Synthesis and Molecular Modelling Studies of New 1,3-Diaryl-5-Oxo-Proline Derivatives as Endothelin Receptor Ligands

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
:
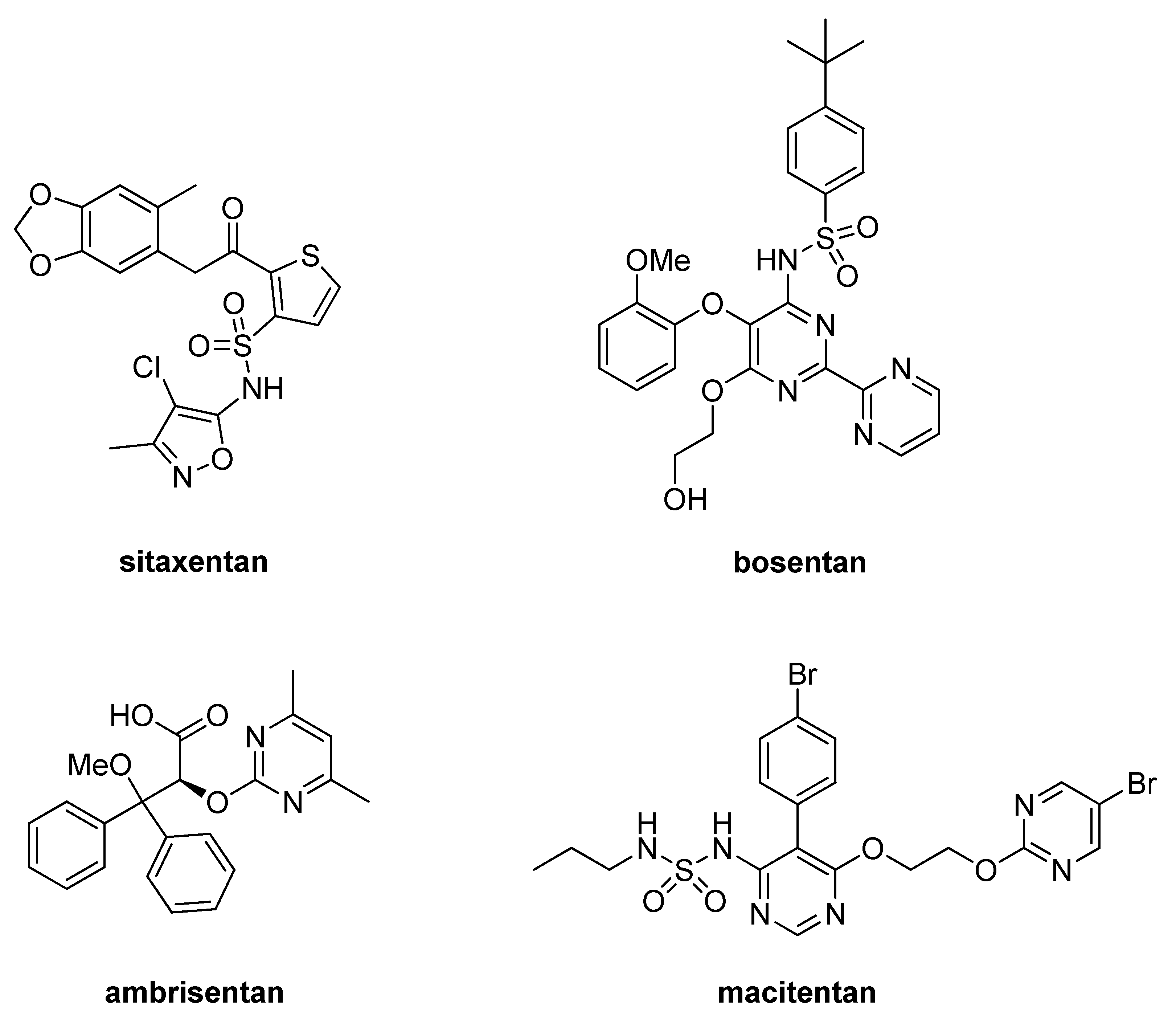
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Chemical Structures Elucidation
2.3. Radioligand Binding Assay
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of (±)-Trans 1,3-substituted-5-oxo-prolines 12a and 15a
3.1.2. General Procedure for the Synthesis of (±)-trans 1,3-Subtituted-5-oxo-prolines (13a, 14a, 16a, 17a)
(±)-trans 1-(1,3-Benzodioxol-5-yl)-3-(4-methoxyphenyl)-5-oxo-proline (13a)
(±)-trans 1,3-Di(1,3-benzodioxol-5-yl)-5-oxo-proline (14a)
(±)-trans 3-(3,4-Dimethoxyphenyl)-1-(4-methoxyphenyl)-5-oxo-proline (16a)
(±)-trans 1-(3,4-Dimethoxyphenyl)-3-(4-methoxyphenyl)-5-oxo-proline (17a)
3.1.3. (±)-Trans 1-(4-Methoxyphenyl)-5-oxo-3-phenyl-proline (12a)
3.1.4. General Procedure for the Synthesis of (±)-Trans 1,3-disubstituted-5-oxo-proline 31a–k
3.2. Radioligand Binding Assay
3.3. Molecular Modeling Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barton, M.; Yanagisawa, M. Endothelin: 30 Years from Discovery to Therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, H.; Hori, S.; Aramori, I.; Ohkubo, H.; Nakanishi, S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 1990, 348, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yanagisawa, M.; Takuwa, Y.; Miyazaki, H.; Kimura, S.; Goto, K.; Masaki, T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 1990, 348, 732–735. [Google Scholar] [CrossRef] [Green Version]
- Pittala, V.; Modica, M.; Romeo, G.; Materia, L.; Salerno, L.; Siracusa, M.; Cagnotto, A.; Mereghetti, I.; Russo, F. A facile synthesis of new 2-carboxamido-3-carboxythiophene and 4,5,6,7-tetrahydro-2-carboxamido-3-carboxythieno[2,3-c]pyridine derivatives as potential endothelin receptors ligands. Farmaco 2005, 60, 711–720. [Google Scholar] [CrossRef]
- Pittala, V.; Romeo, G.; Materia, L.; Salerno, L.; Siracusa, M.A.; Modica, M.; Mereghetti, I.; Cagnotto, A.; Russo, F. Novel (E)-alpha-[(1H-indol-3-yl)methylene]benzeneacetic acids as endothelin receptor ligands. Farmaco 2005, 60, 731–738. [Google Scholar] [CrossRef]
- Salerno, L.; Guerrera, F.; Modica, M.; Romeo, G.; Pittala, V.; Siracusa, M.A.; Mereghetti, I.; Cagnotto, A.; Mennini, T. Synthesis of 1,2,4-triazole derivatives: Binding properties on endothelin receptors. Med. Chem. 2007, 3, 551–560. [Google Scholar] [CrossRef]
- Georgianos, P.I.; Agarwal, R. Endothelin A receptor antagonists in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 338–344. [Google Scholar] [CrossRef]
