
Mechanical and Thermal Properties of Polyether Polytriazole Elastomers Formed by Click-Chemical Reaction Curing Glycidyl Azide Polymer


Abstract
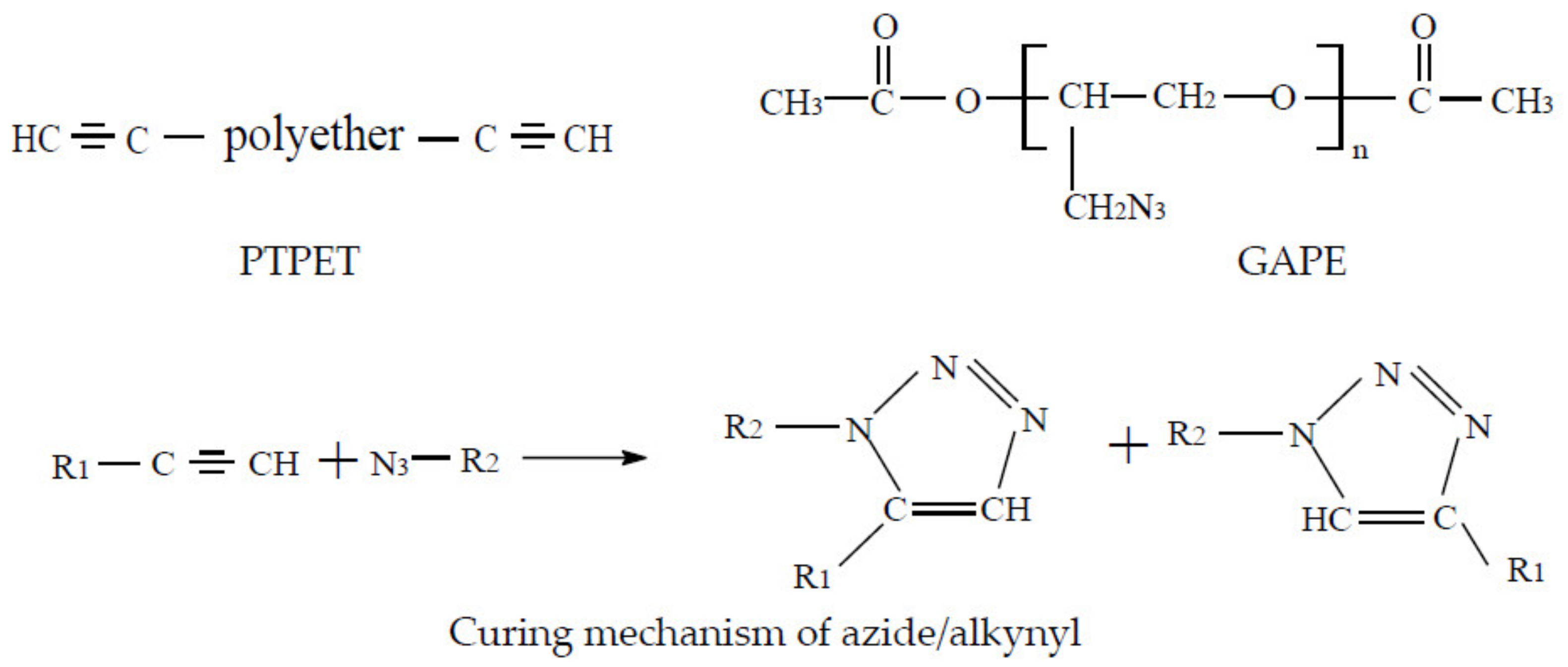
1. Introduction
2. Results and Discussion
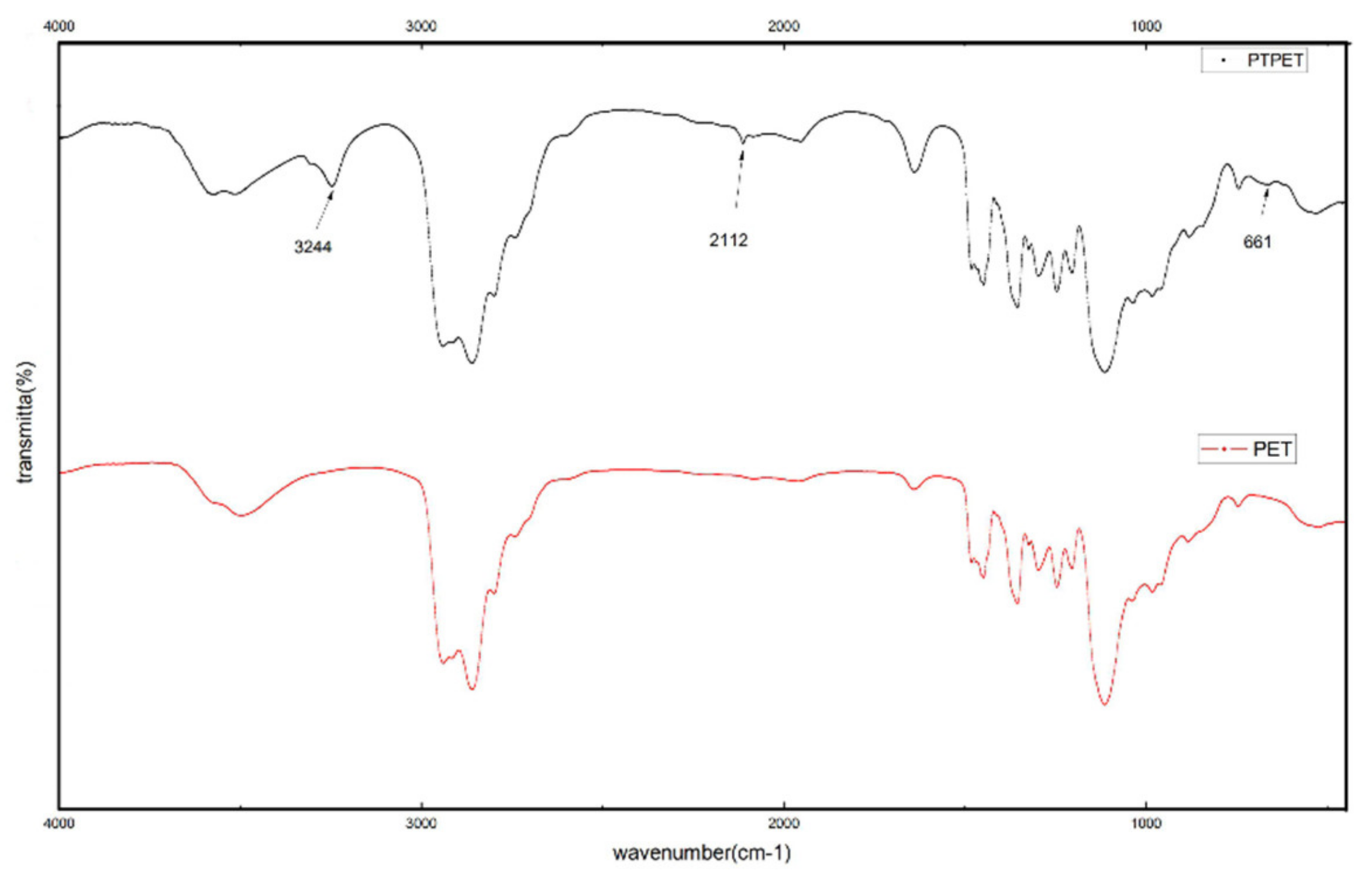
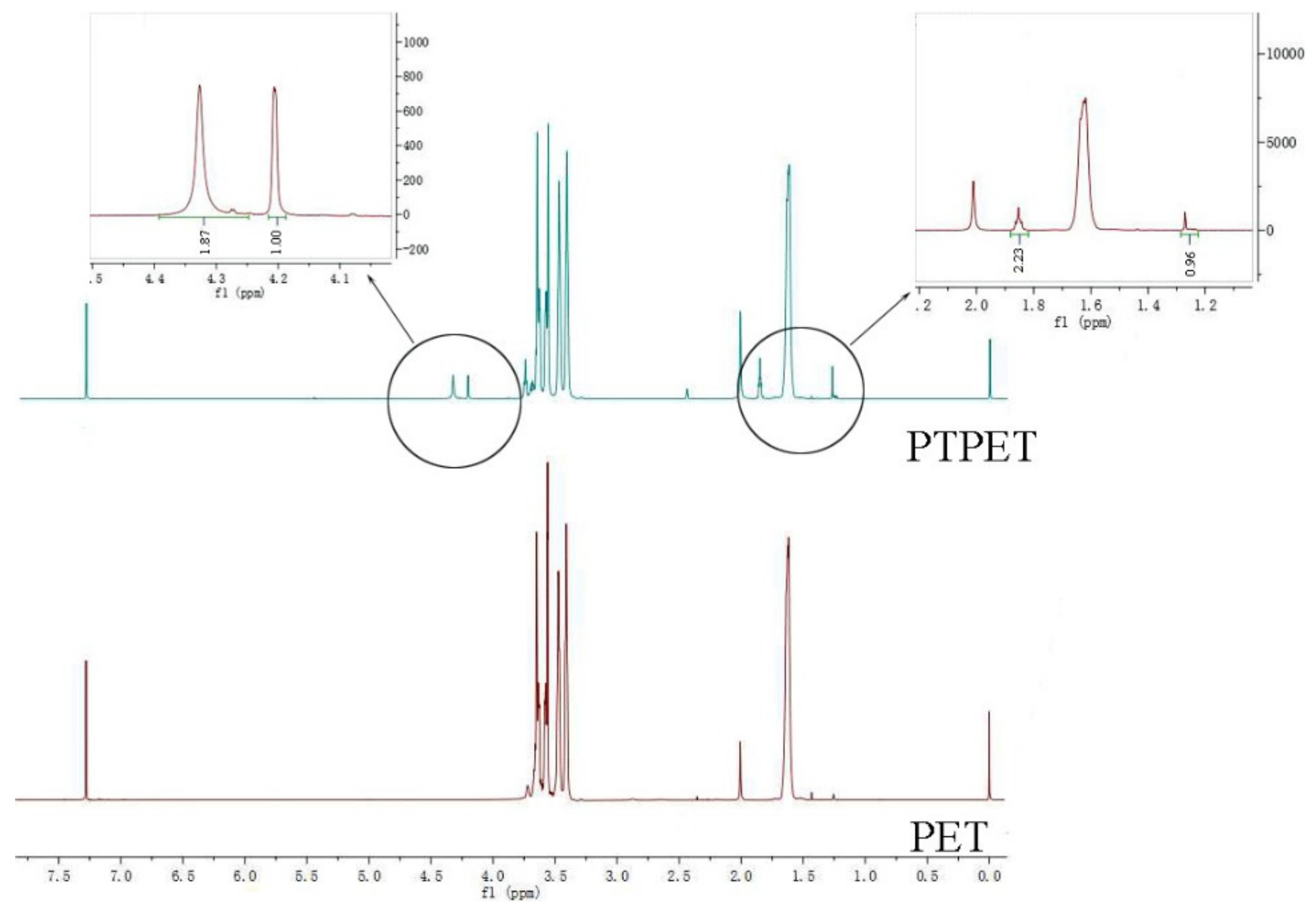
2.1. Structural Characterisation of PTPET
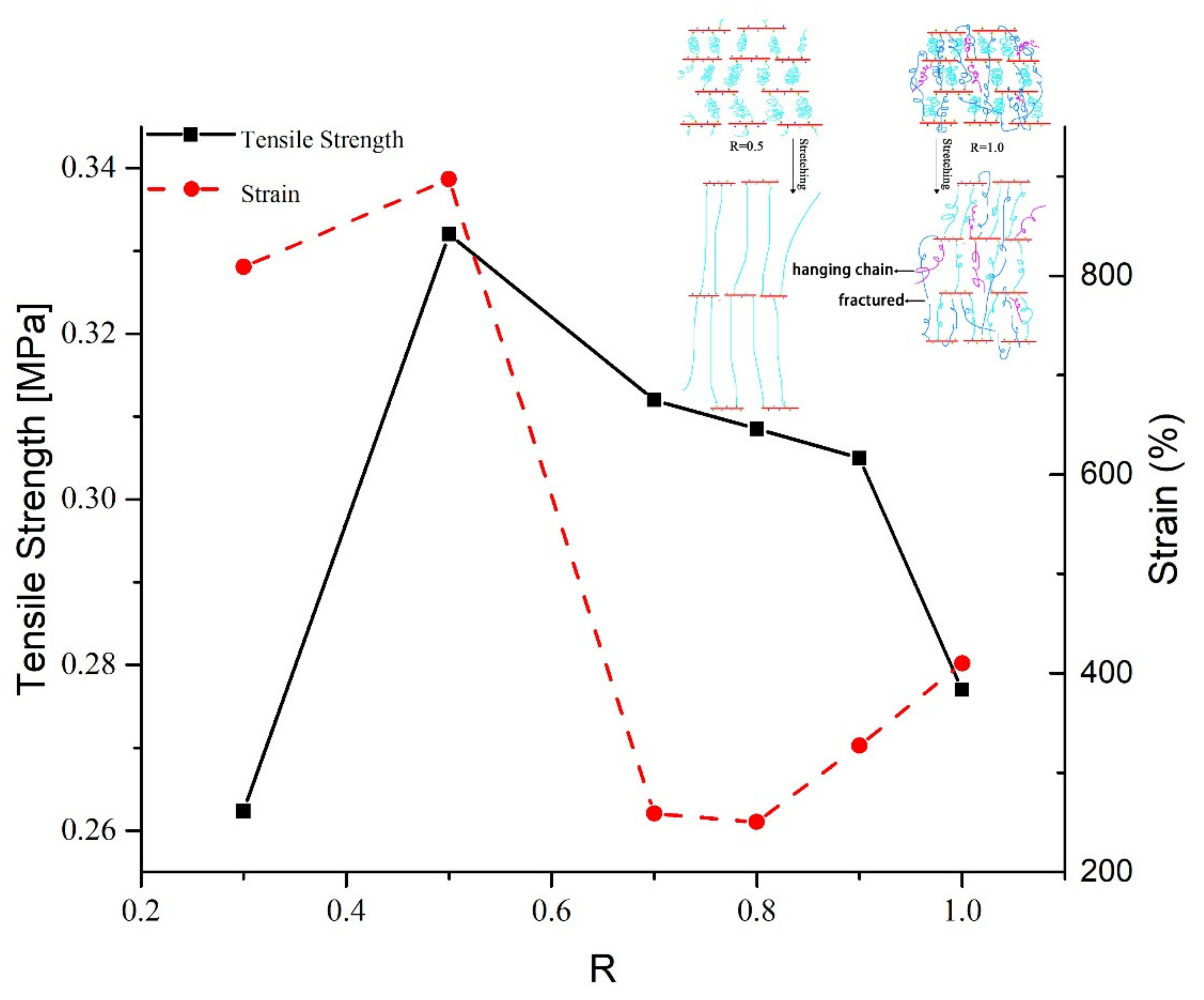
2.2. Mechanical Properties
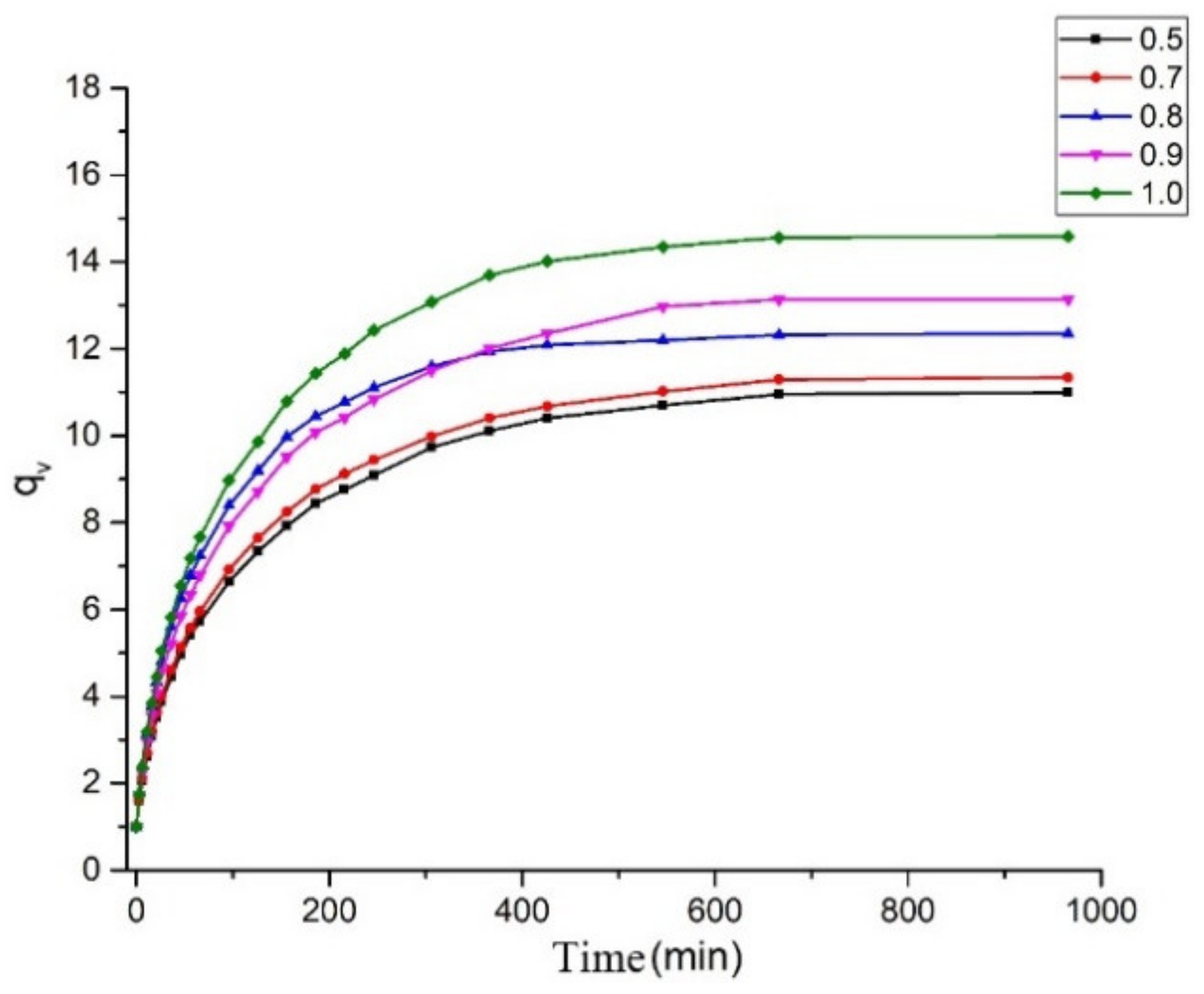
2.3. Network Structure
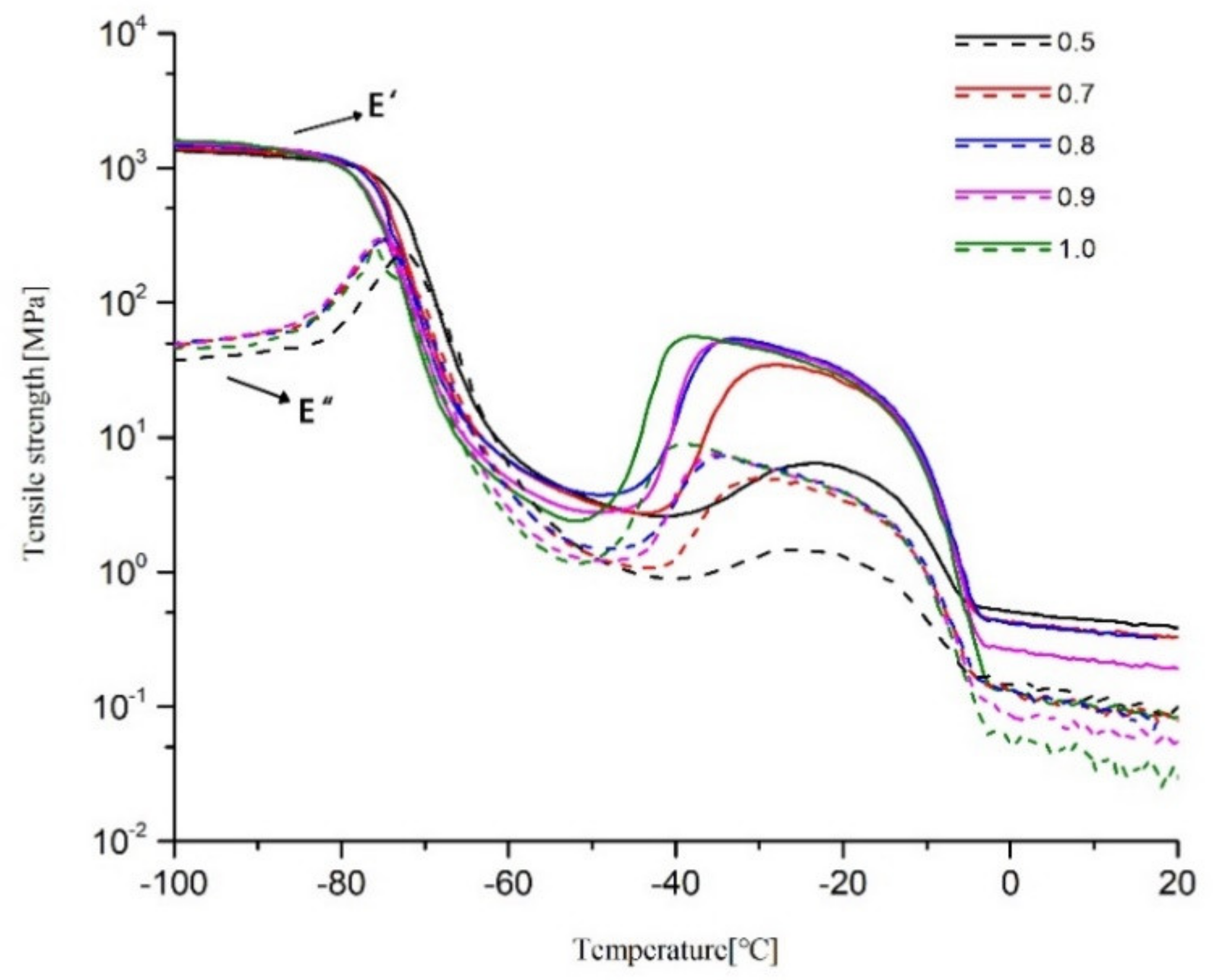
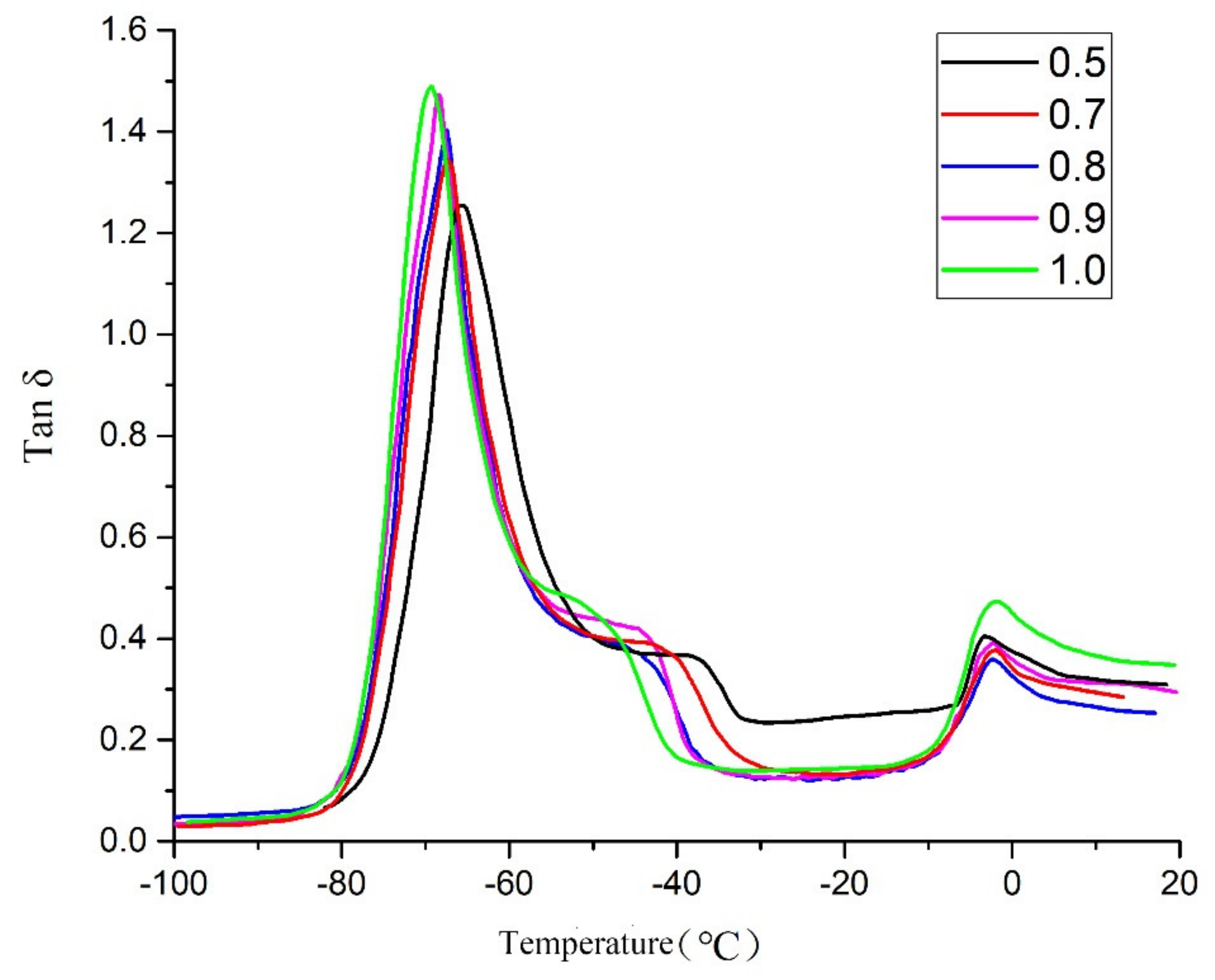
2.4. DMA
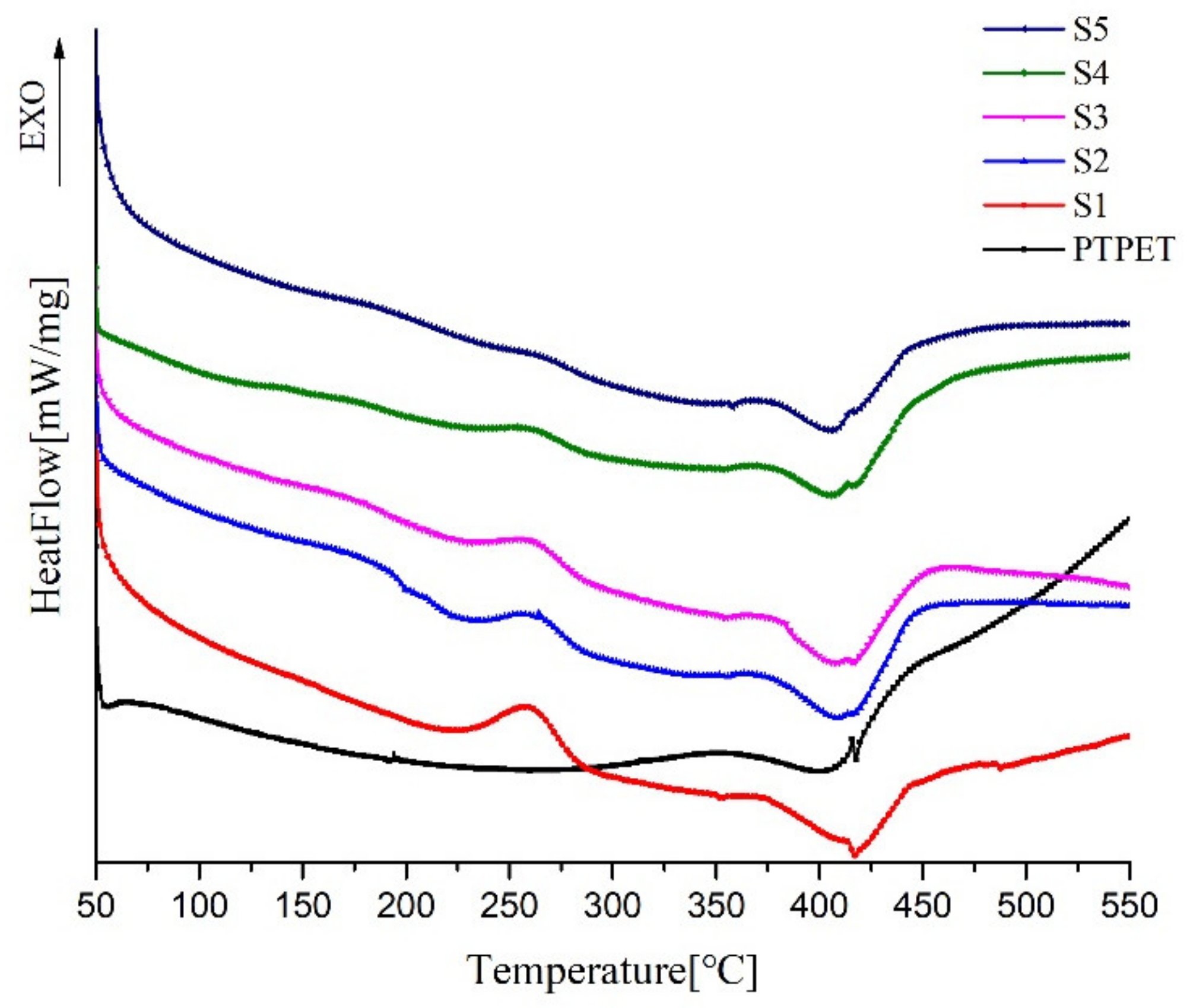
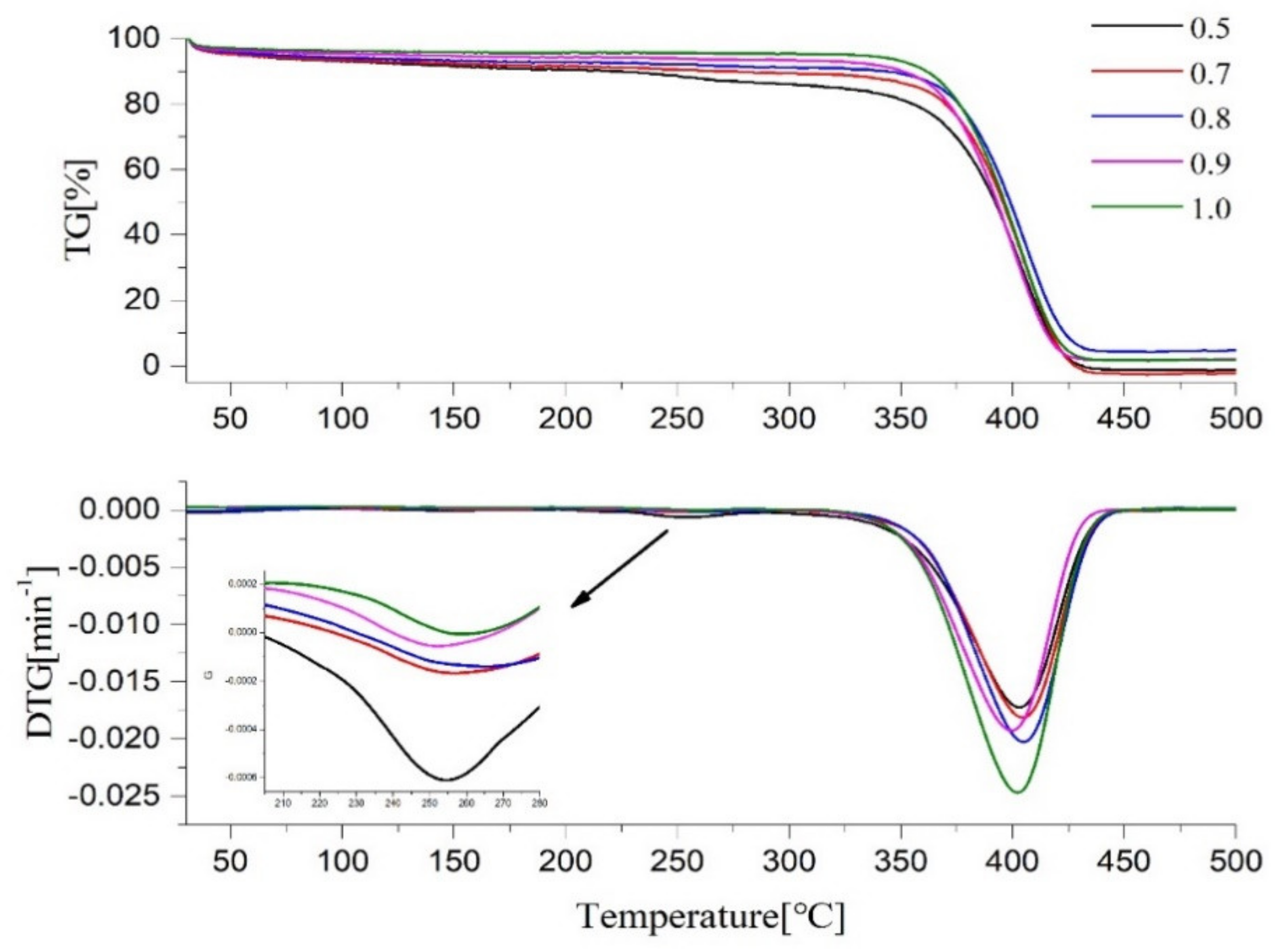
2.5. Thermal Performance
3. Experimental Work
3.1. Materials
3.2. Instrument and Analysis
3.3. Swelling Test
3.4. Preparation of PTPET
3.5. Curing of PTPET with GAP
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Q.; Liu, N.; Mo, H.; Lu, X.; Wang, Y.; Xu, M.; Shu, Y. Facile Preparation and Properties of