Structure–Activity Relationships and Biological Evaluation of 7-Substituted Harmine Analogs for Human β-Cell Proliferation

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
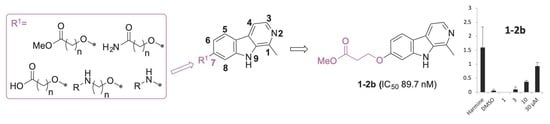
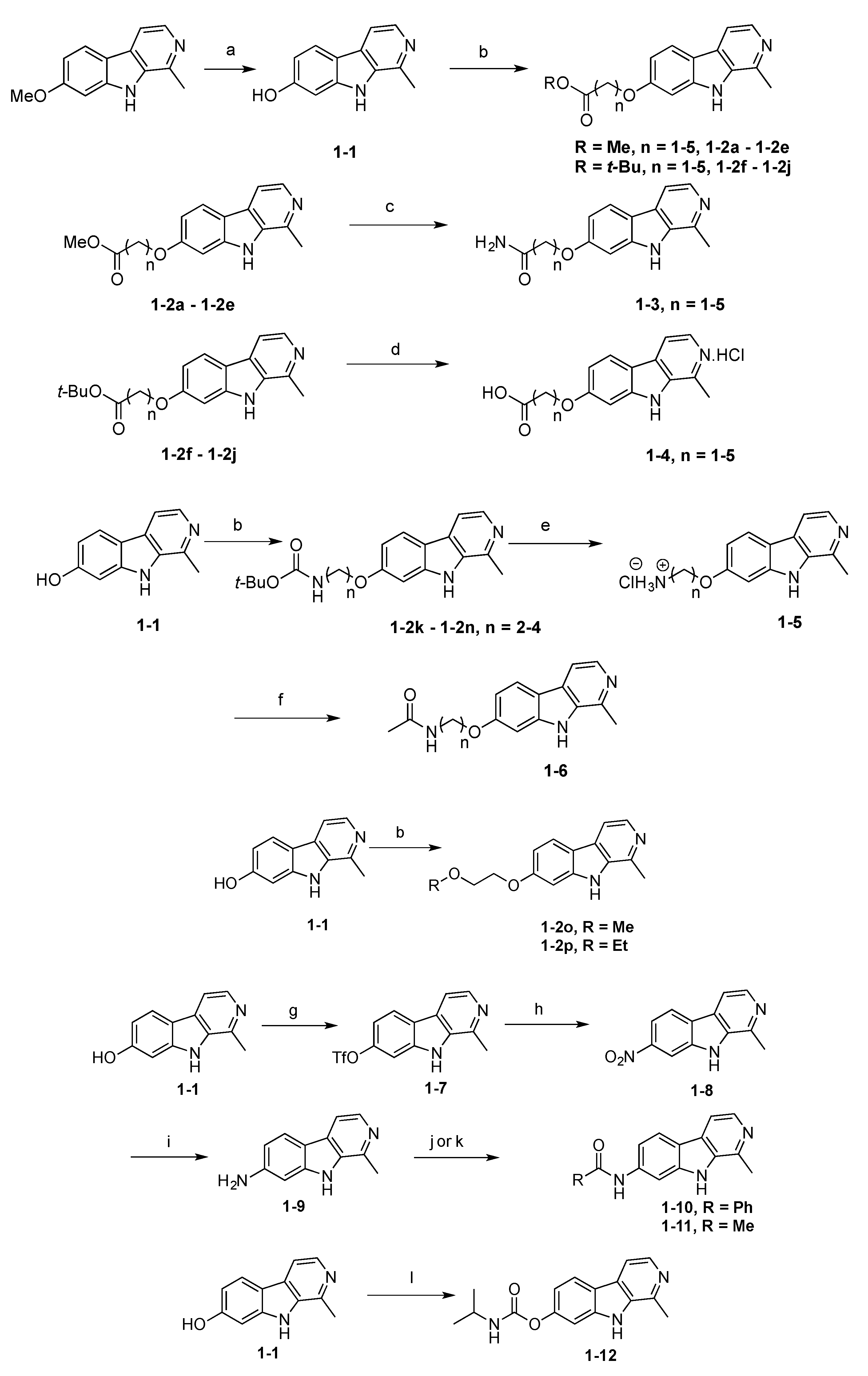
2.1. Synthesis
2.2. Structure–Activity Relationships of 7-Substituted Harmine Analogs
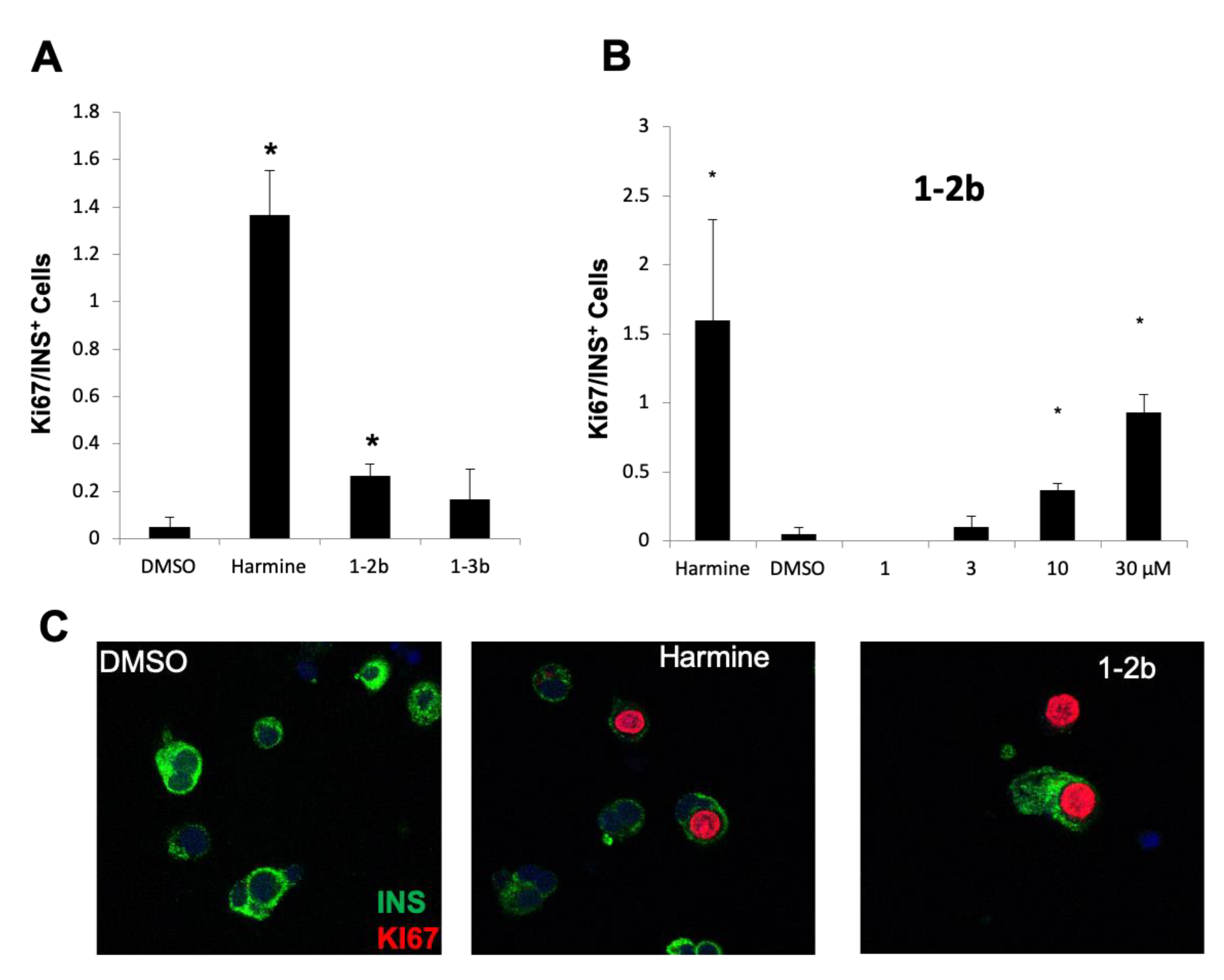
2.3. Effects of Harmine Analogs on Human β-Cell Proliferation
3. Materials and Methods
3.1. Chemistry
3.2. DYRK1A Binding Assays
3.3. β-Cell Proliferation Assay
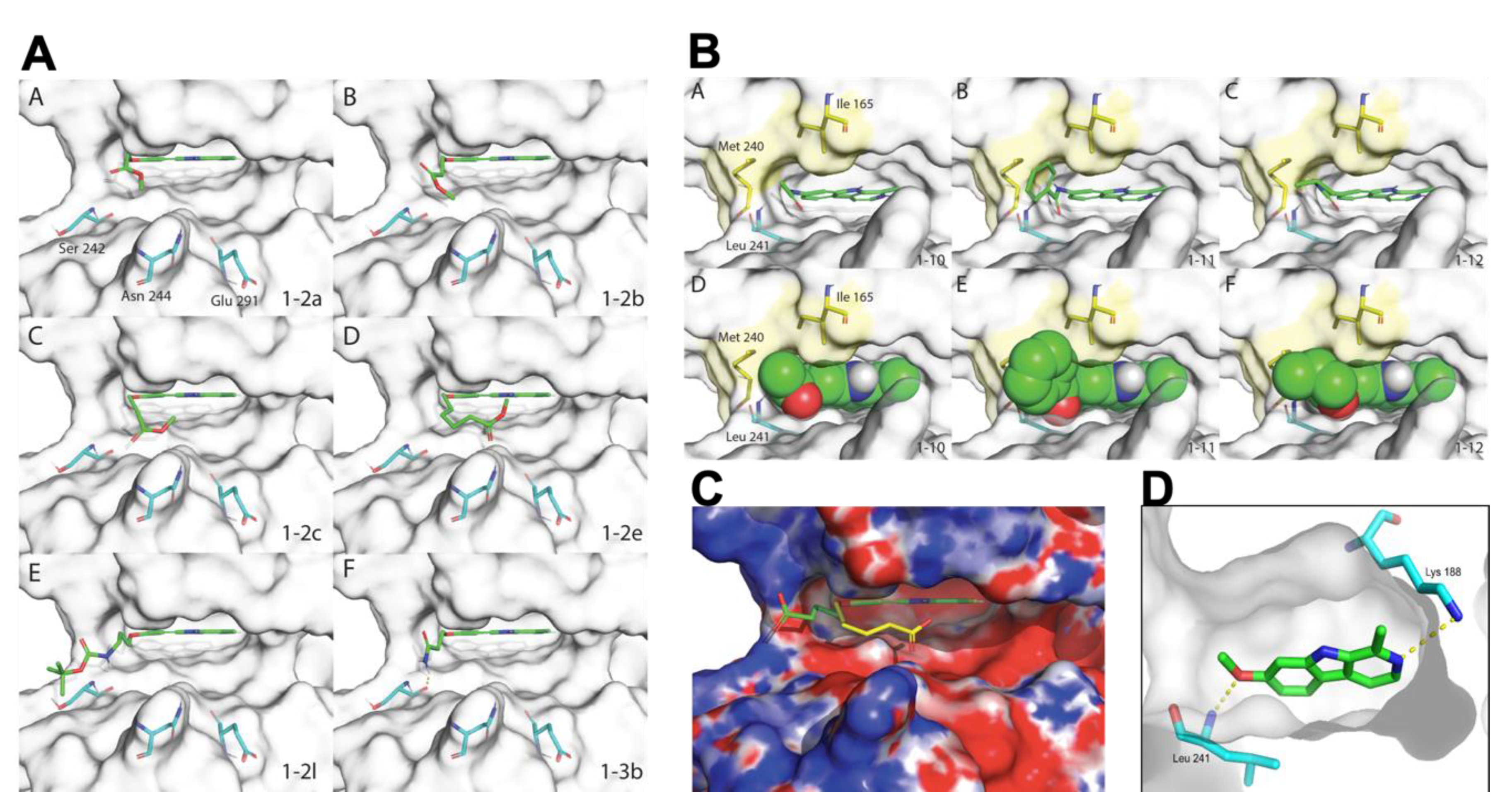
3.4. Docking
3.5. Electrostatic Potential
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wegiel, J.; Gong, C.-X.; Hwang, Y.-W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: A New Avenue for a Disease Modifying Treatment of Alzheimer’s? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ionescu, A.; Dufrasne, F.; Gelbcke, M.; Jabin, I.; Kiss, R.; Lamoral-Theys, D. DYRK1A kinase inhibitors with emphasis on cancer. Mini-Rev. Med. Chem. 2012, 12, 1315–1329. [Google Scholar] [PubMed]
- Fernandez-Martinez, P.; Zahonero, C.; Sanchez-Gomez, P. DYRK1A: The double-edged kinase as a protagonist in cell growth and tumorigenesis. Mol. Cell. Oncol. 2016, 2, e970048. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Alvarez-Perez, J.-C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocana, A.; et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.-Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef] [PubMed]
- Rachdi, L.; Kariyawasam, D.; Aiello, V.; Herault, Y.; Janel, N.; Delabar, J.-M.; Polak, M.; Scharfmann, R. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle 2014, 13, 2221–2229. [Google Scholar] [CrossRef] [Green Version]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dancik, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A stimulates human beta-cell proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef] [Green Version]