- Pollock, J.S.; Pollock, D.M. SONAR propels endothelin A receptor antagonists to success. Nat. Rev. Nephrol. 2019, 15, 461–462. [Google Scholar] [CrossRef]
- Cahn, A.; Cernea, S.; Raz, I. The SONAR study—is there a future for endothelin receptor antagonists in diabetic kidney disease? Ann. Transl. Med. 2019, 7 (Suppl. 8), S330. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, D.B.; Seeburg, P.H. The Role of Molecular Biology in Drug Discovery and Design. In Protein Production by Biotechnolog; Harris, T.J.R., Ed.; Springer: Boston, MA, USA, 1990; pp. 181–190. [Google Scholar]
- Kumar, B.; Singh, S.; Skvortsova, I.; Kumar, V. Promising Targets in Anti-cancer Drug Development: Recent Updates. Curr. Med. Chem. 2017, 24, 4729–4752. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, A.A.; Eynde, J.J.V.; Jampilek, J.; Hadjipavlou-Litina, D.; Liu, H.; Reynisson, J.; Sousa, M.E.; Gomes, P.A.C.; Prokai-Tatrai, K.; Tuccinardi, T.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes-5. Molecules 2019, 24, 2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stock, J.K.; Jones, N.P.; Hammonds, T.; Roffey, J.; Dillon, C. Addressing the right targets in oncology: Challenges and alternative approaches. J. Biomol. Screen. 2015, 20, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Intagliata, S.; Salerno, L.; Ciaffaglione, V.; Leonardi, C.; Fallica, A.N.; Carota, G.; Amata, E.; Marrazzo, A.; Pittala, V.; Romeo, G. Heme Oxygenase-2 (HO-2) as a therapeutic target: Activators and inhibitors. Eur. J. Med. Chem. 2019, 183, 111703. [Google Scholar] [CrossRef]
- Nicholson, H.E.; Alsharif, W.F.; Comeau, A.B.; Mesangeau, C.; Intagliata, S.; Mottinelli, M.; McCurdy, C.R.; Bowen, W.D. Divergent Cytotoxic and Metabolically Stimulative Functions of Sigma-2 Receptors: Structure-activity Relationships of 6-acetyl-3-(4-(4-(4-fluorophenyl) piperazin-1-yl) butyl) benzo [d] oxazol-2 (3H)-one (SN79) Derivatives. J. Pharmacol. Exp. Ther. 2019, 368, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Romeo, G.; Prezzavento, O.; Intagliata, S.; Pittalà, V.; Modica, M.N.; Marrazzo, A.; Turnaturi, R.; Parenti, C.; Chiechio, S.; Arena, E. Synthesis, in vitro and in vivo characterization of new benzoxazole and benzothiazole-based sigma receptor ligands. Eur. J. Med. Chem. 2019, 174, 226–235. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Panchapakesan, U.; Pollock, C. Drug repurposing in kidney disease. Kidney Int. 2018, 94, 40–48. [Google Scholar] [CrossRef]