Crosslinked Copolyether Elastomers with 1,2,3-Triazole and Urethane Subunit via Click Polymerization. ChemistryOpen 2019, 8, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Reshmi, S.K.; Vijayalakshmi, K.P.; Thomas, D.; Arunan, E.; Reghunadhan Nair, C.P. Glycidyl Azide Polymer Crosslinked Through Triazoles by Click Chemistry: Curing, Mechanical and Thermal Properties. Propell. Explos. Pyrot. 2013, 38, 525–532. [Google Scholar] [CrossRef]
- Liu, E.-C.; Topczewski, J.J. Enantioselective Copper Catalyzed Alkyne-Azide Cycloaddition by Dynamic Kinetic Resolution. J. Am. Chem. Soc. 2019, 141, 5135–5138. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.Y.; Zhai, J.X. Synthesis and curing of terminal alkynyl polyethylene glycol. Chin. J. Explos. Propell. 2016, 33, 61–64. [Google Scholar]
- Guan, X.; Li, J.M. Study on the mechanical properties of GAP propellant by azido-alkynyl click chemistry. Chem. Propell. Polym. Mater. 2012, 10, 75–78. [Google Scholar]
- Gong, L.; Yang, R.J. Thermal degradation of polyether polytriazole and polyether polyurethane elastomer. Chin. J. Energetic Materia 2018, 26, 565–571. [Google Scholar]
- Zhu, L.; Zhang, H.; Wang, C.; Chen, Z. Recent Progress in the Synthesis of N2-Substituted 1, 2, 3-Triazoles. Chin. J. Org. Chem. 2018, 38, 1052–1064. [Google Scholar] [CrossRef]
- Qi, Y.X. Research progress in modification of hydroxyl terminated polybutadiene. New Chem. Mater. 2014, 42, 1–9. [Google Scholar]
- Fu, X.-L.; Fan, X.-Z. Curing reaction kinetics of HTPE polymer studied by simultaneous rheometry and FTIR measurements. J. Therm. Anal. Calorim. 2016, 125, 977–982. [Google Scholar] [CrossRef]
- Lemos, M.F.; Bohn, M.A. DMA of polyester-based polyurethane elastomers for composite rocket propellants containing different energetic plasticizers. J. Therm. Anal. Calorim. 2016, 131, 595–600. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Edit. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Hagen, T.H.; Jensen, T.L.; Unneberg, E.; Stenstrøm, Y.H.; Kristensen, T.E. Curing of Glycidyl Azide Polymer (GAP) Diol Using Isocyanate, Isocyanate-Free, Synchronous Dual, and Sequential Dual Curing Systems. Propell. Explos. Pyrot. 2014, 40, 275–284. [Google Scholar] [CrossRef]
- Qu, Z.; Zhai, J.; Yang, R. Comparison between properties of polyether polytriazole elastomers and polyether polyurethane elastomers. Polym. Adv. Technol. 2014, 25, 314–321. [Google Scholar] [CrossRef]
- Reed, R., Jr. Triazole cross-linked polymers. U.S. Patent 6,103,029, 23 June 1997. [Google Scholar]
- Li, X.; Bai, G.; Chen, L. Study on curing reaction kinetics of propargyl triazine with GAP. Chem. Propell. Polym. Mater. 2014, 12, 36–39. [Google Scholar]
- Frankel, M.B.; Grant, L.R.; Flanagan, J.E. Historical development of glycidyl azide polymer. J. Propuls. Power 1992, 8, 560–563. [Google Scholar] [CrossRef]
- Keicher, T.; Kuglstatter, W.; Eisele, S.; Wetzel, T.; Krause, H. Isocyanate-Free Curing of Glycidyl Azide Polymer (GAP) with Bis-Propargyl-Succinate (II). Propell. Explos. Pyrot. 2009, 34, 210–217. [Google Scholar] [CrossRef]
- Xu, Z.; Du, L.; Wan, L.; Huang, F. Novel Functional Polytriazole Elastomer with Thermal Stability and Adjustable Mechanical Properties. Soft Mater. 2013, 11, 353–362. [Google Scholar] [CrossRef]
- Lee, D.-H.; Kim, K.T.; Jang, Y.; Lee, S.; Jeon, H.B.; Paik, H.; Kim, W. 1,2,3-triazole crosslinked polymers as binders for solid rocket propellants. J. Appl. Polym. Sci. 2004, 131, 40594. [Google Scholar] [CrossRef]
- Urayama, K.; Kawamura, T.; Kohjiya, S. Structure–mechanical property correlations of model siloxane elastomers with controlled network topology. Polymer 2009, 50, 347–356. [Google Scholar] [CrossRef]
- Van Krevelen, D.W.; Te Nijenhuis, K. Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Wunderlich, B. Theory of Cold Crystallization of High Polymers. J. Chem. Phys. 1958, 29, 1395–1404. [Google Scholar] [CrossRef]