- Becker, W.; Soppa, U.; Tejedor, F.J. DYRK1A: A Potential Drug Target for Multiple Down Syndrome Neuropathologies. CNS Neurol. Disord.: Drug Targets 2014, 13, 26–33. [Google Scholar] [CrossRef]
- Becker, W.; Sippl, W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef]
- Ahmadu, A.; Abdulkarim, A.; Grougnet, R.; Myrianthopoulos, V.; Tillequin, F.; Magiatis, P.; Skaltsounis, A.-L. Two new peltogynoids from Acacia nilotica Delile with kinase inhibitory activity. Planta Med. 2010, 76, 458–460. [Google Scholar] [CrossRef]
- Akue-Gedu, R.; Debiton, E.; Ferandin, Y.; Meijer, L.; Prudhomme, M.; Anizon, F.; Moreau, P. Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins. Bioorg. Med. Chem. 2009, 17, 4420–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annes, J.P.; Ryu, J.H.; Lam, K.; Carolan, P.J.; Utz, K.; Hollister-Lock, J.; Arvanites, A.C.; Rubin, L.L.; Weir, G.; Melton, D.A. Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc. Natl. Acad. Sci. USA 2012, 109, 3915–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.-S.; Taverne, T.; Girard, A.; et al. A novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Mazzorana, M.; Papinutto, E.; Bain, J.; Elliott, M.; di Maira, G.; Gianoncelli, A.; Pagano, M.A.; Sarno, S.; Ruzzene, M.; et al. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem. J. 2009, 421, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echalier, A.; Bettayeb, K.; Ferandin, Y.; Lozach, O.; Clelment, M.; Valette, A.; Liger, F.; Marquet, B.; Morris, J.C.; Endicott, J.A.; et al. Meriolins (3-(Pyrimidin-4-yl)-7-azaindoles): Synthesis, Kinase Inhibitory Activity, Cellular Effects, and Structure of a CDK2/Cyclin A/Meriolin Complex. J. Med. Chem. 2008, 51, 737–751. [Google Scholar] [CrossRef]
