 2 |  |  | −39.3 | 1.6 | Cys674 Ala683 Leu688 Lys785 |
| 3 |  |  | −4.0 | 0.9 | Cys674 Lys687 Leu688 Asn691 |
| 4 |  |  | −43.4 | 1.9 | Cys674 Ala683 Leu688 Asn691 Met692 Ala749 Lys785 |
| 5 |  |  | −43.0 | 2.5 | Ala179 Val180 Cys674 Leu688 Asn691 Ala749 Lys785 |
| 6 |  |  | −37.2 | 3.0 | Asn100 Ser177 Ala179 Leu681 Lys687 Leu688 Asn691 Lys785 |
| 7 |  |  | −53.7 | 8.7 | Gln10 Asn100 Ser177 Thr178 Cys674 Leu681 Asn682 Ala683 Lys687 Leu688 Asn691 Ala749 Lys785 |
| 8 |  |  | −32.3 | 2.7 | Ser177 Leu681 Ala683 Leu688 Lys785 |
| 9 |  |  | −44.0 | 2.2 | Ala179 Cys674 Ala683 Lys687 Leu688 Asn691 Met693 Ala749 Lys785 |
| 10 |  |  | −41.8 | 2.7 | Ala179 Cys674 Ala683 Leu688 Asn691 Asn782 Lys785 |
| 11 |  |  | −34.7 | 3.0 | Ala179 Cys674 Leu681 Ala683 Lys687 Leu688 Asn691 Lys785 |
| 12 |  |  | −35.2 | 3.3 | Ser177 Ala179 Leu681 Lys687 Leu688 Asn691 Lys785 |
| 13 |  |  | −40.1 | 3.9 | Cys674 Leu681 Ala683 Lys687 Leu688 Asn691 Lys785 |
| 14 |  |  | −34.3 | 3.4 | Val176 Ser177 Thr178 Ala179 Gln680 Leu688 Lys785 |
| 15 |  |  | −43.9 | 2.0 | Asn100 Ser177 Cys674 Leu681 Asn682 Ala683 Lys687 Leu688 Asn691 Lys785 |
| 16 |  |  | −38.3 | 2.0 | Ser177 Thr178 Cys674 Leu681 Ala683 Leu688 Asn691 Lys785 |
| 17 |  |  | −34.8 | 1.9 | Gln10 Ser177 Thr178 Ala179 Asn682 Ala683 Asp684 Lys687 Asn691 Lys785 |
| 18 |  |  | −54.6 | 2.5 | Gln10 Ala99 Asn100 Gly142 Gly143 Thr144 Ser177 Thr178 Ala179 Leu681 |
| 19 |  |  | −23.4 | 2..8 | Ser177 Thr178 Leu681 Ala683 Asp684 Lys687 Asn691 Lys785 |
| 20 |  |  | −59.6 | 2.4 | Gly9 Gln10 Ala11 Asp68 Ala99 Asn100 Gly142 Gly143 Thr144 Gly145 Ser177 Thr178 Lys687 |
| 21 |  |  | −29.8 | 4.9 | Ala179 Val180 Cys674 Leu688 Asn691 Ala749 Lys785 |
 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzywik, J.; Mozga, W.; Aminpour, M.; Janczak, J.; Maj, E.; Wietrzyk, J.; Tuszyński, J.A.; Huczyński, A. Synthesis, Antiproliferative Activity and Molecular Docking Studies of Novel Doubly Modified Colchicine Amides and Sulfonamides as Anticancer Agents. Molecules 2020, 25, 1789. https://doi.org/10.3390/molecules25081789
Krzywik J, Mozga W, Aminpour M, Janczak J, Maj E, Wietrzyk J, Tuszyński JA, Huczyński A. Synthesis, Antiproliferative Activity and Molecular Docking Studies of Novel Doubly Modified Colchicine Amides and Sulfonamides as Anticancer Agents. Molecules. 2020; 25(8):1789. https://doi.org/10.3390/molecules25081789
Chicago/Turabian StyleKrzywik, Julia, Witold Mozga, Maral Aminpour, Jan Janczak, Ewa Maj, Joanna Wietrzyk, Jack A. Tuszyński, and Adam Huczyński. 2020. "Synthesis, Antiproliferative Activity and Molecular Docking Studies of Novel Doubly Modified Colchicine Amides and Sulfonamides as Anticancer Agents" Molecules 25, no. 8: 1789. https://doi.org/10.3390/molecules25081789
APA StyleKrzywik, J., Mozga, W., Aminpour, M., Janczak, J., Maj, E., Wietrzyk, J., Tuszyński, J. A., & Huczyński, A. (2020). Synthesis, Antiproliferative Activity and Molecular Docking Studies of Novel Doubly Modified Colchicine Amides and Sulfonamides as Anticancer Agents. Molecules, 25(8), 1789. https://doi.org/10.3390/molecules25081789