- Vacca, F.; Bagnato, A.; Catt, K.J.; Tecce, R. Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Res. 2000, 60, 5310–5317. [Google Scholar]
- Yamashita, J.; Ogawa, M.; Inada, K.; Yamashita, S.; Matsuo, S.; Takano, S. A large amount of endothelin-1 is present in human breast cancer tissues. Res. Commun. Chem. Pathol. Pharmacol. 1991, 74, 363–369. [Google Scholar] [PubMed]
- Nelson, J.B.; Chan-Tack, K.; Hedican, S.P.; Magnuson, S.R.; Opgenorth, T.J.; Bova, G.S.; Simons, J.W. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996, 56, 663–668. [Google Scholar] [PubMed]
- Nelson, J.; Bagnato, A.; Battistini, B.; Nisen, P. The endothelin axis: Emerging role in cancer. Nat. Rev. Cancer 2003, 3, 110–116. [Google Scholar] [CrossRef]
- Carducci, M.A.; Saad, F.; Abrahamsson, P.A.; Dearnaley, D.P.; Schulman, C.C.; North, S.A.; Sleep, D.J.; Isaacson, J.D.; Nelson, J.B. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer 2007, 110, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.I.; Tangen, C.M.; Hussain, M.; Lara, P.N., Jr.; Goldkorn, A.; Moinpour, C.M.; Garzotto, M.G.; Mack, P.C.; Carducci, M.A.; Monk, J.P.; et al. Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): A randomised phase 3 trial. Lancet. Oncol. 2013, 14, 893–900. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, A.A.; Haura, E.; Rodriguez, F.A.; Boulware, D.; Kapoor, R.; Neuger, A.M.; Lush, R.; Padilla, B.; Burton, M.; Williams, C.; et al. Phase I/II study of atrasentan, an endothelin A receptor antagonist, in combination with paclitaxel and carboplatin as first-line therapy in advanced non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1464–1469. [Google Scholar] [CrossRef] [Green Version]
- Salerno, L.; Modica, M.N.; Romeo, G.; Pittala, V.; Cagnotto, A.; Siracusa, M.A. Synthesis and endothelin receptors binding affinity of new 1,3,5-substituted pyrrole-2-carboxylic acid derivatives. Med. Chem. 2015, 11, 109–117. [Google Scholar] [CrossRef]
- Pittala, V.; Modica, M.; Salerno, L.; Siracusa, M.A.; Guerrera, F.; Mereghetti, I.; Cagnotto, A.; Mennini, T.; Romeo, G. Synthesis and endothelin receptor binding affinity of a novel class of 2-substituted-4-aryl-3-quinolinecarboxylic acid derivatives. Med. Chem. 2008, 4, 129–137. [Google Scholar] [CrossRef]
- Modica, M.; Salerno, L.; Pittala, V.; Romeo, G.; Siracusa, M.; Mereghetti, I.; Cagnotto, A.; Mennini, T. Synthesis and Binding Properties of New Endothelin Receptor Ligands. Lett. Drug Des. Discov. 2007, 4, 232–238. [Google Scholar] [CrossRef]