- Falke, H.; Chaikuad, A.; Becker, A.; Loaec, N.; Lozach, O.; Abu Jhaisha, S.; Becker, W.; Jones, P.G.; Preu, L.; Baumann, K.; et al. 10-Iodo-11H-indolo[3,2-c]quinoline-6-carboxylic Acids Are Selective Inhibitors of DYRK1A. J. Med. Chem. 2015, 58, 3131–3143. [Google Scholar] [CrossRef]
- Foucourt, A.; Hedou, D.; Dubouilh-Benard, C.; Girard, A.; Taverne, T.; Casagrande, A.-S.; Desire, L.; Leblond, B.; Besson, T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, part II. Molecules 2014, 19, 15411–15439. [Google Scholar] [CrossRef] [Green Version]
- Giraud, F.; Alves, G.; Debiton, E.; Nauton, L.; Thery, V.; Durieu, E.; Ferandin, Y.; Lozach, O.; Meijer, L.; Anizon, F.; et al. Synthesis, Protein Kinase Inhibitory Potencies, and in Vitro Antiproliferative Activities of Meridianin Derivatives. J. Med. Chem. 2011, 54, 4474–4489. [Google Scholar] [CrossRef] [PubMed]
- Gourdain, S.; Dairou, J.; Denhez, C.; Bui, L.C.; Rodrigues-Lima, F.; Janel, N.; Delabar, J.M.; Cariou, K.; Dodd, R.H. Development of DANDYs, New 3,5-Diaryl-7-azaindoles Demonstrating Potent DYRK1A Kinase Inhibitory Activity. J. Med. Chem. 2013, 56, 9569–9585. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Sebrie, C.; Rivals, I.; Ledru, A.; Paly, E.; Bizot, J.C.; Smith, D.; Rubin, E.; Gillet, B.; Arbones, M.; et al. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS ONE 2009, 4, e4606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassis, P.; Brzeszcz, J.; Beneteau, V.; Lozach, O.; Meijer, L.; Le Guevel, R.; Guillouzo, C.; Lewinski, K.; Bourg, S.; Colliandre, L.; et al. Synthesis and biological evaluation of new 3-(6-hydroxyindol-2-yl)-5-(Phenyl) pyridine or pyrazine V-Shaped molecules as kinase inhibitors and cytotoxic agents. Eur. J. Med. Chem. 2011, 46, 5416–5434. [Google Scholar] [CrossRef]