- Stephen, R.; Joseph, K.; Oommen, Z.; Thomas, S. Molecular transport of aromatic solvents through microcomposites of natural rubber (NR), carboxylated styrene butadiene rubber (XSBR) and their blends. Compos. Sci. Technol. 2007, 67, 1187–1194. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.Q. Epoxy resin modified polytriazole adhesive. J. Mater. Sci. Eng. 2018, 36, 0788–0794. [Google Scholar]
- Kweon, J.-O.; Lee, Y.-K.; Noh, S.-T. Synthesis and thermal behavior of poly (ethylene oxide) poly (N-substituted urethane). J. Polym. Sci. Pol. Chem. 2001, 39, 4129–4138. [Google Scholar] [CrossRef]
Sample Availability: Samples of all tested compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | R Value | Tensile Strength, σb/MPa | Elongation, εb/% |
|---|---|---|---|
| S0 | 0.3 | 0.262 | 808.9 |
| S1 | 0.5 | 0.332 | 897.1 |
| S2 | 0.7 | 0.312 | 258.7 |
| S3 | 0.8 | 0.3085 | 250.3 |
| S4 | 0.9 | 0.305 | 327.1 |
| S5 | 1.0 | 0.277 | 410.2 |
| Sample. | R Value | χ1 | Qv | V2m | ρ g · cm−3 | Mc g · mol−1 | N0 mmol · cm−3 |
|---|---|---|---|---|---|---|---|
| S1 | 0.5 | 0.383 | 10.996 | 0.091 | 1.029 | 7201.1 | 0.143 |
| S2 | 0.7 | 11.340 | 0.088 | 1.091 | 7925.1 | 0.138 | |
| S3 | 0.8 | 12.348 | 0.081 | 1.048 | 8449.7 | 0.124 | |
| S4 | 0.9 | 13.136 | 0.076 | 1.109 | 9630.4 | 0.115 | |
| S5 | 1.0 | 14.581 | 0.069 | 1.059 | 10416.5 | 0.102 |
| Samples | 1st Peak/℃ | 2nd Peak/℃ | 3rd Peak/℃ |
|---|---|---|---|
| PTPET | -- | -- | 416.0 |
| S1 | 258.4 | 352.7 | 414.0 |
| S2 | 258.8 | 355.4 | 413.7 |
| S3 | 259.2 | 354.0 | 414.0 |
| S4 | 259.4 | 353.1 | 414.0 |
| S5 | 260.5 | 358.0 | 414.8 |
| Sample | Elastomers | GAP | ||||
|---|---|---|---|---|---|---|
| R = 0.3 | R = 0.5 | R = 0.7 | R = 0.8 | R = 1.0 | ||
| Azide content (by weight) | 4.0% | 1.9% | 0.9% | 0.5% | 0 | 37.0% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Zhou, J.; Wang, Y.; Ma, Z.; Chen, C. Mechanical and Thermal Properties of Polyether Polytriazole Elastomers Formed by Click-Chemical Reaction Curing Glycidyl Azide Polymer. Molecules 2020, 25, 1988. https://doi.org/10.3390/molecules25081988
He L, Zhou J, Wang Y, Ma Z, Chen C. Mechanical and Thermal Properties of Polyether Polytriazole Elastomers Formed by Click-Chemical Reaction Curing Glycidyl Azide Polymer. Molecules. 2020; 25(8):1988. https://doi.org/10.3390/molecules25081988
Chicago/Turabian StyleHe, Liming, Jun Zhou, Yutao Wang, Zhongliang Ma, and Chunlin Chen. 2020. "Mechanical and Thermal Properties of Polyether Polytriazole Elastomers Formed by Click-Chemical Reaction Curing Glycidyl Azide Polymer" Molecules 25, no. 8: 1988. https://doi.org/10.3390/molecules25081988
APA StyleHe, L., Zhou, J., Wang, Y., Ma, Z., & Chen, C. (2020). Mechanical and Thermal Properties of Polyether Polytriazole Elastomers Formed by Click-Chemical Reaction Curing Glycidyl Azide Polymer. Molecules, 25(8), 1988. https://doi.org/10.3390/molecules25081988