- Verho, O.; Maetani, M.; Melillo, B.; Zoller, J.; Schreiber, S.L. Stereospecific Palladium-Catalyzed C-H Arylation of Pyroglutamic Acid Derivatives at the C3 Position Enabled by 8-Aminoquinoline as a Directing Group. Org. Lett. 2017, 19, 4424–4427. [Google Scholar] [CrossRef]
- Artico, M.; Nacci, V. Ricerche su composti eterociclici azotati, Nota IV. Acido 1,3-difenilpirrolidin-5-one-2-carbossilico: Forme stereoisomere e derivati. Ann. Di Chim. 1968, 58, 637–650. [Google Scholar]
- Nambi, P.; Elshourbagy, N.; Wu, H.L.; Pullen, M.; Ohlstein, E.H.; Brooks, D.P.; Lago, M.A.; Elliott, J.D.; Gleason, J.G.; Ruffolo, R.R., Jr. Nonpeptide endothelin receptor antagonists. I. Effects on binding and signal transduction on human endothelinA and endothelinB receptors. J. Pharmacol. Exp. Ther. 1994, 271, 755–761. [Google Scholar] [PubMed]
- Shihoya, W.; Nishizawa, T.; Yamashita, K.; Inoue, A.; Hirata, K.; Kadji, F.M.N.; Okuta, A.; Tani, K.; Aoki, J.; Fujiyoshi, Y.; et al. X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat. Struct. Mol. Biol. 2017, 24, 758–764. [Google Scholar] [CrossRef] [PubMed]
- DeLean, A.; Munson, P.J.; Rodbard, D. Simultaneous analysis of families of sigmoidal curves: Application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978, 235, E97–E102. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compd | 1 H | 2 H | 3 H | 4 H | 5 H | 6 H |
|---|---|---|---|---|---|---|
| (±) 6a | δ 4.84 d, J = 5.6 Hz | δ 3.71–3.58 m | δ 3.00 dd, 3J = 9.2 Hz, 2J = 17.0 Hz | δ 2.64 dd, 3J = 7.0 Hz, 2J = 17.0 Hz | δ 4.18–3.98 m | δ 1.04 t, J = 7.0 Hz |
| (±) 6b | δ 5.11 d, J = 8.6 Hz | δ 4.17–4.07 m | δ 3.11 dd, 3J = 12.4 Hz, 2J = 16.6 Hz | δ 2.69 dd, 3J = 8.4 Hz, 2J = 16.6 Hz | δ 3.72–3.53 m | δ 0.66 t, J = 7.4 Hz |
| (±) 7a | δ 4.79 d, J = 5.8 Hz | δ 3.67–3.52 m | δ 2.95 dd, 3J = 8.8 Hz, 2J = 16.8 Hz | δ 2.60 dd, 3J = 7.2 Hz, 2J = 16.8 Hz | δ 4.17–3.98 m | δ 1.05 t, J = 7.0 Hz |
| (±) 7b | δ 5.05 d, J = 8.8 Hz | δ 4.16–4.01 m | δ 3.05 dd, 3J = 12.8 Hz, 2J = 16.6 Hz | δ 2.66 dd, 3J = 8.0 Hz, 2J = 16.6 Hz | δ 3.70–3.55 m | δ 0.71 t, J = 7.2 Hz |
| (±) 8a | δ 4.82 d, J = 6.4 Hz | δ 3.70–3.48 m | δ 2.92 dd, 3J = 9.0 Hz, 2J = 16.8 Hz | δ 2.62 dd, 3J = 7.8 Hz, 2J = 16.8 Hz | δ 4.16–3.96 m | δ 1.03 t, J = 7.2 Hz |
| (±) 8b | δ 5.06 d, J = 8.6 Hz | δ 4.15–4.01 m | δ 3.06 dd, 3J = 13.0 Hz, 2J = 16.6 Hz | δ 2.64 dd, 3J = 8.0 Hz, 2J = 16.6 Hz | δ 3.80–3.63 m | δ 0.78 t, J = 7.0 Hz |
| (±) 9a | δ 4.81 d, J = 6.0 Hz | δ 3.65–3.52 m | δ 2.92 dd, 3J = 8.6 Hz, 2J = 16.6 Hz | δ 2,62 dd, 3J = 7.4 Hz, 2J = 16.6 Hz | δ 4.17–3.99 m | δ 1.03 t, J = 7.0 Hz |
| (±) 9b | δ 5.05 d, J = 8.4 Hz | δ 4.16–4.04 m | δ 3.06 dd, 3J = 12.6 Hz, 2J = 16.4 Hz | δ 2.64 dd, 3 J = 8.4 Hz, 2 J = 16.4 Hz | δ 3.80–3.67 m | δ 0.78 t, J = 7.2 Hz |