- Kii, I.; Sumida, Y.; Goto, T.; Sonamoto, R.; Okuno, Y.; Yoshida, S.; Kato-Sumida, T.; Koike, Y.; Abe, M.; Nonaka, Y.; et al. Selective inhibition of the kinase DYRK1A by targeting its folding process. Nat. Commun. 2016, 7, 11391. [Google Scholar] [CrossRef]
- Kim, N.D.; Yoon, J.; Kim, J.H.; Lee, J.T.; Chon, Y.S.; Hwang, M.-K.; Ha, I.; Song, W.-J. Putative therapeutic agents for the learning and memory deficits of people with Down syndrome. Bioorg. Med. Chem. Lett. 2006, 16, 3772–3776. [Google Scholar] [CrossRef]
- Koo, K.A.; Kim, N.D.; Chon, Y.S.; Jung, M.-S.; Lee, B.-J.; Kim, J.H.; Song, W.-J. QSAR analysis of pyrazolidine-3,5-diones derivatives as Dyrk1A inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2324–2328. [Google Scholar] [CrossRef]
- Naert, G.; Ferre, V.; Meunier, J.; Keller, E.; Malmstrom, S.; Givalois, L.; Carreaux, F.; Bazureau, J.-P.; Maurice, T. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Aβ25-35 peptide administration in mice. Eur. Neuropsychopharmacol. 2015, 25, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Neagoie, C.; Vedrenne, E.; Buron, F.; Merour, J.-Y.; Rosca, S.; Bourg, S.; Lozach, O.; Meijer, L.; Baldeyrou, B.; Lansiaux, A.; et al. Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: A novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012, 49, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, A.S.; Tanega, C.; Shen, M.; Mott, B.T.; Bougie, J.M.; Nguyen, D.-T.; Misteli, T.; Auld, D.S.; Maloney, D.J.; Thomas, C.J. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg. Med. Chem. Lett. 2011, 21, 3152–3158. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Salas, A.P.; Brana, A.F.; Palomino, M.; Pineda-Lucena, A.; Carbajo, R.J.; Mendez, C.; Moris, F.; Salas, J.A. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem. Commun. 2009, 27, 4118–4120. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Mazzorana, M.; Traynor, R.; Ruzzene, M.; Cozza, G.; Pagano, M.A.; Meggio, F.; Zagotto, G.; Battistutta, R.; Pinna, L.A. Structural features underlying the selectivity of the kinase inhibitors NBC and dNBC: Role of a nitro group that discriminates between CK2 and DYRK1A. Cell. Mol. Life Sci. 2012, 69, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, Cocrystal Structures, and Neuroprotective Properties of Leucettines, a Family of Protein Kinase Inhibitors Derived from the Marine Sponge Alkaloid Leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Abdolazimi, Y.; Lee, S.; Xu, H.; Allegretti, P.; Horton, T.M.; Yeh, B.; Moeller, H.P.; McCutcheon, D.; Armstrong, N.A.; Annes, J.P.; et al. CC-401 Promotes β-Cell Replication via Pleiotropic Consequences of DYRK1A/B Inhibition. Endocrinology 2018, 59, 3143–3157. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Man-Un Ung, P.; Wang, P.; Wang, H.; Li, H.; Andrews, M.K.; Stewart, A.F.; Schlessinger, A.; DeVita, R.J. Novel selective thiadiazine DYRK1A inhibitor lead scaffold with human pancreatic β-cell proliferation activity. Eur. J. Med. Chem. 2018, 157, 1005–1016. [Google Scholar] [CrossRef]
- Aamodt, K.I.; Powers, A.C.; Aramandla, R.; Brissova, M.; Powers, A.C.; Brown, J.J.; Fiaschi-Taesch, N.; Wang, P.; Stewart, A.F.; Powers, A.C. Development of a reliable automated screening system to identify small molecules and biologics that promote human β-cell regeneration. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E859–E868. