
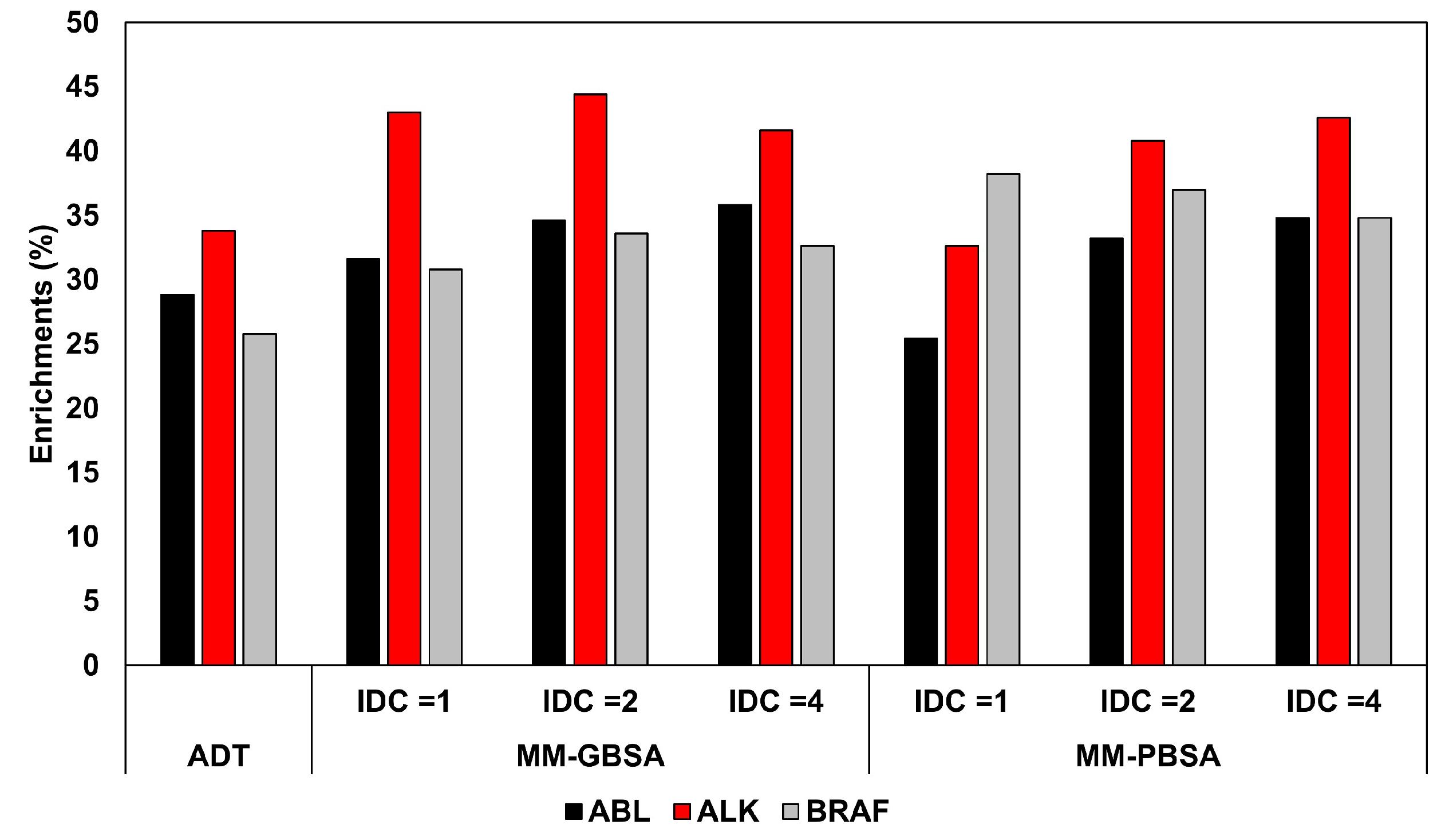
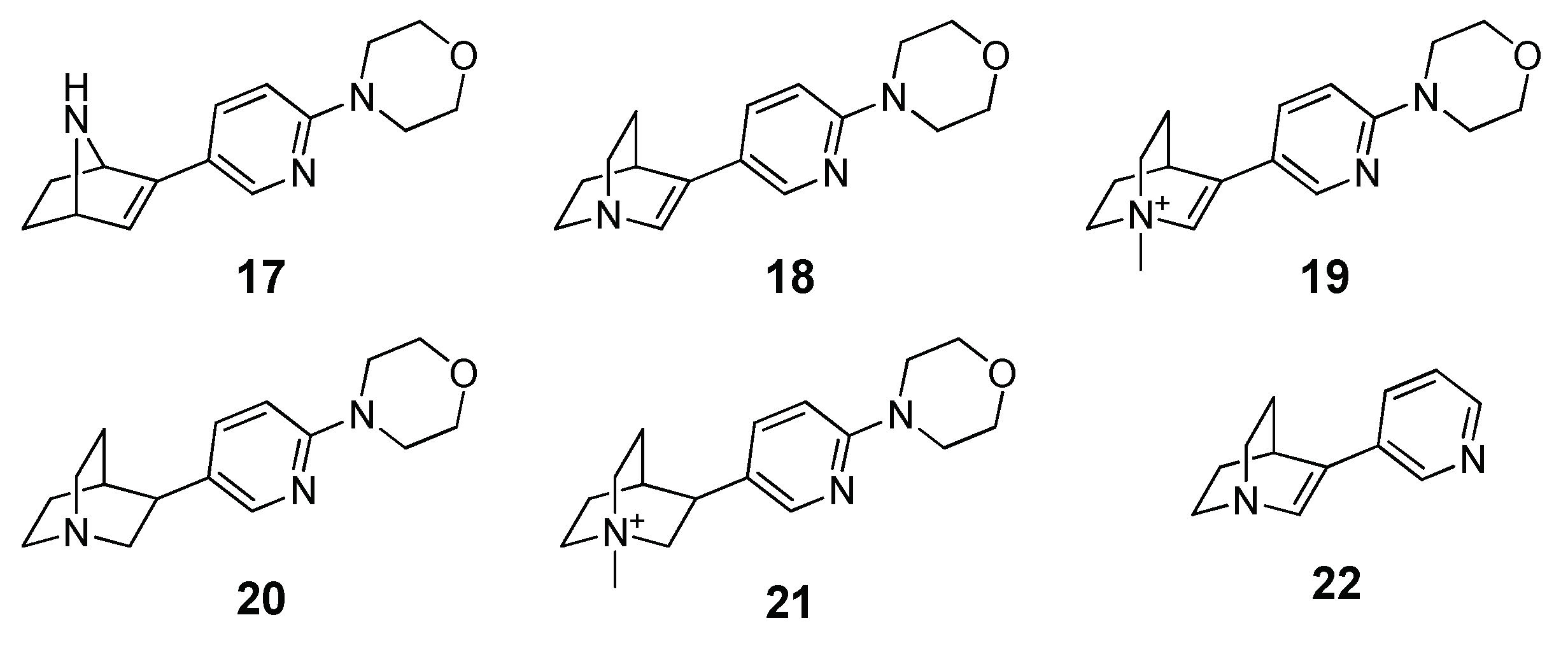
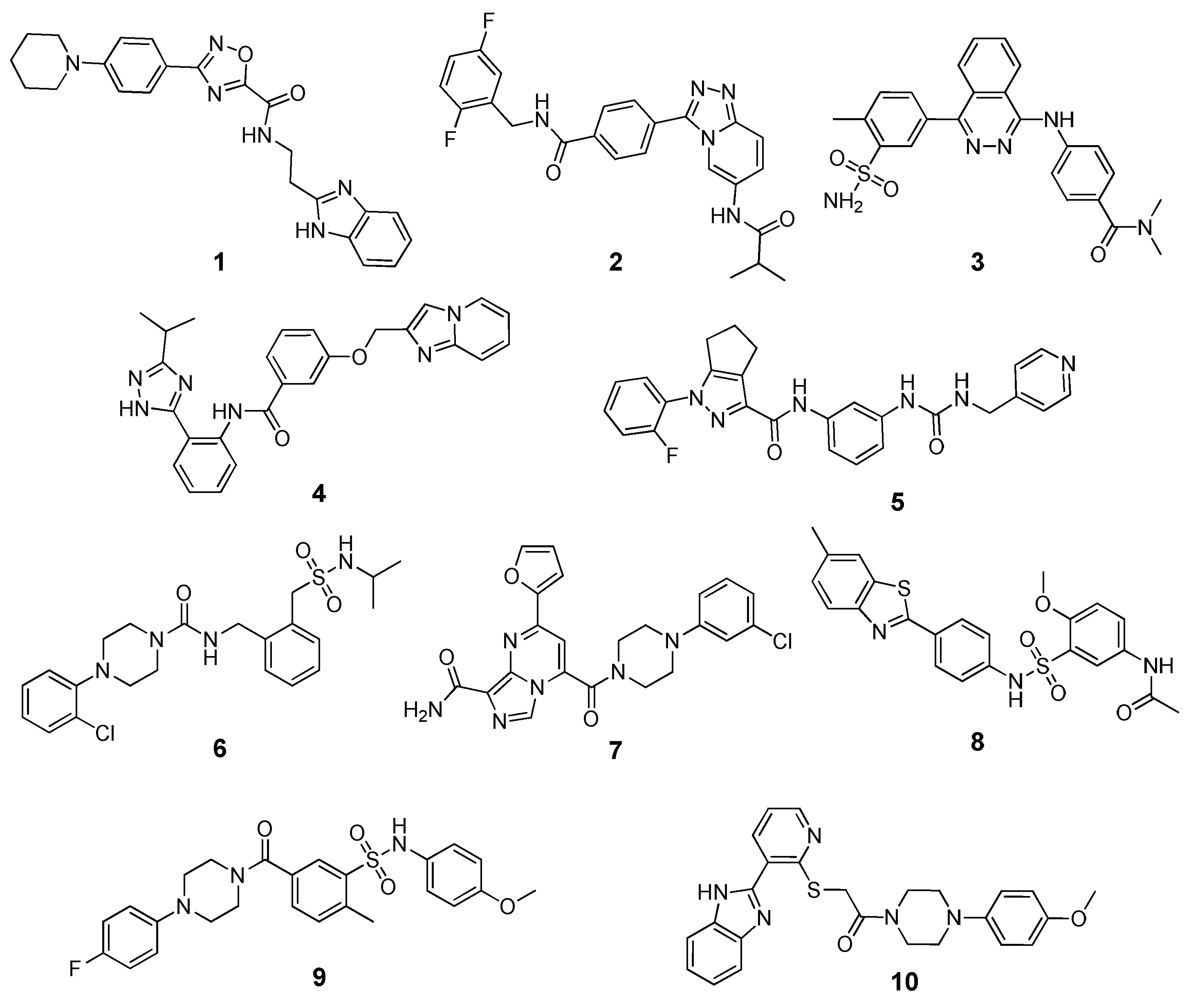
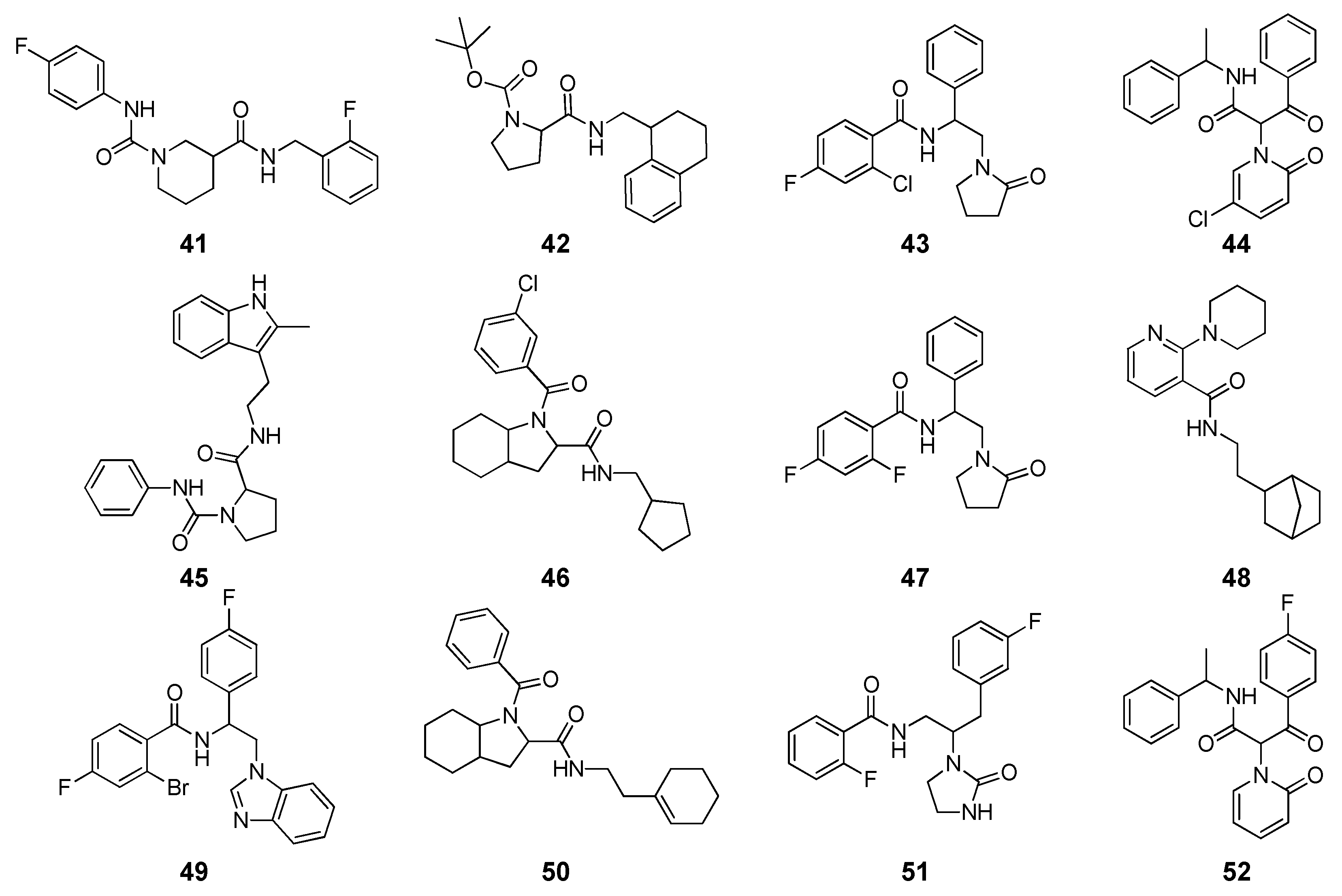
Application of MM-PBSA Methods in Virtual Screening




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. https://doi.org/10.3390/molecules25081971
Poli G, Granchi C, Rizzolio F, Tuccinardi T. Application of MM-PBSA Methods in Virtual Screening. Molecules. 2020; 25(8):1971. https://doi.org/10.3390/molecules25081971
Chicago/Turabian StylePoli, Giulio, Carlotta Granchi, Flavio Rizzolio, and Tiziano Tuccinardi. 2020. "Application of MM-PBSA Methods in Virtual Screening" Molecules 25, no. 8: 1971. https://doi.org/10.3390/molecules25081971
APA StylePoli, G., Granchi, C., Rizzolio, F., & Tuccinardi, T. (2020). Application of MM-PBSA Methods in Virtual Screening. Molecules, 25(8), 1971. https://doi.org/10.3390/molecules25081971