| (±) 10a | δ 4.83 d, J = 6.2 Hz | δ 3.68–3.48 m | δ 2.94 dd, 3J = 8.8 Hz, 2J = 16.2 Hz | δ 2.66 dd, 3J = 8.0 Hz, 2J = 16.2 Hz | δ 4.14–4.00 m | δ 1.04 t, J = 7.0 Hz |
| (±) 10b | δ 5.07 d, J = 8.6 Hz | δ 4.18–4.02 m | δ 3.10 dd, 3J = 12.4 Hz, 2J = 16.4 Hz | δ 2.65 dd, 3J = 8.4 Hz, 2J = 16.4 Hz | δ 3.72–3.63 m | δ 0.73 t, J = 7.2 Hz |
| (±) 11a | δ 4.83 d, J = 5.8 Hz | δ 3.68–3.52 m | δ 2.96 dd, 3J = 8.6 Hz, 2J = 16.8 Hz | δ 2.62 dd, 3J = 7.4 Hz, 2J = 16.8 Hz | δ 4.15–3.94 m | δ 1.05 t, J = 7.0 Hz |
| (±) 11b | δ 5.08 d, J = 8.6 Hz | δ 4.19–4.02 m | δ 3.08 dd, 3J = 12.2 Hz, 2J = 17.0 Hz | δ 2.66 dd, 3J = 8.6 Hz, 2J = 17.0 Hz | δ 3.69–3.59 m | δ 0.73 t, J = 7.2 Hz |
| Compd | ETAh 10−5 M a | ETBh 10−5 M a |
|---|---|---|
| 12a | NA b | 14 |
| 13a | 16 | NA |
| 14a | 11 | NA |
| 15a | >100 | >100 |
| 31a | NA | NA |
| 31b | NA | NA |
| 31c | NA | NA |
| 31d | NA | NA |
| 31e | NA | NA |
| 31f | NA | NA |
| 31g | NA | NA |
| 31h | 3.3 ± 1.1 c | >100 |
| 31i | NA | NA |
| 31j | NA | NA |
| 31k | NA | NA |
| BQ-123 | 0.0054 ± 0.0008 c | NA |
| BQ-788 | NA | 0.0071 ± 0.0023 c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Intagliata, S.; Helal, M.A.; Materia, L.; Pittalà, V.; Salerno, L.; Marrazzo, A.; Cagnotto, A.; Salmona, M.; Modica, M.N.; Romeo, G. Synthesis and Molecular Modelling Studies of New 1,3-Diaryl-5-Oxo-Proline Derivatives as Endothelin Receptor Ligands. Molecules 2020, 25, 1851. https://doi.org/10.3390/molecules25081851
Intagliata S, Helal MA, Materia L, Pittalà V, Salerno L, Marrazzo A, Cagnotto A, Salmona M, Modica MN, Romeo G. Synthesis and Molecular Modelling Studies of New 1,3-Diaryl-5-Oxo-Proline Derivatives as Endothelin Receptor Ligands. Molecules. 2020; 25(8):1851. https://doi.org/10.3390/molecules25081851
Chicago/Turabian StyleIntagliata, Sebastiano, Mohamed A. Helal, Luisa Materia, Valeria Pittalà, Loredana Salerno, Agostino Marrazzo, Alfredo Cagnotto, Mario Salmona, Maria N. Modica, and Giuseppe Romeo. 2020. "Synthesis and Molecular Modelling Studies of New 1,3-Diaryl-5-Oxo-Proline Derivatives as Endothelin Receptor Ligands" Molecules 25, no. 8: 1851. https://doi.org/10.3390/molecules25081851
APA StyleIntagliata, S., Helal, M. A., Materia, L., Pittalà, V., Salerno, L., Marrazzo, A., Cagnotto, A., Salmona, M., Modica, M. N., & Romeo, G. (2020). Synthesis and Molecular Modelling Studies of New 1,3-Diaryl-5-Oxo-Proline Derivatives as Endothelin Receptor Ligands. Molecules, 25(8), 1851. https://doi.org/10.3390/molecules25081851