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.A.; Jin, Q.; Zou, Y.; Ding, Q.; Yan, S.; Wang, Z.; Hao, X.; Nguyen, B.; Zhang, X.; Pan, J.; et al. Selective DYRK1A Inhibitor for the Treatment of Type 1 Diabetes: Discovery of 6-Azaindole Derivative GNF2133. J. Med. Chem. 2020, 63, 2958–2973. [Google Scholar] [CrossRef]
- Kumar, K.; Wang, P.; Sanchez, R.; Swartz, E.A.; Stewart, A.F.; DeVita, R.J. Development of Kinase-Selective, Harmine-Based DYRK1A Inhibitors that Induce Pancreatic Human β-Cell Proliferation. J. Med. Chem. 2018, 61, 7687–7699. [Google Scholar] [CrossRef]
- Kumar, K.W.P.; Wilson, J.; Zlatanic, V.; Berrouet, C.; Khamrui, S.; Secor, C.; Swartz, E.A.; Lazarus, M.; Sanchez, R.; et al. Synthesis and Biological Validation of a Harmine-based, Central Nervous System (CNS)-Avoidant, Selective, Human β-Cell Regenerative Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase A (DYRK1A) Inhibitor. J. Med. Chem. 2020, 63, 2986–3003. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Mueller, T.D.; Yi, C.-X.; Habegger, K.; Schriever, S.C.; Garcia-Caceres, C.; Kabra, D.G.; et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Tiano, J.P.; Tate, C.R.; Yang, B.S.; DiMarchi, R.; Mauvais-Jarvis, F. Effect of targeted estrogen delivery using glucagon-like peptide-1 on insulin secretion, insulin sensitivity and glucose homeostasis. Sci. Rep. 2015, 5, 10211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LanthaScreen® Eu Kinase Binding Assay-Customer Protocol. Available online: https://www.thermofisher.com/us/en/home/industrial/pharma-biopharma/drug-discovery-development/target-and-lead-identification-and-validation/kinasebiology/kinase-activity-assays/lanthascreentm-eu-kinase-binding-assay.html (accessed on 21 April 2020).
- Wang, P.; Karakose, E.; Liu, H.; Swartz, E.; Ackeifi, C.; Zlatanic, V.; Wilson, J.; Gonzalez, B.J.; Bender, A.; Takane, K.K.; et al. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 2019, 29, 638–652.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackeifi, C.; Wang, P.; Karakose, E.; Manning Fox, J.E.; González, B.J.; Liu, H.; Wilson, J.; Swartz, E.; Berrouet, C.; Li, Y.; et al. GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human β cell regeneration. Sci. Transl. Med. 2020, 12, eaaw9996. [Google Scholar] [CrossRef]
- Ackeifi, C.; Swartz, E.; Kumar, K.; Liu, H.; Chalada, S.; Karakose, E.; Scott, D.K.; Garcia-Ocaña, A.; Sanchez, R.; DeVita, R.J.; et al. Pharmacologic and genetic approaches define human pancreatic β cell mitogenic targets of DYRK1A inhibitors. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Synthesis and evaluation of β-carboline derivatives as potential monoamine oxidase inhibitors. Bioorg. Med. Chem. 2011, 19, 134–144. [Google Scholar] [CrossRef]
- Fors, B.P.; Buchwald, S.L. Pd-Catalyzed Conversion of Aryl Chlorides, Triflates, and Nonaflates to Nitroaromatics. J. Amer. Chem. Soc. 2009, 131, 12898–12899. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Liu, Y.; Liu, Y.; Wang, L.; Wang, Q. Synthesis and Antiviral and Fungicidal Activity Evaluation of β-Carboline, Dihydro-β-carboline, Tetrahydro-β-carboline Alkaloids, and Their Derivatives. J. Agric. Food Chem. 2014, 62, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Badura, L.; Swanson, T.; Adamowicz, W.; Adams, J.; Cianfrogna, J.; Fisher, K.; Holland, J.; Kleiman, R.; Nelson, F.; Reynolds, L.; et al. An inhibitor of casein kinase Iε induces phase delays in circadian rhythms under free-running and entrained conditions. J. Pharmacol. Exp. Ther. 2007, 322, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.; Huang, Y.; Hanson, R.M.; Tobias, M.; Krishnan, S.; Li, W.W.; Nielsen, J.E.; Baker, N.A. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J. Comput. Chem. 2011, 32, 1488–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |

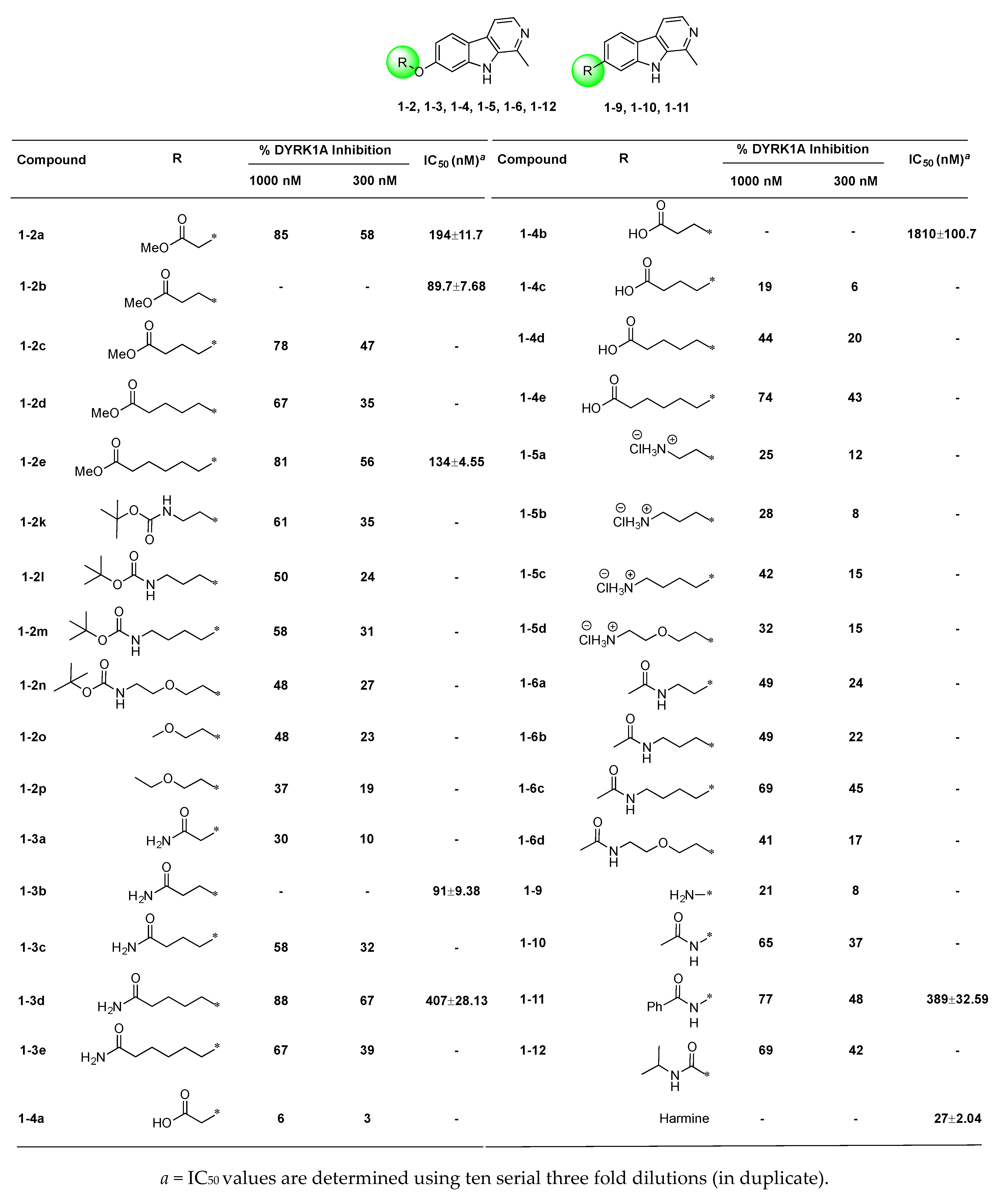
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, K.; Wang, P.; A. Swartz, E.; Khamrui, S.; Secor, C.; B. Lazarus, M.; Sanchez, R.; F. Stewart, A.; DeVita, R.J. Structure–Activity Relationships and Biological Evaluation of 7-Substituted Harmine Analogs for Human β-Cell Proliferation. Molecules 2020, 25, 1983. https://doi.org/10.3390/molecules25081983
Kumar K, Wang P, A. Swartz E, Khamrui S, Secor C, B. Lazarus M, Sanchez R, F. Stewart A, DeVita RJ. Structure–Activity Relationships and Biological Evaluation of 7-Substituted Harmine Analogs for Human β-Cell Proliferation. Molecules. 2020; 25(8):1983. https://doi.org/10.3390/molecules25081983
Chicago/Turabian StyleKumar, Kunal, Peng Wang, Ethan A. Swartz, Susmita Khamrui, Cody Secor, Michael B. Lazarus, Roberto Sanchez, Andrew F. Stewart, and Robert J. DeVita. 2020. "Structure–Activity Relationships and Biological Evaluation of 7-Substituted Harmine Analogs for Human β-Cell Proliferation" Molecules 25, no. 8: 1983. https://doi.org/10.3390/molecules25081983